


Abstract
Background: The major factor limiting the efficacy of breast cancer chemotherapy is multidrug resistance due to overexpression of the breast cancer resistance protein ATP-binding cassette, sub-family G (WHITE), member 2 (ABCG2). We hypothesized that conversion of camptothecin-11 (CPT-11) to its highly cytotoxic metabolite SN-38 by a mutant human carboxyl esterase (hCE1m6) specifically in cancer cells and inhibition of ABCG2 by anti-ABCG2 short hairpin RNA, leads to accumulation of a higher concentration of SN-38, resulting in higher therapeutic efficacy and less toxicity to normal cells. Materials and Methods: A mutant human carboxyl esterase hCE1m6 with human telomerase reverse transcriptase promoter was integrated into the VISA (VP16-Gal4-WPRE) amplification system. The plasmid was transfected into MCF-12A, MDA-MB-231, and MCF-7 cells using JetPRIME®. Cancer-specific expression of hCE1m6 in breast cancer cell lines was tested by real-time polymerase chain reaction (real time-PCR) and western blot. In vitro conversion of CPT-11 to SN-38 was evaluated on lysates of transfected cells. Cytotoxicity of CPT-11 against cells transfected with the plasmid was evaluated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Results: Real-time PCR and western blot analysis revealed that hCE1m6 was expressed only in breast cancer cells, MCF-7 and MDA-MB-231, but not in the normal MCF-12A breast cell line. From the CPT-11 conversion assay on cell lysates, it was found that expressed hCE1m6 in cancer cells was able to effectively convert CPT-11 to SN-38. Conclusion: Breast cancer cell lines transfected with hCE1m6 showed an increased susceptibility to CPT-11 in comparison to MCF-12A cells.
- Transcriptional targeting
- human carboxylesterase
- CPT-11
- BCRP-shRNA
- transfection
- breast cancer cells
- drug resistance
Despite recent advances in early detection, surgical, chemotherapy and radiation interventions, malignant breast cancer remains incurable, and the survival period of patients with breast cancer is usually less than two years (1, 17). This is partly because of the limitations of current therapies such as toxicity and lack of specificity, and development of drug resistance. Novel therapies that can overcome these limitations are in urgent need and gene therapy is one such therapy that holds promise for targeted delivery, as well as for targeting specific cancer pathways. However, one of the main bottlenecks of gene therapy is tumor-specific expression of the transgene. Transcriptional targeting using tumor-specific promoters to limit expression of a transgene that can be potentially cytotoxic by itself or by activation of prodrugs is one method which is being explored to enhance the specificity of cancer gene therapy.
Nucleic acid therapy is based on two different approaches: i) introducing oligonucleotides or plasmids to express target genes for adding, correcting, or replacing genes in transformed cells; ii) introducing small interfering RNA (siRNA) to suppress gene expression. The first approach has been used to express genes to induce apoptosis or to activate prodrugs in tumor cells. The approaches reported for tumor cell-specific activation of prodrugs include: i) enzyme delivery by using enzymes linked with antibodies against receptors expressed on the tumor surface; ii) gene delivery by transfecting cells with cDNAs or plasmids to express active enzymes, using promoters for targeted expression (3, 4, 9, 11, 17, 18).
Cancer cells by-pass normal cellular senescence by up-regulating the expression of the enzyme telomerase that replicates telomeric DNA. Telomerase contains a catalytic unit with reverse-transcriptase activity (human telomerase reverse transcriptase, hTERT) and an RNA part that provides template for telomere extension. Approximately 85% of carcinomas have high levels of telomerase activity (5, 7, 8, 15). hTERT promoter has been used to target other tumor-killing factors, such as caspase 8, TNF-related apoptosis-inducing ligand and BCL2-associated X protein, and subsequently induces tumor-specific apoptosis. Studies using hTERTp/thymidine kinase (TK2) and Cytomegalovirus (CMV)/TK promoters have shown efficacy in various tumor models, with better efficacy with the CMV/TK construct.
The bête noire of cancer therapeutics, multidrug resistance (MDR), is a complex phenomenon whereby tumor cells acquire resistance to a variety of structurally and functionally unrelated drug molecules. It is commonly known that some forms of MDR arise from the overexpression of ATP-binding cassette transporters such as p-glycoprotein, MDR-associated protein 1, and breast cancer resistance protein (BCRP, hereafter referred to as ABCG2). Clinically, it has been reported that a correlation exists between ABCG2 expression and patient outcome for certain solid tumor types. ABCG2 is a transporter of 655 amino acids and is believed to function as a homodimer or a multimer. As an efflux transporter, overexpression of ABCG2 in tumor cells has been correlated with resistance to topotecan, 7-ethyl-10-hydroxycamptothecin (SN-38), mitoxantrone, and flavopiridole. Use of ABCG2 transport inhibitors (such as fumitremorgin C, and novobiocin) and siRNA to suppress ABCG2 expression have been used to circumvent ABCG2-mediated MDR (2, 10, 13, 14, 22, 23). One of the most promising strategies to inhibit ABCG2 employs siRNA for the specific knockdown of ABCG2. Both synthetic and vector-based siRNAs were shown to markedly down-regulate the expression of genes contributing for multidrug resistance and restore drug sensitivity in cancer cells (6). Although combination gene therapy and MDR reversal strategies have been investigated, lack of efficient gene delivery technology and poor target specificity have resulted in limited success.
CPT-11 (irinotecan-7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin), which is United States Food and Drug Administration-approved for use against colon cancer, is hydrolyzed by carboxylesterase to yield SN-38, a potent inhibitor of topoisomerase I. Recently, an active mutant of human liver carboxylesterase (hCE1m6) was reported for intracellular expression to efficiently convert CPT-11 to SN-38 for enzyme/prodrug therapy based on carboxylesterase/CPT-11combination (19). However, transcriptional targeting of this enzyme for tumor-specific activation of CPT-11, which is necessary for use in carboxylesterase/CPT-11 therapy, has not been investigated. In the present study, attempts were made for targeted expression of hCE1m6 for carboxylesterase/CPT-11 therapy and simultaneous suppression of ABCG2 by short hairpin RNA (shRNA) using a single plasmid. We used VISA (VP16-GAL4-WPRE integrated systemic amplifier)-driven hTERT promoter (T) for transcriptional targeting of hCE1m6, and U1 promoter for anti-ABCG2 shRNA expression.
Materials and Methods
Materials. Irinotecan HCl was purchased from LC laboratories (Woburn, MA, USA). SN-38 was purchased from AK Scientific (Union City, CA, USA). 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) was purchased from Sigma Aldrich (St. Louis, MO, USA). Phosphate-buffered saline (PBS, pH 7.4) and fetal bovine serum (FBS) were purchased from Mediatech Inc. (Manassas, VA, USA). Mouse monoclonal antibodies to human carboxylesterase-1 (hCE1) and β-actin, and secondary antibody to mouse IgG-HRP were purchased from Santacruz Biotechnologies (Santa Cruz, CA, USA). hCE1m6 plasmid was kindly provided by Dr. Philip Potter, St. Jude Children's Research Hospital, Memphis, TN (19). pUK21-T-VISA-E1A plasmid was a kind gift from Dr. Mien-Cheng Hung, The University of Texas MD Anderson Cancer Center, Houston, TX, USA (21). Anti-ABCG2 shRNA plasmid was obtained from Integrated DNA Technologies (Coralville, Iowa, USA).
Cell culture. The human epithelial mammary cell line, MCF-12A; human epithelial breast cancer cell lines, MCF-7 and MDA-MB-231 were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured with Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F-12), MEM, and DMEM/F-12 media, respectively. All the media were supplemented with 10% heat inactivated FBS and 1% penicillin-streptomycin solution. MCF-12A medium was supplemented with 20 ng/ml human epidermal growth factor and 0.01 mg/ml bovine insulin, whereas MCF-7 and MDA-MB-231 media were supplemented with 0.01 mg/ml bovine insulin.
Construction of pUK21-T-VISA-hCE1m6 expression-targeted gene delivery system. Considering the success of the T-VISA system used earlier for tumor-specific transgene expression (19671744), the same system was used for transcriptional targeting of hCE1m6 (an active mutant of human liver carboxylesterase) in breast cancer cells using pUK21-T-VISA-E1A as T-VISA-providing plasmid. In order to construct vectors, firstly, the BglII site located in pUK21 multiple cloning site (MCS) present in pUK21-T-VISA-E1A was removed by: (i) separating vector (3090 bp) and T-VISA-E1A (ca 5000 bp) fragments using NotI/SalI digestion; (ii) redigestion of linearized vector fragment with XhoI which cuts after 64 bp of SalI site to remove BglII; and (iii) ligation of NotI/XhoI-digested vector with NotI/SalI-digested T-VISA-E1A. This construct was designated as pSP. pSP1 was digested with BglII/NheI to excise the E1A, and linearized vector was either blunted and self-ligated to produce control plasmid (pUK21-T-VISA), or ligated with BglII/NheI-digested hCE1m6-coding region amplified from pCIhCE1m6 using hCE1:F/hCE1:R primer sets to construct enzyme expressing plasmid (pUK21-T-VISA-hCE1m6). Recombinant plasmids, pUK21-T-VISA and pUK21-T-VISA-hCE1m6, were confirmed by restriction digestion and sequencing and designated as pSP2 and pSP3, respectively.
Integration of anti-ABCG2 shRNA and Monster Green® Fluorescent Protein gene (hMGFP) into pUK21-T-VISA-hCE1m6. To circumvent the ABCG2-mediated efflux of SN-38, a vector that can co-express T-VISA-driven hCE1m6 and U1 promoter-driven anti-ABCG2 was constructed. Oligonucleotides were cloned for random and anti-ABCG2 shRNAs using Random:F/Random:R and anti-ABCG2:F/anti-ABCG2:R oligonucleotide sets, respectively, in pGeneClip™ hMGFP vector (Promega corporation, Madison, WI, USA) and designated as pSP4 and pSP5. A fragment of ca 4 kb encompassing shRNA and hMGFP coding regions were excised from both of the plasmids and ligated with NotI-digested and blunted pSP2 or pSP3 to make four different plasmids pSP6, pSP7, pSP8 and pSP9. Figure 1 shows the various components present in the plasmid pSP9. Table I shows the polymerase chain reaction (PCR) primers used for amplification of hCE1 and endogenous control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Table II describes the different plasmids used.

Schematic presentation of components present in the plasmid constructs.
Transfection. Transfections were performed using jetPRIME (Polyplus transfection Inc, Illkirch, France), according to the manufacturer's protocol with a few modifications in DNA and reagent ratio. Briefly, 4.5×105 to 5×105 cells were plated in 25 cm2 flasks and allowed to grow for 24 h in 5 ml of appropriate growth medium. MCF-12A, MDA-MB-231 and MCF-7 cells were exposed to 5, 8 and 10 μg of plasmid using 1:3, 1:3 and 1:4 ratio of DNA (μg):jetPRIME (μl) reagent ratios, respectively. Media were changed after 5 to 6 h and transfection efficiency was monitored after 24 h by confocal microscopy and flow cytometry.
Confocal microscopy and flow cytometry. Transfection was monitored by visualization of GFP fluorescence in transfected cells by confocal microscopy using the Nikon Eclipse Ti (Nikon Instruments Inc., Melvill, NY, USA) confocal laser scanning microscope (CLSM). Transfection efficiency was quantified by counting the number of fluorescent cells on a flow cytometer (BD Accuri C6 Flow Cytometer; BD Biosciences, San Jose, CA, USA).
Real-time PCR. Total RNA was isolated from cells using RNeasy Mini Kit, and On-column DNase digestion was performed using RNase-Free DNase I (Qiagen, Gaithersburg, MD, USA) according to the manufacturer's instructions. RNA yield and purity were quantified by using Nanodrop 1000 UV-Vis Spectrophotometer (Nanodrop Technologies, Inc, Wilmington, DE, USA). Every RNA sample was used as template with DNA Taq polymerase to check DNA contamination. For quantitative mRNA expression analysis, 2 μg of the RNA samples were reverse-transcribed using AffinityScript Multiple Temperature cDNA Synthesis Kit (Startagene, Agilent Technologies, CA, USA), and cDNA was used as template with appropriate primer sets (Table I) to perform real-time quantitative PCR (qPCR) using FastStart Universal SYBR Green Master (ROX) (Roche Applied Science, USA) and a 7300 Real-Time PCR System instrument (Applied Biosystems, Foster city, CA, USA). GFP and GAPDH were used as the endogenous controls for hCE1m6 and ABCG2, respectively.
Polymerase chain reaction primers for human carboxyl esterase 1 (hCE1) and endogenous control Glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
Western blot analysis. Protein samples were prepared after 24 h of transfection by using M-PER Mammalian Protein extraction reagent (Thermo Scientific, Rockford, IL, USA). Proteins were resolved by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electroblotted onto polyvinylidene difluoride membrane (Whatman, GE Healthcare Biosciences, Pittsburg, PA, USA) and the membranes were blocked with blocking solution containing 5% non-fat dried milk and 0.02% sodium azide in PBS supplemented with 0.02% of Tween 20 and then exposed to primary antibodies by incubating the membranes with mouse monoclonal antibodies (anti-hCE1: 1:800, anti-β-actin: 1:1000) in blocking solution at room temperature for 2 h with gentle shaking and washed three times with PBS. The membranes were exposed to secondary antibody by incubation with rabbit anti-mouse IgG-HRP diluted (1:2000) in tris-buffered saline supplemented with 0.02% Tween 20 (TBS-T) containing 5% nonfat dried milk, washed three times with TBS, and specific immunoreactivity was detected by chemiluminescence (Pierce ECL western blotting substrate, Thermo fisher, Rockford, IL, USA).
CPT-11 conversion assay. Conversion of CPT-11 into SN-38 was monitored as described elsewhere (19). Briefly, cell sonicates were incubated with 10 μM CPT-11 for 2 h in 50 mM HEPES (pH 7.4) at 37°C. The reaction was terminated by addition of an equal volume of acidified methanol. Particulate matter was removed by centrifugation at 18000 ×g for 5 min at 4°C. The amount of SN-38 was measured with a NOVOstar microplate reader (BMG Labtechnologies, Inc. Durham, NC, USA) using an excitation wavelength of 377 nm and emission wavelength of 538 nm. Analysis of known SN-38 standards (from 1 ng/ml to 10 μg/ml) indicated that lower limit of detection of this instrument for SN-38 was approximately 10 ng/ml.
Description of the four plasmids constructed in the study.
Growth-inhibition assays. Ttransfected cells (4×104) were exposed to different concentrations (0.01 to 6 μM) of CPT-11 in 24-well plates for 24 h. The cells were washed 2 times with PBS and growth inhibition was monitored by MTT assay. The results are expressed as the percentage of cell viability as compared to the control cells (cells grown with medium only) and the Half-maximal inhibitory concentrations (IC50) was determined using Graphpad Prism 5 software (San Diego, CA, USA). Student's t-test was used to compare IC50 values and a value of p<0.05 was considered statistically significant.
Results and Discussion
Appreciable attempts have been made to separately circumvent both the following problems in cancer therapeutics: lack of tumor-specific expression and ABCG2-mediated MDR using gene therapy strategies. Different permutations such as chemotherapeutic agents with a chemical inhibitor of ABCG2 (combination of chemotherapies), and anti-ABCG2 shRNA (combination of chemotherapy and gene therapy) have been used to find an effective combination therapy of MDR tumor cells (12, 19, 21). However, a practically applicable combination of therapies that can simultaneously activate prodrugs and suppress efflux transporter expression is rare. In order to explore a more effective and practical approach to implementing enzyme/prodrug therapy for MDR tumor cells, we designed and constructed a plasmid for simultaneous expression of hCE1m6 in a tumor-specific manner using the T-VISA system, and ABCG2 suppression using U1 promoter-driven anti-ABCG2 shRNA.
Transient transfection of cell lines. Non-viral vectors have been widely used for their well-known advantages including ease of scale-up, storage stability, improved quality control, reduced/non-immunogenicity, and surface modification ability (14). Recombinant plasmids were mobilized into cells by non-viral transfection methods, as described in the Materials and Methods section. In order to enhance the transfection efficiency, the DNA:transfection reagent ratio and the cell numbers were optimized. Maximum efficiency was found with 6, 8 and 12 μg of plasmid using 1:3, 1:4 and 1:4 DNA:Reagent ratios with 24 h pre-grown (45×104 cells in 25 cm2 flasks) MCF-12A, MDA-MB-231 and MCF-7 cells, respectively. Since all of the plasmids used for transfection had CMV promoter-driven hMGFP, confocal microscopy and flow cytometry were used to monitor the transfection efficiencies (Figure 2). With our standardized protocol, we were able to achieve 90%, 80% and 75% transfection of MCF-12A, MDA-MB-231 and MCF-7 cells, respectively. These percentages were recorded after 18 to 20 h of transfection and remained almost constant up to 44 h after transfection. We used this time, approximately 24 h, during which the number of cells expressing hMGFP was constant to perform all the analyses of this study, assuming that the cells expressing hMGFP would also express hCE1m6/anti-ABCG2 shRNA, since all these components were present in the same plasmid.
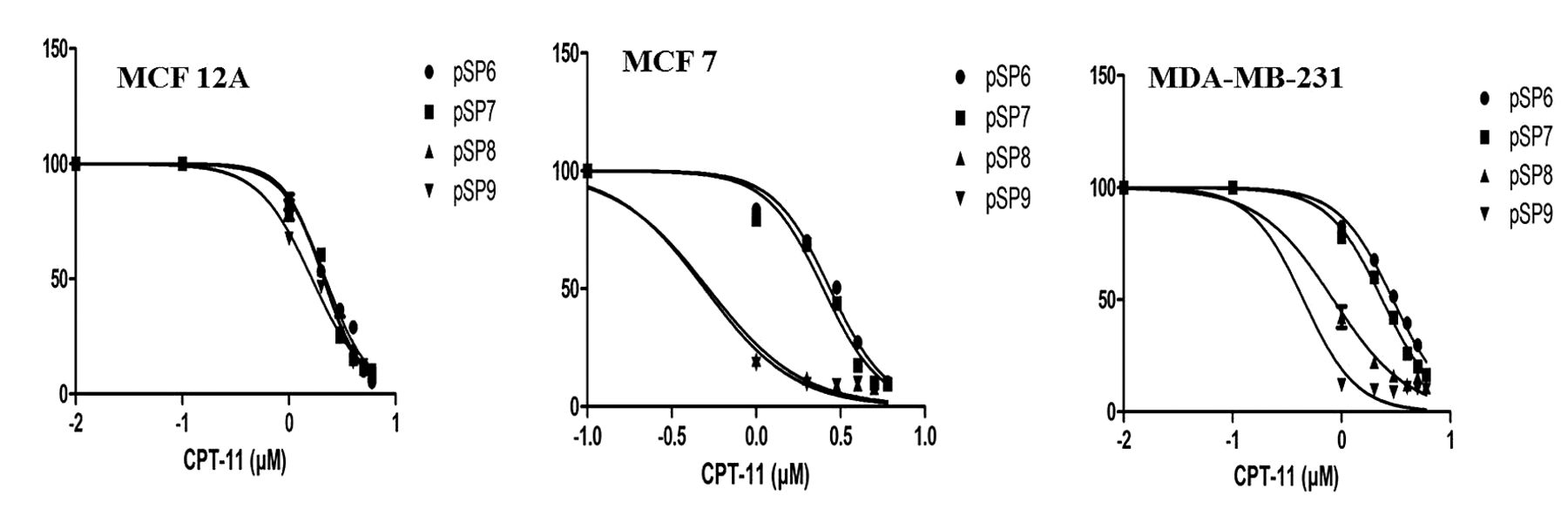
Half-maximal inhibitory concentrations (IC50) values (μM) for MCF-12A, MCF-7, and MDA-MB-231 cells transfected with pSP6, pSP7, pSP8, and pSP9, following treatment with CPT-11.
T-VISA efficiently expresses hCE1m6 specifically in cancer cells. To examine whether transfected cells expressed hCE1m6 in a cancer cell-specific manner, western blotting was performed to detect hCE1m6 in the protein samples prepared from pSP6 or pSP8 transfected MCF-12A, MDA-MB-231 and MCF-7 cells. Figure 3A shows that a band was detected in protein samples of pSP8-transfected MDA-MB-231 and MCF-7 cells but not in those of MCF-12A cells. This band was absent from samples prepared from any cell line transfected with pSP6. We performed real-time qPCR to compare the expression level of hCE1m6 in these three cell lines using hMGFP as an endogenous control because only transfected cells produce mRNA for these two proteins. Cancer cell lines expressed higher levels of hCE1m6 mRNA relative to the mRNA level in pSP8-transfected MCF-12A cells (Figure 3B). The relative expression level of hCE1m6 mRNA (2−ΔΔCT) in pSP8-transfected MDA-MB-231 and MCF-7 cells compared to that in pSP8-transfected MCF-12A cells was 27- and 30-fold, respectively.
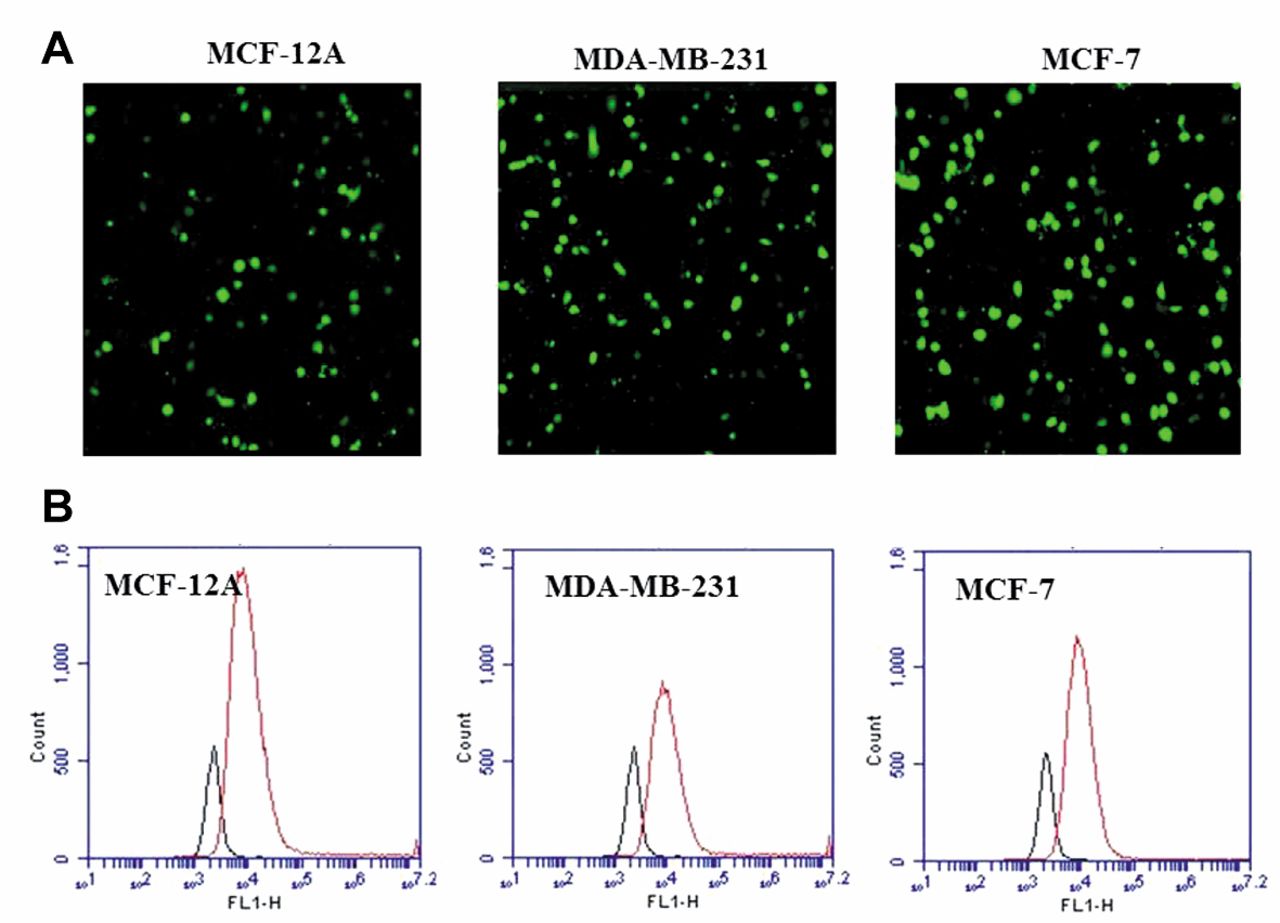
Transfection of MCF-12A, MDA-MB-231, and MCF-7 cells (18 to 20 h post-transfection). A: Confocal microscopic visualization. B: Flow cytometric histogram showing the percentage of fluorescent cells.

Expression of hCE1 protein by western blot (A) and relative expression levels of hCE1 mRNA by real-time PCR, in MCF-12A, MDA-MB-231, and MCF-7 cells transfected with pSP6 and pSP8 plasmids (B).
Conversion of CPT-11 to SN-38 by transfected cancer cells. To examine whether our construct expresses an active hCE1m6 enzyme, we prepared cell extracts by sonicating the pSP6-, and pSP8-transfected cells incubated with 10 mM of CPT-11 for 2 h at 37°C. Sonicates prepared from pSP8-transfected MDA-MB-231 and MCF-7 cells produced significant amounts of SN-38 in comparison to the amount produced by sonicates prepared from both pSP8-transfected MCF-12A cells and pSP6-transfected cell lines. Quantification of CPT-11 and SN-38 in assay samples using standard graph for CPT-11 and SN-38 revealed that MDA-MB-231 and MCF-7 sonicates converted 2.7 μg and 3.2 μg of CPT-11 into SN-38 in 2 h (Figure 4). These results indicate that pSP8 transfection results in expression of enzymatically active hCE1m6 enzyme only in cancer cells. The results are consistent with the results obtained from western blot and real-time PCR.

Quantification of CPT-11 conversion to SN-38 in MCF-12A, MDA-MB-231, and MCF-7 cells transfected with pSP6 and pSP8.

Growth-inhibition curve for MCF 12A, MCF 7, and MDA-MB-231 cells transfected with pSP6, pSP7, pSP8, and pSP9, following treatment with CPT-11.
Human telomerase activity has been shown to be elevated in cancer cells in comparison to normal cells (16). T promoter has been used for ovarian cancer cell-targeted expression of E1A, an adenoviral type 5 transcription factor that possesses anticancer properties using the VISA expression system which can amplify the transcriptional activity of T promoter by utilizing two-step transcriptional amplification (TSTA) system and elevate the expression of the transgene by woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) (20, 21). In this study, this system worked specifically and efficiently in breast cancer cells (in vitro), as we observed that hCE1m6 was expressed only in cancer cells not in normal cells.
T-VISA regulated expression of hCE1m6 sensitizes cancer cell lines to CPT-11. Cancer cell-specific sensitivity to CPT-11 due to T-VISA-regulated expression of hCE1m6 was monitored by measuring the growth inhibition of pSP6-, and of pSP8-transfected cells exposed to different concentrations of CPT-11 for 14 to 16 h. The IC50 values are given in Table III. Growth-inhibition studies revealed that expression of hCE1m6 sensitized both the cancer cell lines to CPT-11 but it could not sensitize MCF-12A cells, consistent with the lack of CPT-11 conversion to SN-38 by sonicates of pSP8-transfected MCF-12A cells (Figure 5). T-VISA-regulated expression of hCE1m6 (pSP8 transfection) resulted in approximately 6- and 14-fold reduction in the IC50 value of CPT-11 for MDA-MB-231 and MCF-7 cells, respectively, compared to the corresponding cells transfected with pSP6. No difference was observed in the IC50 value of CPT-11 for MCF-12A cells transfected with either of these plasmids.
The results obtained from this study show that pSP8 and the construct bearing hCE1m6 was specifically expressed in cancer cells in vitro and can efficiently convert CPT-11 to SN-38, imparting significant cytotoxicity to cancer cells. Further studies are in progress to evaluate the efficiency of pSP7 (bearing anti-ABCG2 shRNA) and pSP9 (with both hCE1m6 and anti-ABCG2 shRNA) in highly MDR breast cancer cells.
Acknowledgements
This work was partially supported by the Texas A&M Health Science Center Cancer Research Council Seed Grant to SP.
- Received July 12, 2014.
- Revision received August 21, 2014.
- Accepted August 25, 2014.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.