Article Text

Statistics from Altmetric.com
Apoptosis, the morphologically defined form of programmed cell death, is a cellular process that is of tremendous current interest to clinicians who study and treat cancer.1 Nowhere is this more true than in the area of colorectal cancer and its management. Abnormalities in apoptotic function contribute to both the pathogenesis of colorectal cancer and its resistance to chemotherapeutic drugs and radiotherapy, both of which act, at least in part, by killing cancer cells. In this article, current knowledge of the mechanisms of apoptosis and their place in the pathogenesis of colorectal cancer will be reviewed together with the progress that has been made in the development of therapies designed to target apoptosis.
STRUCTURE OF THE NORMAL COLONIC EPITHELIUM
Before discussing apoptosis itself it is important to understand the hierarchical organisation of intestinal epithelium as this is central to any appreciation of the pathogenesis of colorectal cancer. Colonic epithelial cells are configured in deep invaginations into the wall of the colon called crypts. These cells arise from stem cells that are located at the base of the crypt and migrate to the luminal surface of the crypt where they are shed.2 Evidence is accumulating that stem cells do not have unique intrinsic properties but rather are epithelial cells that acquire self renewal together with related properties as a result of being located within a specialised niche.3 To date, no markers for stem cells have been identified although significant advances have been made towards this holy grail.4 Stem cells divide asymmetrically, with newly synthesised DNA donated to daughter cells that migrate up the crypt ultimately to be shed while “old” DNA is retained in the stem cell population.5 This renders the stem cell particularly vulnerable to developing mutations that might evolve into a malignant clone. To counteract this possibility, cells at the base of crypts, and therefore presumably stem cells, are highly prone to apoptosis, an altruistic form of cell death that rids the organism of cells harbouring dangerous mutations.6
APOPTOSIS IN THE NORMAL INTESTINE
In the unstimulated state there is a relatively low background rate of apoptosis that is restricted to the base of the crypt where stem cells are believed to reside. These epithelial cells have been shown to have a marked tendency to undergo apoptosis following DNA damage.6 Food constituents that are known to prevent the development of colorectal cancer have been shown to enhance apoptosis following DNA damage and this may reflect an important mechanism of cancer prevention.7 These constituents include butyrate,8,9 flavonoids,10 and glucosinate breakdown products from brassicas.11 The mechanism by which DNA damage induces apoptosis in the intestine has not been not fully elucidated though the DNA glycosylase MDB4 plays an important role in detecting damage and coupling this to apoptosis thereby suppressing neoplasia in APCMin/+ mice.12 Furthermore, knockout experiments in mice have shown that apoptosis induced by low dose gamma radiation (<10 Gy) is mediated by both BAX and p53 and is antagonised by bcl-2 and bcl-w and is restricted to epithelial cells.13–16
High dose (>10 Gy) radiation to mice not only induces apoptosis in epithelial cells but also in the endothelial cells of blood vessels supplying the intestinal mucosa. Ceramide is an important mediator of endothelial apoptosis following high dose radiation damage and is antagonised by basic fibroblast growth factor.17 The ischaemic damage caused by endothelial apoptosis may be a key mechanism of action of radiotherapy.18 Although it is possible to induce substantial amounts of apoptosis in crypt epithelial cells it does not appear possible to kill all cells through apoptosis alone. There remains a population of cells that seems resistant to induction of apoptosis. Total cell loss is only achieved if a cell cycle arrest mechanism is imposed in addition to induction of apoptosis. The sterilised cells migrate out of the crypt but are not replaced, resulting in destruction of the mucosa.19
A fascinating feature of apoptosis in the intestine is that epithelial cells on the villus of the small intestine and the table of the colon appear to be highly resistant to apoptosis.20 This is despite the fact these same villus cells having migrated from the crypt only hours earlier. The molecular mechanism underlying this loss of ability to undergo apoptosis is unknown. The only stimulus known to induce apoptosis on the villus is ischaemia/reperfusion injury.21 Villus apoptosis has also been observed in the artificial situation of mice lacking N-cadherin in their epithelial cells.22
APC, WNT SIGNALS, AND THE STEM CELL NICHE IN NORMAL INTESTINE
The molecular signals that create the stem cell niche at the base of the colonic crypt are currently being identified and have already been implicated in the regulation of apoptosis. Central to this niche is regulation of β-catenin/T cell factor (Tcf) activity by the Wnt signalling pathway.23 In the absence of WNT signals, β-catenin is held in a complex with glycogen synthase kinase 3β (GSK3β), axin/conductin, and adenomatous polyposis coli (APC) that is rapidly degraded.24 GSK3β functions to target β-catenin for destruction by ubiquitination. Wnt proteins, of which 19 have been identified in humans, are secreted from myofibroblasts surrounding the base of the crypt.25 On binding to receptors called “Frizzled” on crypt epithelial cells, GSK3β activity is inhibited and degradation of β-catenin prevented (fig 1). The accumulating β-catenin translocates from the cytosol to the nucleus where it binds to members of the Tcf/Lef1 (lymphoid enhancer factor 1) family. Activation of Tcf/Lef dependent transcription leads to transactivation of a genetic programme that has a number of consequences. Firstly, Tcf/β-catenin activity acts as a switch, which when “on” promotes proliferation and suppresses differentiation and when “off” suppresses proliferation and promotes differentiation. Transgenic expression of the secreted Wnt inhibitor dickkopf-2 results in loss of crypts and a reduction in proliferation.26 c-MYC is a target gene of TCF transactivation and is one of the mediators of this switch and drives proliferation. Downregulation of c-Myc leads to cell cycle arrest through an increase in activity of the cell cycle inhibitor p21WAF1/CIP1.27 A second output is the control of cell migration in which cells that will differentiate into goblet, absorptive, and endocrine cells migrate up the crypt and Paneth cells migrate downwards. This is achieved through inverse expression of EphB receptors and their ligands ephrin-B in opposite directions along the crypt axis.28 Current data do not explain the extreme tendency of cells at the crypt base to undergo apoptosis following DNA damage. It seems reasonable to speculate that this property may be regulated ultimately by Wnt signalling.
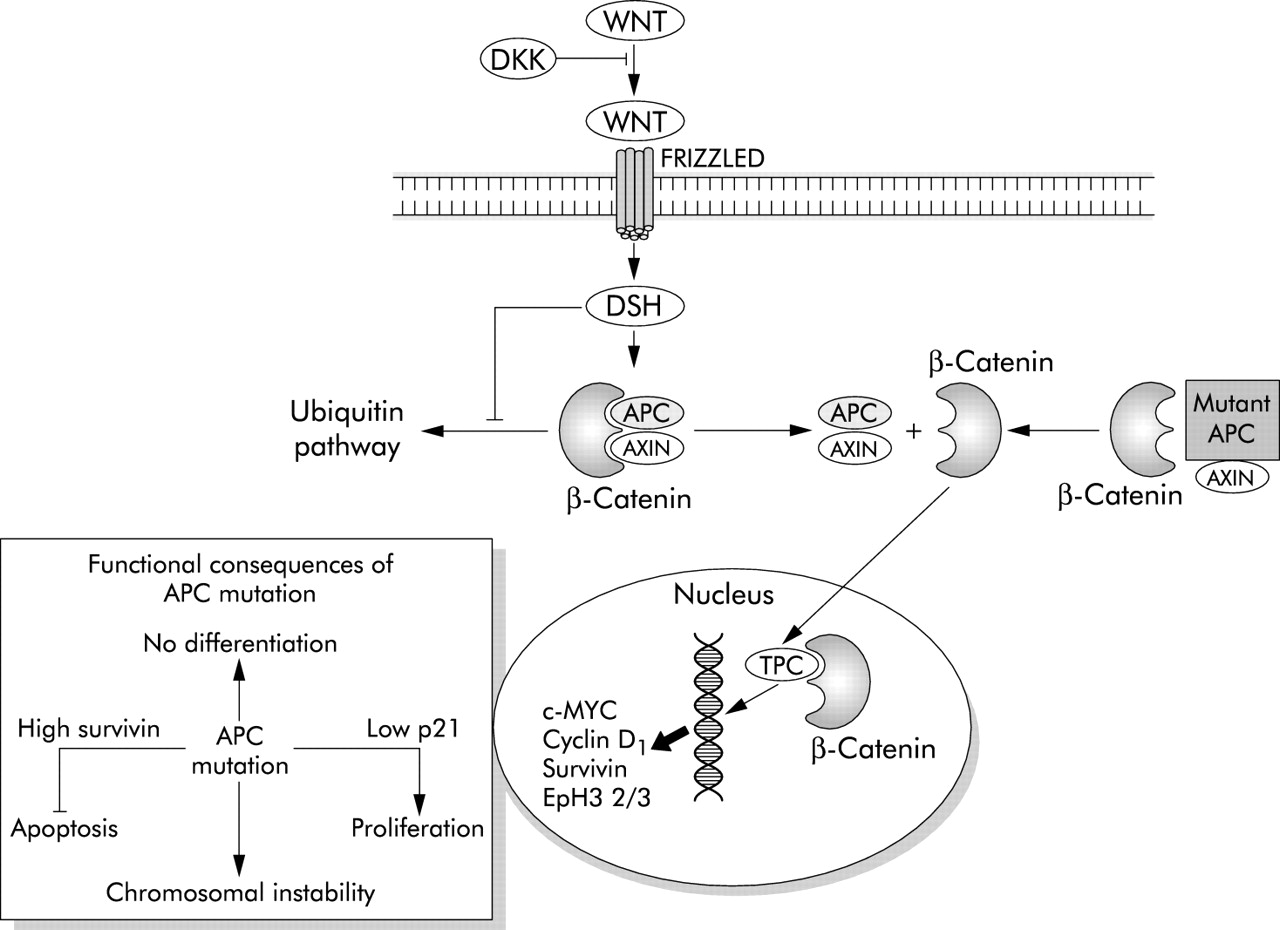
A simplified diagram of the Wnt/APC pathway. DKK, dickkopf-1; DSH, dishevelled; APC, adenomatous polyposis coli; TCF, T cell factor; Eph, ephrin.
INTRINSIC AND EXTRINSIC PATHWAYS OF APOPTOSIS (TABLE 1)
Apoptosis pathways
A remarkable feature of apoptosis is that the essential features of its regulation are conserved in all metazoans.29 Study of cell death pathways in the nematode worm Caenorhabditis elegans has proved enormously helpful in identifying the key elements of cell death mechanisms. During its development, 131 of its 1090 cells invariably die.30 Pioneering experiments by Horvitz established that two genes ced-3 and ced-4 were invariably required for death while ced-9 prevented cell death. Ced-3 proved to be a protease and a homologue of a mammalian protease family called caspases that utilise a cysteine nucleophile to cleave aspartate motifs in target proteins.31 Caspases are synthesised as inactive zymogens that must be activated in order to function.32 Two types are recognised. Upstream initiator caspases, such as caspase 9, are capable of autocatalytic activation while downstream effector caspases such as caspase 3 and 7 can only be activated by initiator caspases. The effector caspases are responsible for all of the morphological features of apoptosis such as chromatin condensation, membrane blebbing, and DNA degradation.
Experiments proved that ced-4 was upstream of ced-3 in C elegans and proved to be the homologue of a mammalian adapter molecule APAF-1 (apoptotic protease activating factor 1).33 In mammals, APAF-1 binds to procaspase 9 and cytochrome c to form a protein complex called the apoptosome.34 This is achieved through binding of caspase recruitment domains in both APAF-1 and caspase 9. When cytochrome c binds to APAF-1 on its WD40 domain in the presence of ATP/dATP, procaspase 9 is activated by autocatalyic cleavage and proceeds to activate caspase 3 and thence the rest of the caspase cascade.35
Ced-9 functions to inhibit ced-4, and remarkably has been found to be a homologue of the mammalian protein Bcl-2.36 Therefore, in order for cell death to take place in C elegans, ced-9 must be inhibited. This is achieved by egl-1 which has homology to the BH3 domain of mammalian bcl-2.37 This four gene pathway constitutes the functional stages of what is now known as the “intrinsic” or “mitochondrial” apoptotic pathway in mammals. In mammals, this pathway is far more elaborate but the functional stages still remain and retain remarkable parallels with C elegans (fig 2).

The intrinsic, or mitochondrial, pathway (after Danial and Korsmeyer29). See text for details.
Release of cytochrome c from mitochondria is a key stage in the intrinsic pathway and this is controlled by the bcl-2 family of proteins. These can be divided into three main subclasses based on their primary structure.38 The antiapoptotic members bcl-2, bcl-XL, and bcl-w all have four BH domains whereas the proapoptotic members bax and bak contain only BH domains 1–3.29 Bax and Bak act as gateways to the intrinsic pathway.39 Death signals direct bax from the cytosol to the mitochondrial outer membrane and bak, which resides in the mitochondrial membrane in an inactive form, to be activated.40 Together they allow release of cytochrome c from the intermembrane space of the mitochondria.41 A third subclass contains only the BH3 domain and these serve as upstream sentinels responding to specific death signals. For example, following DNA damage, p53 controls the transcription of Puma and Noxa,42,43 Bid is activated by the death receptors of the extrinsic pathway,44 while Bad only responds to absence of growth factors or glucose.45 Activation of these BH3 only molecules leads to activation of Bax and/or Bak. The antiapoptotic proteins Bcl-2 and Bcl-XL inhibit apoptosis primarily, although not exclusively, by sequestering BH-3 only molecules thereby preventing activation of Bax and Bak. This arrangement of molecules is the same as in C elegans where the BH3 only homologue egl-1 serves to inhibit the multidomain ced-9 molecule.
Higher organisms have developed a second “extrinsic” apoptosis pathway that is activated by the death receptor Fas (APO-1/CD95) and other members of the tumour necrosis factor (TNF) receptor family (fig 3).29 This pathway is of importance in colorectal cancer as it is inactivated creating a state of immune privilege.46 Binding of their cognate ligand triggers a series of intermediate proteins including FADD/Mort1 and RIP that bind to the death receptor.47 When activated, a signalling complex called death inducing signalling complex is formed by the recruitment of procaspase 8 which is processed autocatalytically and activated. In turn this activates by cleavage the effector caspases 3 and 7.48 Two classes of cell have been defined by the differential effect of bcl-2 on the extrinsic pathway. In type I cells such as thymocytes, bcl-2 does not prevent Fas induced apoptosis whereas in type II cells such as hepatocytes, bcl-2 blocks Fas induced apoptosis. This is because in type II cells Fas ligation also triggers the intrinsic mitochondrial pathways through activation of the BH3 only protein BID by caspase 8.49,50 This secondary activation of the mitochondrial pathway acts to create an amplification loop that appears to be necessary as caspase 8 is not able to activate sufficient effector caspase activity by itself to kill the cell.

The extrinsic, or death receptor, pathway (see text for details).
The TNF receptor can activate both apoptosis and proliferation. The decision appears to be determined by the degree of nuclear factor κB (NFκB) activation that occurs. If sufficient NFκB transcriptional activity is generated, the inhibitory protein c-FLIP is transactivated. c-FLIP inhibits caspase 8 and cell survival is ensured. However, failure to sufficiently activate NFκB leads to caspase 8 mediated apoptosis. This ingenious mechanism ensures only the survival of cells with robust NFκB responses.51
HUMAN CANCER DEVELOPMENT
It is important to review current ideas about how human cancers develop and what phenotypes must be acquired by a cell to become malignant in order to appreciate fully the contribution of apoptosis to the development of colorectal cancer. In contrast with the huge number of genes mutated in cancer, probably only a few phenotypes are required for cancer development, each of which can be achieved by a large number of alternative genetic changes.* Many lines of evidence have been synthesised by Hahn and Weinberg into a number of simple “rules” stating the limited number of phenotypes that a human cell must acquire to become malignant (table 2).52 Firstly, some of the normal DNA repair mechanisms must be disabled, creating a state of genetic instability so that the cell can accumulate sufficient mutations to develop all of the phenotypes essential for malignancy.53 Other mandatory phenotypes include: resistance to growth inhibition, immortalisation, independence from mitogenic stimulation, ability to gain a blood supply or angiogenesis, ability to metastasise and invade and, the subject of this review, the ability to evade apoptosis.
Phenotypes proposed by Hahn and Weinberg to be essential for human malignancy52
CHROMOSOMAL INSTABILITY PATHWAY
In the case of colorectal cancer, two distinct types of genetic instability are recognised; chromosomal (CIN) and microsatellite (MIN) instability.54 They are characterised by distinct types of genetic abnormality and create mutations in distinct sets of genes that control cell division, differentiation, and apoptosis. Of course, abnormalities can develop within any gene but the majority of these abnormalities will either not create a growth advantage or be so catastrophic to cell function that cell division becomes impossible. In CIN there is loss and gain of chromosomes, rearrangements, and a loss of heterozygosity which has been estimated to occur at a rate 105 times greater than in normal cells (fig 4).55 Approximately 60–80% of colorectal cancers display CIN.56 It has long been assumed that CIN is the result of mutations in specific genes that control mitosis. Recently, inactivation of hCDC4 has been identified as an important cause of CIN in colorectal cancers. CDC4 is an E3 ubiquitin ligase that regulates the G1-S checkpoint by targeting cyclin E for destruction. Upregulation of cyclin E following inactivation of hCDC4 produces the CIN phenotype.57 It is intriguing to speculate whether inactivation of hCDC4 is coupled with inactivation of apoptosis in order to prevent elimination of cells that have developed the CIN phenotype.
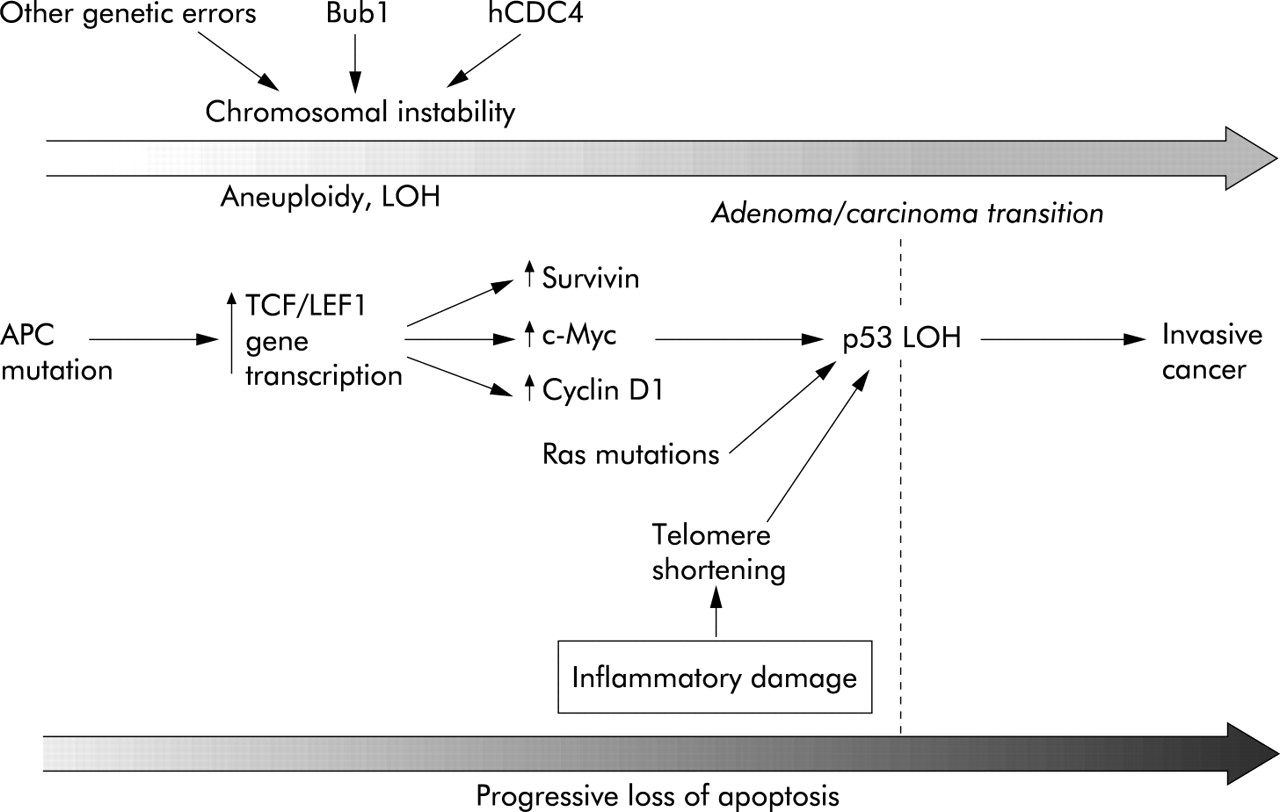
The chromosomal instability (CIN) pathway (see text for details).
APC
Inactivation of the APC (adenomatous polyposis coli) gene is among the earliest genetic events in the development of adenomas that arise via the CIN pathway into colorectal cancer.58 Truncation mutations of APC prevent the ubiquitation and breakdown of β-catenin which thus stabilised migrates to the nucleus activating Tcf/Lef1 with the subsequent transactivation of target genes.23,59 The functional consequences of this are similar to Wnt signalling with one important exception (fig 1). Cells subject to normal Wnt signalling are highly prone to apoptosis whereas Tcf/Lef1 activation as a result of APC mutation renders the cell highly resistant to apoptosis. It may be that the only APC mutant cells that can survive are those in which apoptosis has been inactivated. The molecular basis of apoptosis resistance following mutation of APC is starting to be understood as a number of important apoptosis regulating genes have been identified as Tcf/Lef1 targets. Survivin is one such gene and is a member of the IAP (inhibitor of apoptosis) family. It is a cytosolic protein that may function to inhibit activation of effector caspases and also regulates spindle microtubule function.60 It thereby simultaneously reduces apoptosis and promotes mitotic progression. β-catenin/TCF pathway activation also reduces expression of the initiator procaspase 9 together with the effector caspases 3 and 7.61 Microarray studies suggest that APC mutation also reduces cytochrome c expression.61 Together, these data suggest an explanation for the observation that mutation of APC alters the balance between pro- and antiapoptotic proteins rendering the cell resistant to apoptotic stimuli such as radiotherapy and chemotherapeutic drugs. APC has also been shown to bind directly to microtubules and APC mutation disrupts mitosis, promoting chromosomal instability thus contributing to the emergence of CIN.62
c-Myc
c-Myc, a target of the β-catenin/TCF signalling pathway, has two functional outputs: cell division and apoptosis.27 The capacity of c-Myc to induce cell division is potent but is not unleashed in normal cells unless apoptotic mechanisms are simultaneously inactivated. c-Myc sensitises cells to many apoptotic stimuli, including DNA damage, which are sensed through the p53 pathway and mediated by the BH3 only proteins Puma and Noxa together with Bax.63 Loss of activity of PUMA, Noxa, or Bax will give c-Myc expressing cells a significant growth advantage and drives the development of a malignant clone. c-Myc also augments the effectiveness of the death receptors Fas and Trail induced apoptosis.64
Adenoma/carcinoma transition, telomere shortening, and p53
A fundamental biological problem is how DNA strand breaks which must be repaired are distinguished from the ends of chromosomes which under no circumstances should be ligated to other chromosomes. This vital task is achieved by telomeres, which are stretches of repetitive DNA at the end of the chromosome. Telomeres shorten with each cell division such that once the telomeres are lost, cell cycle arrest is triggered by recognition of a double strand break by protective mechanisms, including p53.65 As telomeres shorten, the risk of CIN increases with chromatin bridge breakage and the fusion of chromosomal ends.52 This phenomenon of telomere shortening and resultant CIN has been observed in patients with ulcerative colitis who subsequently develop colorectal cancer.66 Of course, normally such a serious genetic error would be expected to trigger apoptosis and thus eliminate the aberrant cell. Cells with non-functional p53 are tolerant to CIN arising from telomere shortening and have a powerful selection advantage. The adenoma/carcinoma transition of colorectal cancer is one of the events where failure of apoptosis is decisive in the development of a malignant clone.67
The tumour suppressor gene p53 is mutated in 70% of colorectal cancers. p53 functions to integrate a variety of cellular stresses into a range of responses that include apoptosis.68 It is a transcription factor that binds to specific sequences in DNA and regulates expression of a number of proapoptotic genes. These include Bax and the BH3 only proteins puma and noxa.69 As discussed above, these inactivate bcl-2 and bcl-xL and trigger release of cytochrome c from mitochondria. p53 also increases expression of components of apoptosis effector mechanisms such as APAF-1 and caspase 6.70,71 Furthermore, p53 has important elements of the extrinsic apoptosis pathway as transcriptional targets such as the death receptor Fas and DR5 as well as the BH3 only protein Bid that couples the extrinsic pathway to activation of the intrinsic pathway.72 Although less thoroughly studied, p53 also transrepresses the important IAP gene survivin which may directly inhibit caspase activity.73 In addition to regulation of apoptosis genes, p53 also turns off survival pathways that counteract apoptosis such as the PI3 kinase/AKT survival pathway by increasing expression of the PI3 kinase inhibitor PTEN and in doing so prevents inhibition of p53 by MDM2 (fig 5).74 Little of the data above have been confirmed in colonic epithelial tissue and it is possible that p53 will regulate other important genes in this cell type.

Mitogenic signals are transduced by Ras which inhibits the retinoblastoma (Rb) protein allowing E2F and Myc to promote cell cycle progression. p16INK4a is a tumour suppressor whose mutation permits cyclin D dependent kinase (CDK) to inhibit Rb. High levels of E2F or Myc activates p14ARF and thereby p53 which has multiple outputs. A series of proapoptotic bcl-2 family members are stimulated and via PTEN the antiapoptotic actions of AKT are inhibited. Note that p53 can still respond to DNA damage, and therefore chemotherapy, if the Ras/Rb pathway is disabled.
p53 can initiate cell cycle arrest, DNA repair, and senescence, in addition to apoptosis. Induction of apoptosis, rather than any of its other effects, mediates the functional action of p53 in tumours in vivo.75 A variety of factors determine which outcome is triggered by p53 activation although this is not well understood. Cell type and cellular microenvironment are known to be important. Also, the context of other intracellular signals can be crucial. For example, Myc can switch p53 signalling from cell cycle arrest to apoptosis through prevention of p21 activation by Miz1.76
The mechanism by which p53 is inactivated early in the evolution of cancer can have important consequences on responsiveness to treatment. For example, lymphomas that have lost p53 function through mutation of the Ink4a/ARF pathway,77 which couples mitogenic signals to p53 activation, have a better response to anticancer therapy than tumours that have direct p53 mutations. This is because tumours with the Ink4/ARF mutation can still respond to DNA damage even though the apoptotic response to excessive mitogenesis is disabled.78 These observations may explain why the functional status of p53 has unpredictable effects on the response of cancer cells to commonly used chemotherapeutic drugs. For example, p53 mutation renders HCT116 colon carcinoma cells more sensitive to adriamycin and radiation but less sensitive to 5-fluorouracil.79 A further complication is that p53 appears to respond to RNA damage rather than DNA damage in response to 5-fluorouracil treatment.79,80
MICROSATELLITE INSTABILITY (MIS) PATHWAY AND APOPTOSIS
Defective repair of mismatches between nucleotide bases of DNA constitutes a second pathway to colorectal cancer. This can occur as an inherited syndrome called hereditary non-polyposis colorectal cancer in which mutations of the human mismatch repair genes hMLH1, hMSH2, hMSH6, hPMS1, and hPMS2 results in frameshift mutations of a number of cancer associated genes.81,82 The MIN pathway to cancer is also followed in up to 15% of sporadic colorectal cancers where mismatch repair gene hMLH1 (human mut-L homologue) expression is abolished through silencing of its promoter by methylation.83,84 Such aberrant methylation occurs in what has been termed the CpG island phenotype.85 Some promoters have extensive stretches of cytosine/guanine dinucleotide sequences or CpG “islands”. The principal enzyme responsible for this aberrant methylation is DNMT1.86 A wide range of important genes are silenced in this way, including the DNA repair enzyme MGMT (O-6-methylguanine-DNA methyltransferase), hMLH1, and p14ARF (a regulator of p53) (fig 6).87 Defective repair of mismatch errors in DNA targets genes that contain mononucleotide repeats such as poly(A)n. Genes that are characteristically disabled in this way are transforming growth factor β receptor II (TGFβRII),88 insulin-like growth factor 2 receptor (IGF2R),89 and the proapoptosis gene BAX.90 Tumours that have developed along this pathway tend to have wild-type APC and p53 genes and occur typically in the right colon.87

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The microsatellite instability (MSI) pathway (see text for details).
Jass et al have proposed that colorectal cancers arising through the MSI pathway, particularly those with a high degree of microsatellite instability (MSI-H) which constitutes some 10% of sporadic colorectal cancers, arise from large hyperplastic polyps in the right colon (lesions called “serrated” adenoma).87,91 On the basis of shared molecular defects, Jass and others have argued that the serrated adenoma is a specialised type of hyperplastic polyp. This is frequently misdiagnosed as a villous or tubulovillous adenoma.92 Studies from the 1970s have suggested that hyperplastic polyps occur as a result of defective cell shedding from surface epithelium.93 As a result, it have been suggested that cells within the crypt accumulate causing distortion with increased folding, assuming a saw tooth or serrated appearance. Increased crypt fission results, leading to polyp formation. Many authors claim that cell shedding from the epithelial surface is the result of apoptosis.94 A critical evaluation of the literature shows there is little evidence for this although once detached from the basement membrane apoptosis is inevitable. Nevertheless, it is possible that epithelial apoptosis is defective in hyperplastic polyps and serrated adenomas as mutation of K-RAS or its downstream kinase BRAF are frequent.95–97 The Ras pathway can regulate apoptosis through downregulation of the death receptor Fas.98 Together, these observations suggest that defective apoptosis plays a critical role in the MSI pathway both at its initiation and once fully established through inactivation of Bax.
FUTURE THERAPEUTICS DIRECTIONS
Innumerable studies have demonstrated that anticancer drugs and gamma radiation induce apoptosis in cancer cells grown in culture. It is therefore a surprising that there are few data available on whether apoptosis is an important mechanism of action in vivo. This question was thrown into sharp focus by Waldman et al who showed that while cancer cells undergo cell death in response to anticancer drugs in vitro, when the same cells were grown as xenografts the capacity of the cells to circumvent cell cycle arrest was a stronger determinant of treatment response.99 A major obstacle to investigating this question more thoroughly is the lack of fully developed technologies for the non-invasive detection and measurement of apoptosis in patients. Currently, most studies rely on histological techniques for measurement of apoptosis in biopsy samples. This approach has obvious limitations. The tissue has to be accessible to biopsy and serial measurements are usually impossible because of the need for repeated biopsies. These problems are now being addressed with the development of isotope based imaging techniques employing labelled reagents such as annexin V.100 PET scanning holds particular promise because of its high resolution and capability of scanning multiple parameters such as cell division, blood flow, glucose consumption, and drug distribution.101
Cyclooxygenase, NSAIDS, and apoptosis
Perhaps the most clinically important discovery in colorectal cancer biology in the past 15 years is the role of cyclooxygenase (COX) enzymes in tumour progression. COX catalyses the conversion of arachadonic acid to prostaglandin H2 that is subsequently converted to a large number of other structurally related prostaglandins. There are two principal isoforms: COX-1, which is constitutively expressed, and COX-2, which is not normally expressed in most tissues but induced by a wide range of growth factors and cytokines.102 COX-2 is overexpressed in 40% of adenomas and 85% of colorectal cancers.103 This occurs in a range of cell types within cancer, including the neoplastic epithelial cells, endothelial cells, infiltrating host fibroblasts, and inflammatory cells.104 Decisive evidence of their importance is that disruption of either the COX-1 or COX-2 gene reduces the development of colonic tumours in mice.105,106 There is also a large body of evidence from both clinical trials and animal models that non-steroidal anti-inflammatory drugs (NSAIDs) that inhibit either COX-1 or COX-2 reduce the development of colorectal cancer and even have potential for the treatment of metastatic disease (table 3).107 Induction of apoptosis in adenomas is likely to be an important mechanism.108 There is also accumulating evidence that COX-2 inhibitors can treat fully established cancers without toxicity to the gastrointestinal tract.109
Targets for apoptosis directed therapy
Inhibition of COX enzyme activity can induce apoptosis by altering the balance between pro- and antiapoptotic members of the bcl-2 family, particularly through increasing expression of bcl-2.110 Furthermore, the COX product prostacyclin can activate the nuclear hormone receptor peroxisome proliferator activated receptor (PPAR)-δ and thereby inhibit apoptosis and accelerate adenoma growth.111 Inhibition of COX also increases appropriate cell cycle control, decreases angiogenesis and invasiveness, and modulates the immune response.110 A number of groups have also established that NSAIDs can induce apoptosis through mechanisms independent of inhibition of COX. These include an increase in BAX expression,112 inhibition of IκB kinase beta thereby preventing activation of NFκB,113 and an increase in the sensitivity to cell death receptor induced apoptosis.114
Novel apoptosis based therapies
Naturally, many investigators are starting to exploit the recent discoveries about apoptosis to develop new treatments (table 3). A variety of strategies have been taken. It has been estimated that about half of all cancers express the antiapoptotic proteins Bcl-2 or Bcl-xL.115 Retinoid, PPARγ, and vitamin D receptor agonists all show potential for reducing bcl-2 or Bcl-XL expression in specific circumstances.116 Caution must be exercised as PPARδ agonists stimulate colonic neoplasia.111 Small molecule drugs have been developed that mimic BH3-only proteins; these can bind to and antagonise Bcl-2 and Bcl-xL.117 Death receptor pathways often remain intact in cancer. Attempts to use TNF and FasL have been thwarted by induction of NFκB mediated inflammation and fulminant hepatic failure respectively. However, (TNF) related apoptosis inducing ligand (TRAIL) is well tolerated and is now in phase 1 trials, although not for colorectal cancer.118,119 Some tumours are resistant to death receptor induced apoptosis. This is being tackled by the development of synthetic triterpenoids that antagonise the extrinsic pathway inhibitor c-Flip thus triggering the intrinsic pathway via a Ca2+ mechanism.120 The downstream effector caspases represent a further therapeutic target. These key enzymes are inhibited by IAPs such as survivin that are overexpressed in colorectal cancer. The endogenous antagonists SMAC (Diablo) and HtrA2 (Omi) can by mimicked by synthetic peptides and show promise in tumour xenograft models.121,122
CONCLUSION
There has been remarkable progress in our understanding of apoptosis in cancer and how it is related to genetic instability, cell cycle control, and other crucial processes. As more research is reported, a recurring theme is the importance of the genetic and environmental context in determining the precise behaviour of apoptotic pathways and their functional outcome. This will be a central focus of future research in colorectal cancer whose results will shape the development of therapies that target apoptosis.
Acknowledgments
I thank Mark Boyd and Mark Pritchard for their critical comments on the manuscript and Phil Shering and Dan Tackley of the North Cheshire Concert Band for their mathematical assistance.
REFERENCES
Footnotes
-
↵* If we assume there are 30 000 human genes, each of which can exist in at least two forms, then there are a minimum of 230 000 or 109031 possible combinations of genes. This illustrates that the number of genetic changes potentially related to malignant transformation is hugely greater that the minimum number of phenotypic changes required.