


Abstract
It has long been thought that the G1/S cell cycle checkpoint allows time for DNA repair by delaying S-phase entry. The p53 tumor suppressor pathway regulates the G1/S checkpoint by regulating the cyclin-dependent kinase inhibitor p21Waf1/Cip1, but p53 also regulates the nucleotide excision DNA repair protein XPC. Here, using p53-null cell lines we show that additional mechanisms stabilize XPC protein and promote nucleotide excision repair (NER) in concert with the G1/S checkpoint. At least one mechanism to stabilize and destabilize XPC involves ubiquitin-mediated degradation of XPC, as the ubiquitin ligase inhibitor MG-132 blocked XPC degradation. The retinoblastoma protein RB, in its unphosphorylated form actually stabilized XPC and promoted NER as measured by host cell reactivation experiments. The data suggest that XPC protein and XPC-mediated NER are tightly linked to the G1/S checkpoint, even in cells lacking functional p53.
Studies have shown that the XPC DNA repair protein is required for the recognition of DNA damage in global genomic nucleotide excision repair (G-NER) and for the recruitment of other repair factors to the site of damage (1, 2). A number of investigations have focused on the relationship of cellular DNA repair to cell cycle checkpoints. The G1/S checkpoint in particular is regulated by p53 on two levels. Firstly, the XPC DNA repair protein is p53 regulated (3). Secondly, the induction of G1 cell cycle arrest by the cyclin-dependent kinase inhibitor p21Waf1/Cip1 (hereafter named p21) is also p53 regulated. While XPC is a true nucleotide excision DNA repair (NER) protein expected to enhance the rate of cellular NER, p21 has no biochemical role in NER (4). Nonetheless, p21 was shown to enhance the rate of cellular NER in several studies (5-8). While the role of XPC and its relationship with the G1/S cell cycle checkpoint has been speculated upon, little is known of the role of G1/S mediators in NER.
Here, we used a human lung cancer cell line H1299 that carries deletions of both p53 alleles (9). We found that co-transfection of plasmids encoding RB and XPC led to XPC stabilization at the protein level which corresponded to enhanced NER as measured in host cell reactivation (HCR) studies. Interestingly, XPC protein appears to exist in a latent form unable to activate HCR, and an active form that is stabilized by RB.
RB mutants defective for G1 cell cycle arrest were defective in stabilizing or activating XPC. Cyclin E, which phosphorylates RB among other proteins and triggers the G1 to S phase transition, abrogated XPC stabilization by RB. Thus, the stabilization of XPC by RB is a feature of the G1/S checkpoint role of RB. It is likely that XPC is stabilized and destabilized by post-translational modifications and that XPC interaction with RB signals post-translational modifications of XPC to occur. As such, XPC activity in NER, measured by HCR assays, is tightly linked to the G1 phase of the cell cycle even in cells lacking functional p53.
Materials and Methods
Cell lines. H1299 lung carcinoma cells (National Cancer Institute) and Saos2 osteosarcoma cells (National Cancer Institute) were cultured in DMEM 4500 mg/L glucose supplemented with 10% fetal bovine serum (FBS) and 1% L-glutamine and gentamicin. Cells were plated for transfection at sub-confluency. H1299 and Saos2 carry deletions in both p53 alleles and do not express p53 protein (9). Extensive characterization as part of the NCI anticancer drug screen showed absence of functional p53 and absence of the G1/S checkpoint in these cell lines (9).
Cell transfection and plasmids. Cells were evenly seeded at 50% confluency onto cell culture dishes and incubated at 37°C to allow adhesion overnight. Cells were transiently transfected using either Fugene HD transfection reagent (Roche Applied Science, Indianapolis, IN, USA) or Turbofect transfection reagent (Fermentas Life Science, Ontario, Canada). Transfection mixtures were added to each plate drop-wise. The plate was then gently rocked back and forth to achieve even distribution. The cells were incubated for 48 hours at 37°C before collection. All plasmids were pCMV3 derivatives and encoded full-length cDNAs driven by the CMV promoter. Empty pCMV3 or pCMV6 (Origene, Rockville, MD, USA) plasmids were used as vector controls. Full-length cDNA encoding XPC, RB, or cyclin E were in pCMV6 (Origene). RB mutants driven by CMV plasmids were a gift from Dr. Nick Dyson, Massachusetts General Hospital.
Immunoblotting. Samples were lysed by directly adding 2X SDS loading dye and collected by scraping and transferred into 1.5 ml Eppendorf tubes. The samples were boiled for approximately 30 minutes and 15-20 μl of sample were added to precast 4-20% Tris-Glycine gels (Invitrogen, Carlsbad, CA, USA) and ran at 100-150 V. Gels were transferred to nitrocellulose membranes at 5 V <10 A overnight. Nitrocellulose membranes were blocked with 4% milk buffer prior to probing with primary and secondary antibodies. Primary antibodies XPC A-5 mouse monoclonal or XPC H-300 rabbit polyclonal (Santa Cruz Biotechnology, Santa Cruz, CA, USA.) were used in most experiments. Anti-actin rabbit polyclonal (Thermo Scientific, Waltham, MA, USA), Anti-beta galactosidase mouse monoclonal BG-02 (Abcam, Cambridge, MA, USA), and others were used as indicated in the Figure legends. Secondary antibodies were peroxidase conjugates from Santa Cruz Biotechnology. Immunoblots were detected by ECL (Pierce Thermo Scientific, Rockville, IL, USA) and exposed to Kodak Biomax XAR film (Sigma Aldrich, St. Louis, MO, USA).
Host cell reactivation. The host cell reactivation assay (HCR) is used to measure DNA repair in cells. HCR is an established assay in use in several laboratories (10-13). A plasmid encoding a chloramphenicol acetyltransferase (CAT) reporter gene was damaged by cisplatin to produce 1 to 5 adducted bases per plasmid molecule. NER-defective fibroblasts from XPC patients are defective in reactivating and thereby expressing the reporter gene (10-13). NER-proficient cells repair the gene and thereby express the reporter (10-13). One advantage of the assay is that only the damaged reporter plasmid and not the cells receive DNA damage. Thus, cellular effects e.g. apoptosis are not an issue.
CAT ELISA. The CAT ELISA immunoassay (Roche Applied Science) was used for the quantitative determination of chloramphenicol acetyltransferase (CAT) enzyme (10-12). Briefly, equivalent protein concentrations of lysed cell extracts were added to an anti-CAT-coated 96-well microplate and refrigerated overnight. Microplate wells were then washed and a digoxigenin-labeled antibody to CAT was added to wells at room temperature for at least 1 hour. Wells were then washed and an antibody to digoxigenin conjugated to peroxidase was added for 1 hour. The peroxidase substrate 2,2′-azino-di-[3-ethylbenzthiazoline sulfonate (ABTS) was added after washing. The plate was then incubated and read at 405 nm in a Tecan Spectra plate reader at multiple time intervals.
Immunoprecipitation. Cells were harvested and lysed using immunoprecipitation (IP) lysis buffer containing 1% Triton X-100 and protease inhibitors. Samples were transferred to a 1.5 ml microcentrifuge tube and pelleted by microcentrifugation. The supernatant was then transferred to a new tube containing 50 μl of Protein A/Protein G agarose beads (Santa Cruz Biotechnology) and 1 μg of XPC clone D-10 antibody (Santa Cruz Biotechnology). Rb antibodies XZ104 and XZ155 were used in some experiments as indicated. The mixture was then allowed to incubate at 4°C on a rotator overnight. After incubation, the beads were collected by microcentrifugation and washed 4 times in lysis buffer. Beads were resuspended in 50 μl of 2× SDS loading buffer and boiled prior to loading on 4-20% Tris-glycine gels and subjected to immunoblotting.

RB enhances XPC-mediated host cell reactivation of a cisplatin-damaged reporter gene. A: Plasmids were transfected at the indicated amounts. The total amount of plasmid DNA was kept constant by adding empty CMV plasmid where necessary. H1299 cells have low endogenous XPC. RB and XPC were readily detected where encoded by the respective plasmids (Western blots, lanes 2 and 4). B: Mixing RB and XPC plasmids resulted in a clear increase in XPC protein (Western blot, lane 8). XPC was detected with antibody A5, while RB was detected with antibody AF-11. Actin served as a loading control. C: Host cell reactivation corresponding to the same lanes as in A and B. Neither RB alone, nor XPC alone (bar graph, lanes 2 and 4 respectively) resulted in increased host cell reactivation. When combined (bar graph, lane 8), a significant increase in host cell reactivation was observed (*p=0.0005).
Pulse chase analysis. After H1299 cells were transfected with XPC and RB plasmids, cellular proteins were labeled with an L-methionine analog that can be subsequently labeled with biotin (Invitrogen). The free label was removed by placing the cells in complete medium and cells were harvested at 0, 4, and 8, hours. XPC was immunoprecipitated as described above and was then labeled by biotinylation (Invitrogen). The L-methionine analog has a reactive azide group that is then covalently modified by an alkyne-biotin reagent (Invitrogen) and detected using a mouse monoclonal anti-biotin antibody (Sigma).
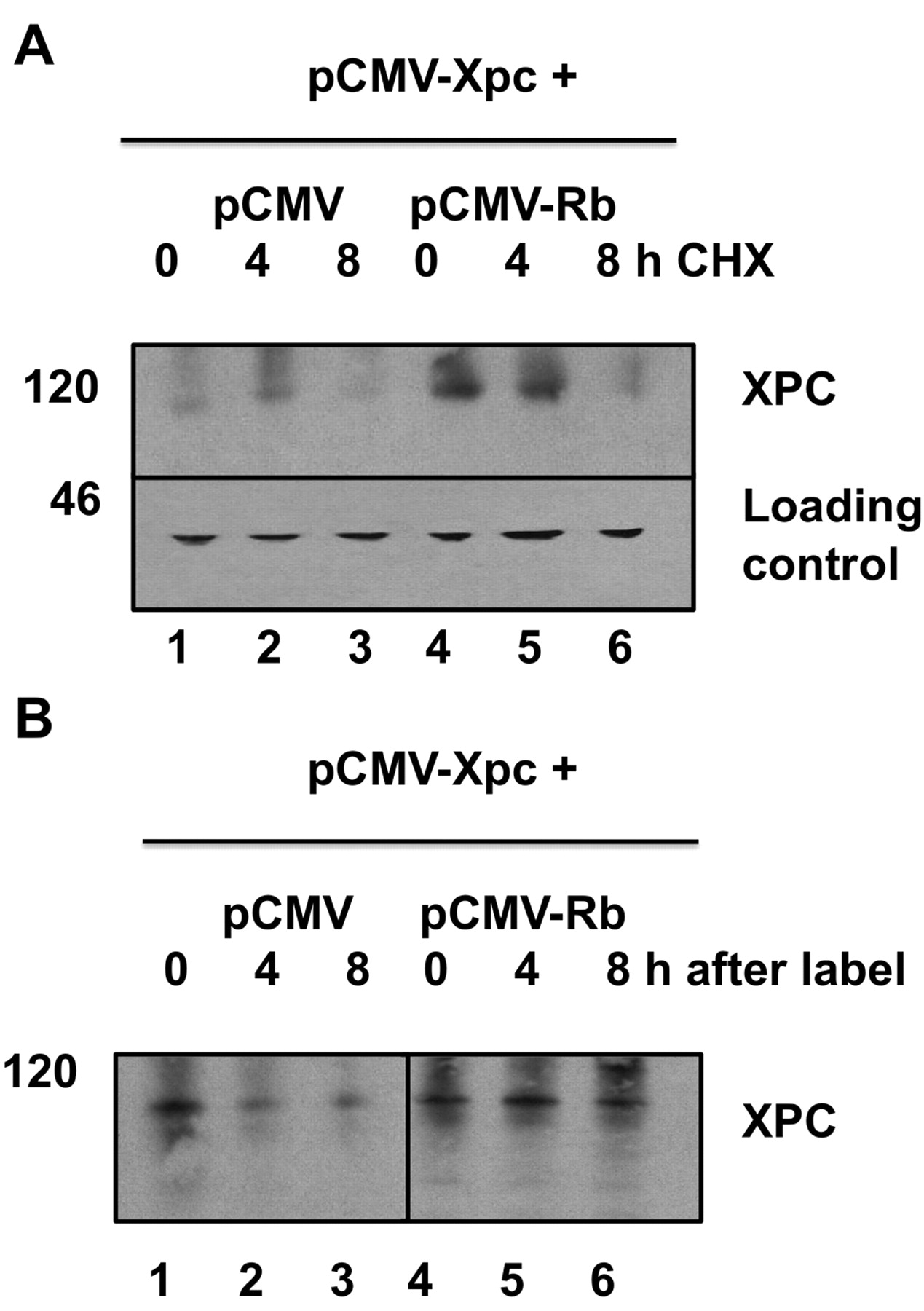
RB stabilizes XPC in the presence of cycloheximide or in pulse-chase experiments. A: H1299 cells were transfected as previously with plasmids encoding XPC and RB. After 48 h in culture, cycloheximide (CHX; 10 μM) was added for 15 h. Western blots show accumulation of XPC protein where RB was co-transfected with XPC (lanes 4 and 5). XPC was detected by anti-XPC monoclonal antibody A5. Actin was not affected by CHX and served as a loading control. B: Metabolic labeling of XPC with an L-methionine analog. Cells were transfected with XPC and RB plasmids as indicated, then labeled (pulsed) after 48 h in culture. The label was removed and cells returned to complete medium and chased for 0, 4, or 8 h. Immunoprecipitated XPC was detected using an anti-biotin antibody. Accumulated XPC is evident in lanes that were co-transfected with RB (lanes 5 and 6).
Statistical analysis. All data are presented as mean or relative units±standard error. Data were analyzed as indicated in the Figure legends using one way ANOVA or paired t-test with Microsoft Office Excel 2007 (Seattle, WA, USA) or Graph Pad Prism (San Diego, CA, USA) software.
Results
We chose H1299 human lung cancer cells because they have low endogenous XPC, thereby making it easier to detect transfected XPC in transient expression experiments. The plasmids used, pCMV-RB and pCMV-XPC, showed appreciable expression of their respective proteins when introduced to H1299 cells compared to empty CMV plasmid (Figure 1A). We combined pCMV-RB and pCMV-XPC in all possible combinations. The amount of plasmid DNA was kept constant across all the lanes. When combined with pCMV-RB, the abundance of XPC protein was markedly increased (Figure 1B, lane 8). Most importantly, combining pCMV-RB with pCMV-XPC markedly enhanced DNA repair of a cisplatin-damaged reporter plasmid, pCMV-CAT (Figure 1C, lane 8). Thus, the co-transfection of RB together with XPC significantly (p=0.0005) enhanced the DNA repair activity of XPC. It is likely that the observed enhancement of XPC-mediated DNA repair is due to i) interaction of XPC with RB and/or ii) activating modifications of XPC protein that are triggered by interaction with RB.

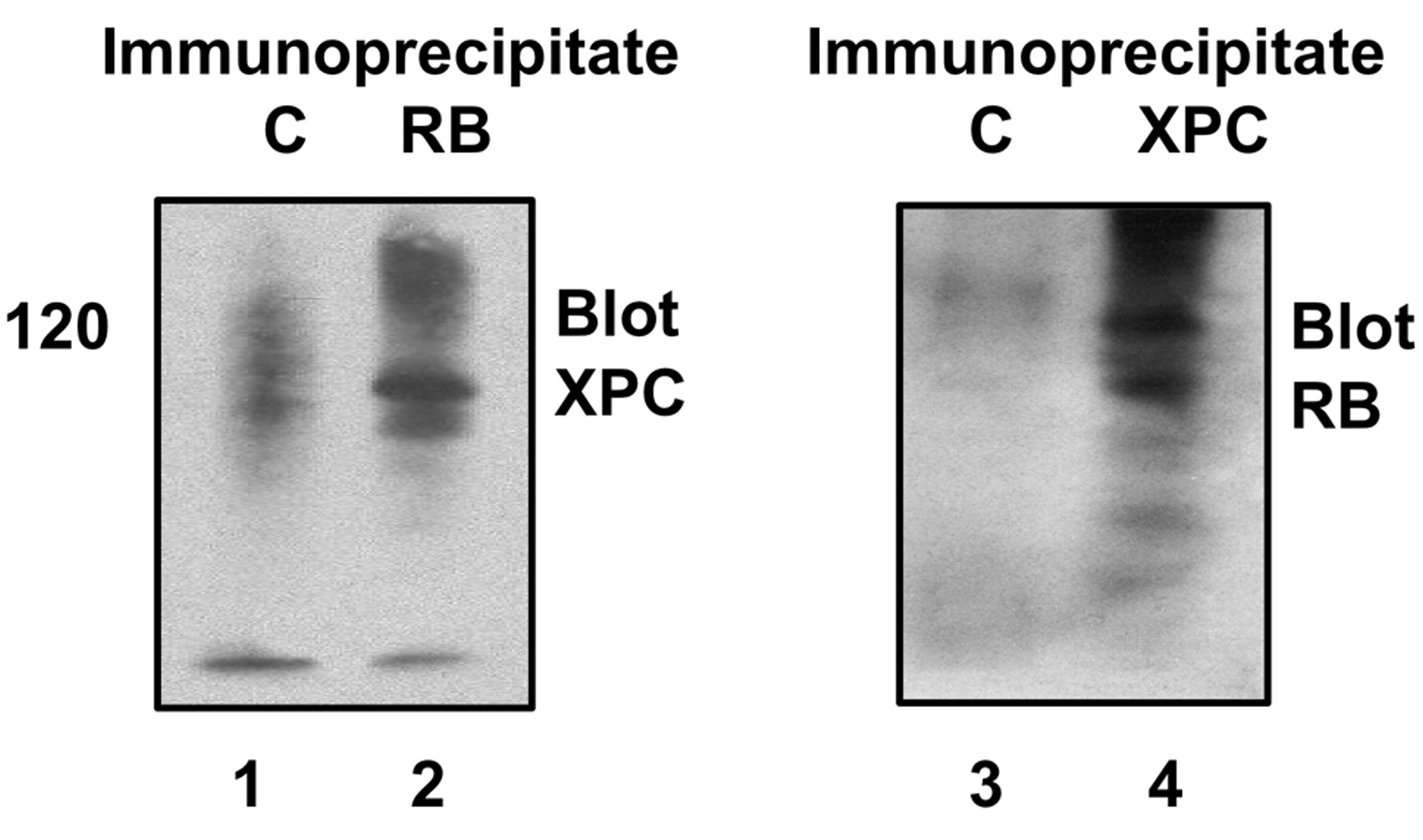
Putative interaction between RB and XPC. H1299 cells were transfected with RB and XPC plasmids. Immune complexes containing RB or XPC were probed on Western blots to detect possible interaction. Left panel, immune complexes obtained using a negative control, MOPC21 IgG, or anti-RB monoclonal antibodies XZ104 and XZ155; right panel, immune complexes obtained using a negative control, nonimmune rabbit IgG, or anti-XPC rabbit polyclonal antibody H300. The left panel was probed by Western blot with polyclonal rabbit anti-XPC. The right panel was probed by Western blot with monoclonal anti-RB clone AF-11.
A third possibility was that RB enhanced the translation of XPC protein. However, if this were the case, it would result in active XPC protein. As can be seen in Figure 1A, simply increasing the amount of XPC did not necessarily lead to an increase in actual DNA repair as measured by HCR (Figure 1A, lane 4 compared to Figure 1C, lane 4). We conducted experiments with cycloheximide to block protein synthesis to address this possibility. Co-transfection with RB led to increased XPC levels in the presence of cycloheximide (Figure 2A). Therefore, the observed enhancement of XPC by RB does not require ongoing protein synthesis. We also conducted pulse-chase experiments, in which cells co-transfected with RB and XPC were labeled with an L-methionine analog to label methionine residues during a 4-hour pulse. Cells were returned to complete medium with excess unlabeled L-methionine (chase) and were harvested 0, 4, or 8 hours later. The co-transfection of RB extended the half-life of XPC beyond 8 hours, compared to <4 hours in the control transfectants (Figure 2B). Thus, RB appeared to increase the half-life of XPC.
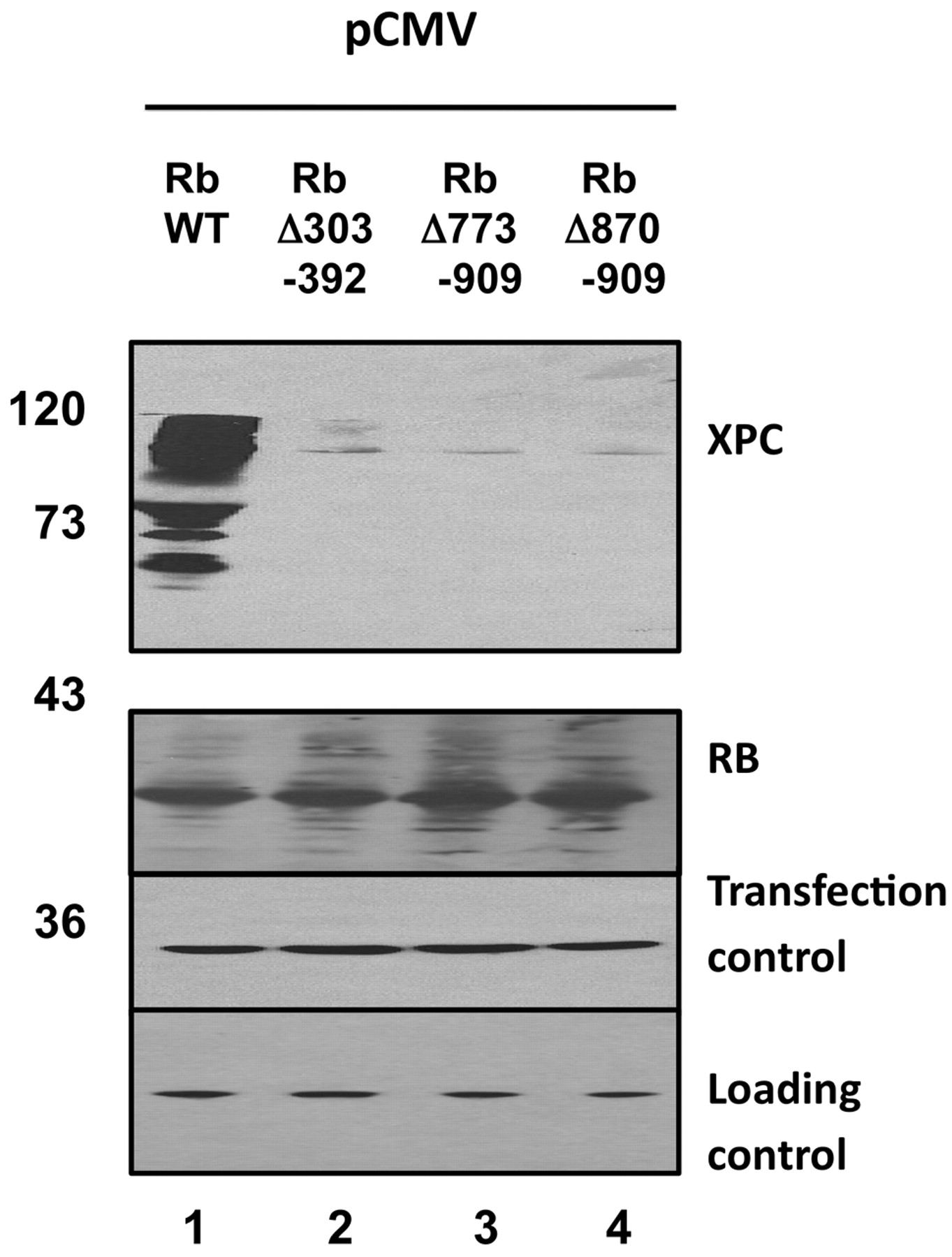
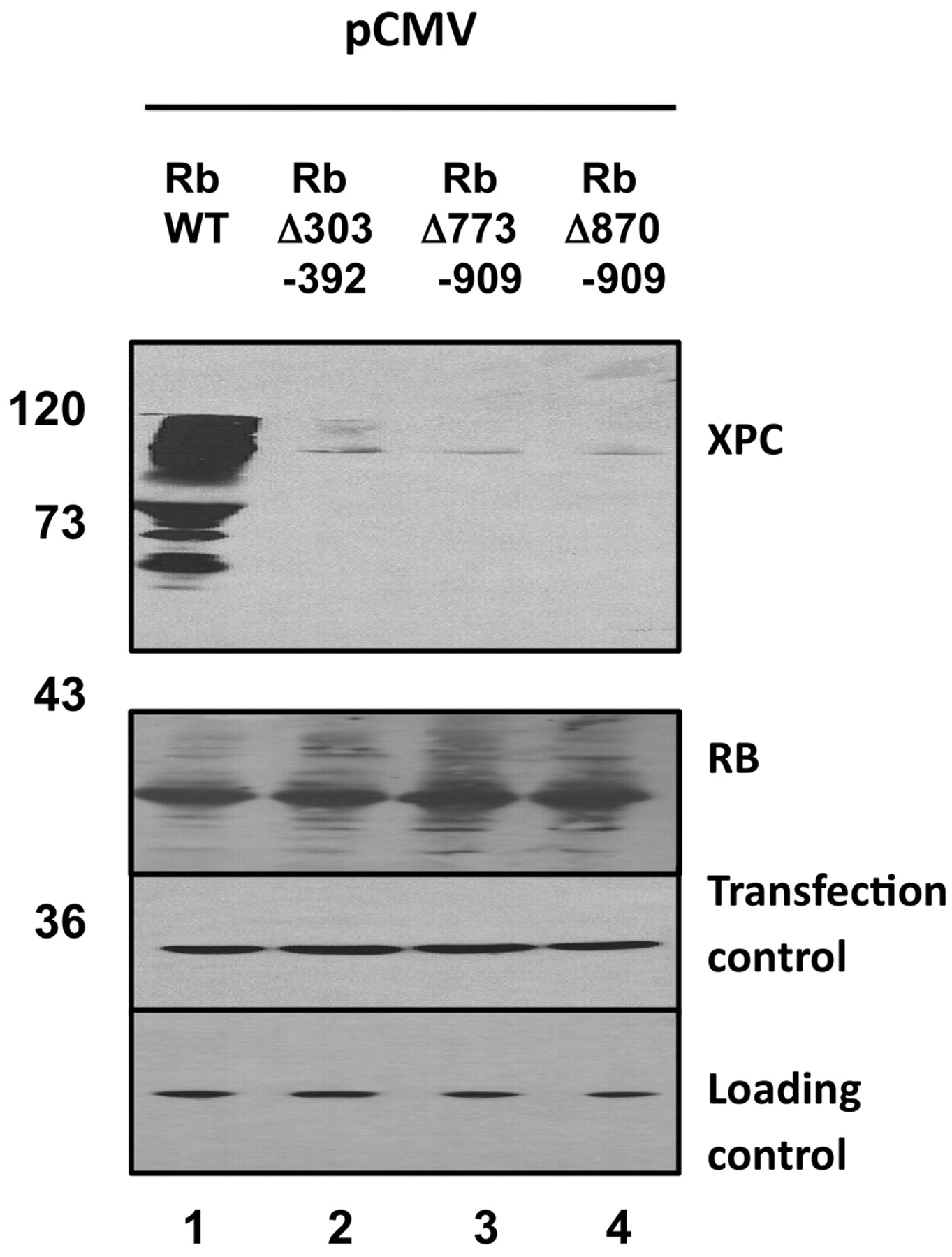
RB mutants fail to stabilize XPC. Wild-type RB or three deletion mutants were co-transfected together with XPC as previously. Saos2 cells were used. Robust stabilization of XPC was observed by wild-type RB (lane 1). Each of the mutants failed to stabilize XPC (lanes 2, 3, 4). Beta galactosidase served as a co-transfection control. Actin served as a loading control. XPC was detected by anti-XPC monoclonal antibody A5.
Given these results, we hypothesized a possible protein protein interaction between RB and XPC. Toward this end, we used recombinant proteins but failed to detect an interaction (results not shown). One possibility is that the interaction requires protein folding concurrent with translation. If this were the case, the two proteins would interact when co-expressed in the same cell. When mixed as full-length proteins, they would not. To investigate this likelihood, immune complexes containing RB were probed for the presence of XPC and vice versa. XPC was detected in RB immune complexes, and RB was detected in XPC immune complexes (Figure 3). Thus, there is a putative interaction.

Cyclin E counteracts (wild-type) RB stabilization of XPC suggesting that stabilization of XPC is related to the cell cycle role of RB. Saos2 cells were transfected as previously with the indicated plasmids, again keeping the total amount of plasmid DNA constant. Co-transfection of cyclin E strongly reduced XPC protein expression, suggesting that cyclin E counteracts any stabilizing effect of RB on XPC. Beta galactosidase served as a co-transfection control. Actin served as a loading control. XPC was detected by anti-XPC monoclonal antibody A5. Cyclin E was detected using monoclonal antibody HE-12 (Calbiochem).
We tried to map the interaction domain of RB with XPC. We used a series of RB deletion mutants for co-transfection experiments, the idea being that we could map the domain by determining which mutants did or did not stabilize XPC. Surprisingly, each of the three deletion mutants failed to stabilize XPC (Figure 4). Each of the mutants are partially defective in G1 cell cycle arrest, thus, perhaps a G1 arrest is required. Alternatively, each of the mutants may be improperly folded such that they cannot interact with XPC. Each of the mutants is expressed similar to wild-type (Figure 4) but we cannot exclude a defect in protein folding.
As RB is a key mediator of the G1/S transition in cells, we tested whether the effect of stabilizing XPC was linked to the cell cycle role of RB, or whether it could be a novel function of RB. Cyclin E serves as the signal for S-phase entry by phosphorylating RB at the G1/S transition. We co-transfected a CMV plasmid encoding cyclin E, together with all possible combinations of RB and XPC-encoding plasmids. Cyclin E completely abolished the stabilization of XPC by RB (Figure 5, lane 8). Thus, it is likely that RB in its underphosphorylated G1 form stabilizes XPC, but not in its cyclin E-phosphorylated form. Cyclin E as expected reduced DNA repair in HCR assays, again suggesting that cyclin E plays a negative role in regulating XPC (Figure 6).

Ubiquitin ligase inhibitor MG-132 rescues XPC from cyclin E-triggered degradation. Cyclin E inhibited XPC expression (left panel) and host cell reactivation (HCR; right panel; p=0.004 comparing lanes 1 and 2). MG-132 was added (10 μM) 15 h prior to cell lysis. MG-132 restored XPC and HCR to control levels. H1299 cells were utilized for both panels.
The simplest hypothesis to explain the data is that RB protects XPC from degradation and that cyclin E reverses that protection from degradation. The proteosome inhibitor MG132 rescued XPC from cyclin E-mediated turnover (Figure 6). Thus, it is likely that XPC is tightly linked to the G1/S cell cycle checkpoint by proteosome-mediated stabilization and destabilization mechanisms.
Discussion
In response to DNA damage, XPC protein is phosphorylated, ubiquitinated, and SUMOylated however the amino acid residues involved, and the net effects on XPC function are not well-known (14-16). Presumably, some modifications promote XPC-mediated NER while others inhibit XPC-mediated NER. It has long been thought that NER occurs readily in G1 but cannot occur in S phase. Therefore, protein modifications and protein protein interactions probably restrict XPC function to G1 while inactivating XPC upon S phase entry.
One important aspect of the G1/S checkpoint is that p53 transcriptionally activates XPC (15). This important finding suggested that p53 and XPC could actively promote NER in cells that carry functional p53. We used human lung cancer H1299 cells, which carry deletions in both p53 alleles and have low endogenous levels of XPC. One often-ignored feature of the HCR assays that we and others have used extensively is that it measures NER of a damaged reporter gene. The cells do not receive DNA damage. In this assay, we detected a latent XPC protein that did not promote HCR (Figure 1A and C, lanes 4). When combined with RB, XPC was in some way activated for HCR (Figure 1B and C, lanes 8). Given evidence that RB may interact with XPC (Figure 3), it is likely that the interaction of XPC with RB promotes NER as measured by HCR. Post-translational modification(s) of XPC in the presence of RB is in evidence (Figure 1A, lane 8).
Several studies showed that p21Waf1/Cip1 promoted HCR in p53-defective cell lines, although the mechanistic basis was not known (5-8). The simple view was that simply arresting the cell cycle by p21 promoted HCR. Our data provide a mechanistic insight that p21 would lead to accumulation of underphosphorylated RB, which would in turn stabilize XPC in a form that is active for HCR (Figure 1B and C, lanes 8). Our finding that cyclin E destabilized XPC and led to a decrease in HCR is also consistent with the published studies on p21 (Figure 5).
The studies herein were conducted in a p53-null cancer cell line, H1299, which carries deletions in both p53 alleles (9). Although it is expected that p53 will contribute to XPC expression and cellular NER in cells with functional p53, our study raises the possibility that p53-defective cancer cells might still retain some degree of XPC regulation and the capacity to regulate cellular NER. We used a cisplatin-damaged reporter gene for the HCR assays. Possibly, cancer cells lacking functional p53 could evade or resist cisplatin therapy by repairing the cisplatin DNA damage. From a chemotherapeutic perspective, it may be necessary to target additional components of the G1/S checkpoint, such as RB, in order to maximize cancer cell killing by cisplatin.
- Received March 24, 2010.
- Revision received May 21, 2010.
- Accepted May 27, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.