Article Text

Abstract
The products of the two mammalian Axin genes (Axin1 and its homologue Axin2) are essential for the degradation of β catenin, a component of Wnt signalling that is frequently dysregulated in cancer cells. Axin is a multidomain scaffold protein that has many functions in biological signalling pathways. Overexpression of axin results in axis duplication in mouse embryos. Wnt signalling activity determines dorsal–ventral axis formation in vertebrates, implicating axin as a negative regulator of this signalling pathway. In addition, Wnts modulate pattern formation and the morphogenesis of most organs by influencing and controlling cell proliferation, motility, and fate. Defects in different components of the Wnt signalling pathway promote tumorigenesis and tumour progression. Recent biochemical studies of axins indicate that these molecules are the primary limiting components of this pathway. This review explores the intriguing connections between defects in axin function and human diseases.
- aa, amino acids
- AD, Alzheimer’s disease
- APC, adenomatous polyposis coli
- CKI/II, casein kinase I/II
- DIX, Dishevelled and axin binding domain
- DVL, Dishevelled
- Frat, frequently rearranged in activated T cells
- GSK3, glycogen synthase kinase 3
- HB, hepatoblastoma
- HCC, hepatocellular cancer
- JNK, Jun N-terminal kinase
- LEF-1, lymphoid enhancer binding factor 1
- MB, medulloblastoma
- MEKK1, mitogen activated protein/extracellular regulated kinase kinase kinase 1
- MID, MEKK binding domain
- PP2A, protein phosphatase 2A
- RGS, regulator of G protein signalling
- SAPK, stress activated protein kinase
- SCC, oesophageal squamous cell carcinoma
- TCF, T cell specific factor
- TGFβR, transforming growth factor β receptor
- Axin
- Axin1
- Axin2
- carcinogenesis
- colon cancer
Statistics from Altmetric.com
- aa, amino acids
- AD, Alzheimer’s disease
- APC, adenomatous polyposis coli
- CKI/II, casein kinase I/II
- DIX, Dishevelled and axin binding domain
- DVL, Dishevelled
- Frat, frequently rearranged in activated T cells
- GSK3, glycogen synthase kinase 3
- HB, hepatoblastoma
- HCC, hepatocellular cancer
- JNK, Jun N-terminal kinase
- LEF-1, lymphoid enhancer binding factor 1
- MB, medulloblastoma
- MEKK1, mitogen activated protein/extracellular regulated kinase kinase kinase 1
- MID, MEKK binding domain
- PP2A, protein phosphatase 2A
- RGS, regulator of G protein signalling
- SAPK, stress activated protein kinase
- SCC, oesophageal squamous cell carcinoma
- TCF, T cell specific factor
- TGFβR, transforming growth factor β receptor
Axin was originally identified as the product of the mouse gene called “fused” or fu (renamed Axin), and has since been shown to play a crucial role in controlling axis formation during embryonic development. Axin overexpression in frog embryos inhibits dorsal axis formation. Furthermore, mutation of the mouse gene “fused” was found to cause axis duplication in homozygous mouse embryos.1 Wnt signalling activity determines dorsal–ventral duplication in vertebrates and these results suggested that Axin somehow negatively regulates this signalling pathway. The subsequent demonstration of the effect of Axin on β catenin concentrations, together with its biallelic inactivation in some human hepatocellular carcinomas (HCCs), indicated that Axin is a tumour suppressor gene.2 To date, axin has been implicated in at least three different signalling pathways: the stress activated protein kinase (SAPK), transforming growth factor β (TGFβ), and Wnt signalling pathways (fig 1). The multimeric nature of axin complexes suggests that axin might play an important role in other cell signalling systems, in addition to the coordination of these signals.

At least three different signalling pathways are regulated by axin. (A) In the absence of Wnt ligands, axin stimulates β catenin phosphorylation and subsequent protease mediated degradation limits its transcriptional activity. (B) In the presence of transforming growth factor β (TGFβ) signals, axin stimulates Smad phosphorylation by TGFβ receptors (TGFβ receptors I and II). The activated Smads then translocate to the nucleus and activate transcription of downstream target genes. (C) In cells subjected to stress, axin binds to mitogen activated protein/extracellular regulated kinase kinase kinase 1 (MEKK1) and stimulates stress activated protein kinase (SAPK)/JNK (Jun N-terminal kinase) mediated apoptosis. GSK3, glycogen synthase kinase 3.
“The effect of Axin on β catenin concentrations, together with its biallelic inactivation in some human hepatocellular carcinomas, indicated that Axin is a tumour suppressor gene”
WNT SIGNALLING
The Wnt signalling pathway regulates cellular proliferation, differentiation, and motility and is essential for development and morphogenesis.3–5 Alterations in protein phosphorylation status are central to the regulation of Wnt signalling. Several components of the Wnt signalling pathway—including axin, adenomatous polyposis coli (APC), glycogen synthase kinase 3 (GSK3; both GSKβ and GSKα), and β catenin—are phosphoproteins that are regulated through phosphorylation. In unstimulated cells, GSK3α and GSK3β phosphorylate cytoplasmic β catenin, which creates a recognition motif for β transducin repeat containing protein, an E3 ubiquitin ligase.6 The ubiquitinylated β catenin is rapidly degraded by the 26S proteasome, ensuring that cytoplasmic concentrations of β catenin are very low. Secreted Wnt ligands act on the cell surface receptor Frizzled and activate Dishevelled (DVL) through a poorly understood mechanism. Activated DVL binds and inhibits the phosphorylation of β catenin by GSK3β/α, blocking β catenin degradation (fig 2), so that β catenin accumulates and translocates to the nucleus, where it interacts with the T cell specific factor (TCF)/lymphoid enhancer binding factor 1 (LEF-1) transcription factor and induces the transcription of target genes such as c-jun, c-myc, and cyclin D1.7–11 Several in vitro and in vivo studies suggest that axins serve as scaffold proteins that bind directly to many proteins involved in the Wnt signalling pathway, and promote the phosphorylation of β catenin by driving the formation of a complex with APC and GSK3.1,12–14
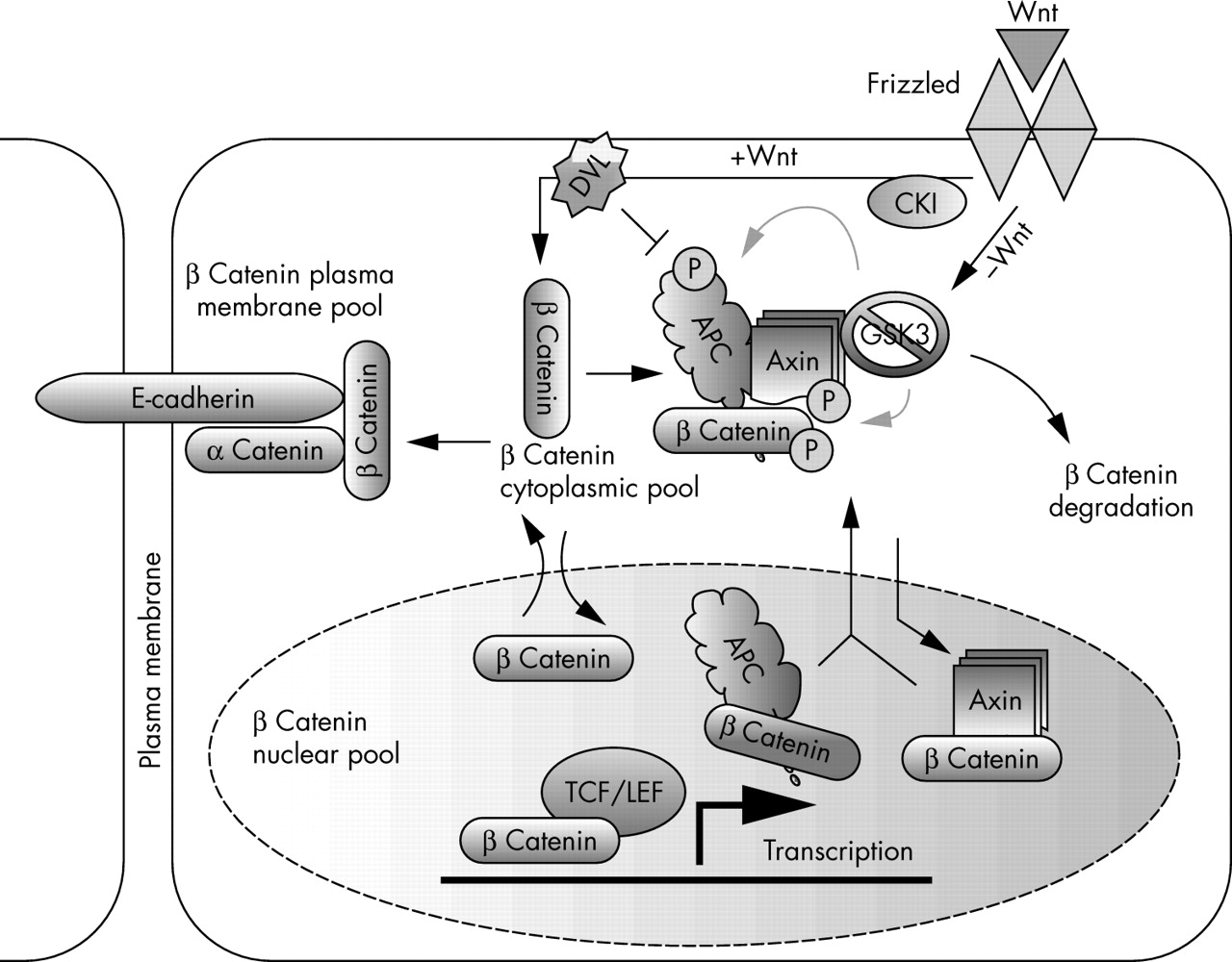
The role of axin in Wnt signalling. The Wnt signalling pathway plays an important role in the regulation of cellular proliferation, differentiation, motility, and morphogenesis. Axin serves as a scaffold protein that binds many of the proteins involved in this pathway. In the absence of Wnt ligands, the β catenin destruction complex, which is composed of adenomatous polyposis coli (APC), glycogen synthase kinase 3 (GSK3), and axin, is formed and leads to the phosphorylation and degradation of β catenin. In the presence of Wnt ligands, the formation of this complex is inhibited and the now stabilised β catenin is translocated into the nucleus and activates the transcription of downstream target genes. Axin is also implicated in shuttling β catenin out of the nucleus. However, β catenin can move in and out of the nucleus independently of axin. It is unclear whether axin participates in the regulation of all of the different pools of β catenin in the cell. Deregulation of many components of the Wnt pathway has been found in human cancer. DVL, Dishevelled; LEF, lymphoid enhancer binding factor; P, organic phosphate; TCF, T cell specific factor.
The β catenin molecule is crucial in this pathway and shows abnormal expression and localisation in a wide variety of human cancers.15 In recent years, so called “canonical” Wnt signalling has received considerable attention because many components of this signalling cascade have been shown to play a role in tumorigenesis.1 The concentration of β catenin in cells is tightly regulated by the APC–axin–GSK3 destruction complex.16 APC, another component of Wnt signalling, is mutated in most (∼ 70%) human colorectal cancers and is also linked to familial adenomatous polyposis.17 Inappropriate activation of Wnt signalling by mutation of different components of this pathway has been seen in a large number of other human cancers including colon carcinoma,18,19 medulloblastoma,20,21 melanoma,22 hepatocellular carcinoma,23 and ovarian and uterine cancer.24–26 Although both β catenin and Wnt are positive regulators of the pathway and have been identified as protooncogenes, APC and axin are considered as negative effectors of the pathway and function as tumour suppressors.27 This model asserts that because axin–APC–GSK3 mediated degradation of β catenin is inhibited by either Wnt signals or mutation of components of the Wnt signalling pathway, the concentration of β catenin in cells is raised, leading to increased cell proliferation and cancer.
TGFβ SIGNALLING
Similar to the Wnt signalling pathway, TGFβ signalling regulates several cellular functions, often in concert with Wnt signalling, including proliferation, differentiation, migration, and apoptosis.28 TGFβ also plays an important role in carcinogenesis. Upon stimulation, TGFβ receptors phosphorylate Smads—the TGFβ effector proteins (fig 1B).29 The phosphorylated Smads then translocate into the nucleus, where they regulate the transcription of target genes. Although TGFβ can often appear to have different effects in different cell types, its primary effect on colonic epithelial cells is to reduce proliferation and induce differentiation.30 Mutations of the TGFβ receptors are often found in cancer cells with defects in mismatch repair systems.31 Axin has been shown to regulate the TGFβ signalling pathway by acting as an adaptor for Smad3, one of the TGFβ effectors (fig 1B).32 Both axin1 and axin2 physically interact with Smad3 through interaction with a domain that is located between the β catenin and DVL binding domains within the C-terminal region. The binding of DVL to axin has been shown to inhibit axin mediated downregulation of β catenin. Although both Smad3 and DVL interact with the same region of axin, biochemical studies have shown that the interaction of DVL with axin does not compete with Smad3 binding to axin. Colocalisation of axin with Smad2 and Smad3 in the cytoplasm has been observed. Upon receptor activation, Smad3 bound to axin is efficiently phosphorylated by TGFβ receptor 1 and dissociates from the axin complex. Thus, axin may facilitate the phosphorylation and transcriptional activity of Smad3.32 TGFβ is one of the anti-oncogenic factors that inhibit nuclear β catenin signalling in the Wnt pathway. Smad4, another effector of TGFβ, has been shown to facilitate Wnt signalling through interaction with β catenin and TCF/LEF.33 Although axin is a negative regulator of the Wnt signalling pathway, it facilitates TGFβ signalling. These results indicate that axin has a dual function in signal transduction—acting as a negative regulator of the Wnt signalling pathway and as a positive regulator of the TGFβ signalling pathway. Deregulation of both pathways is frequently detected in human cancer cells.
SAPK/JNK SIGNALLING
The SAPK/JNK (stress activated protein kinase/Jun N-terminal kinase) signal transduction pathway is activated in cells in response to stress. It is also involved in many normal physiological processes including tissue morphogenesis, cell proliferation, cell survival, and cell death.34 Depending on the cell type or the context of activation of other signalling pathways, SAPK/JNK can help mediate cell survival or apoptosis.35,36 Overexpression of axin1 in cells stimulates SAPK/JNK and can induce apoptosis through the activation of MEKK (mitogen activated protein/extracellular regulated kinas kinase kinase).37,38 A MEKK1 interacting domain (MID) on axin is flanked by the APC and GSK3 binding sites (fig 3A), and the C-terminal region of axin, which contains an oligomerisation domain, is necessary for SAPK activation (fig 3A). The effect of axin on the induction of apoptosis is mediated by SAPK/JNK activation and the destabilisation of β catenin.39 The importance of the axin–MEKK interaction and subsequent SAPK/JNK activation has been shown by the Axin1 mutations found in cancer cells that are resistant to apoptosis.2,39–41 Induced expression of Axin in transgenic mice leads to massive cell death in different organs as a consequence of the increased capacity to degrade β catenin.42 The introduction of wild-type Axin1 into heptocellular and colorectal cancer cells induces apoptosis in a SAPK/JNK dependent manner. GSK3 competes with a MEKK binding site on axin1 and thereby inhibits SAPK/JNK induced apoptosis. This is consistent with observations that GSK3 can prevent cell death under certain circumstances.43 Thus, axin can bind to either GSK3 or MEKK1 and form distinct complexes with opposing effects on apoptosis. Although axin can be “switched” to activate the SAPK/JNK pathway instead of Wnt signalling, it is still capable of downregulating β catenin.44 Axin domains essential for Wnt signalling activation are distinct from those domains required for SAPK/JNK activation.
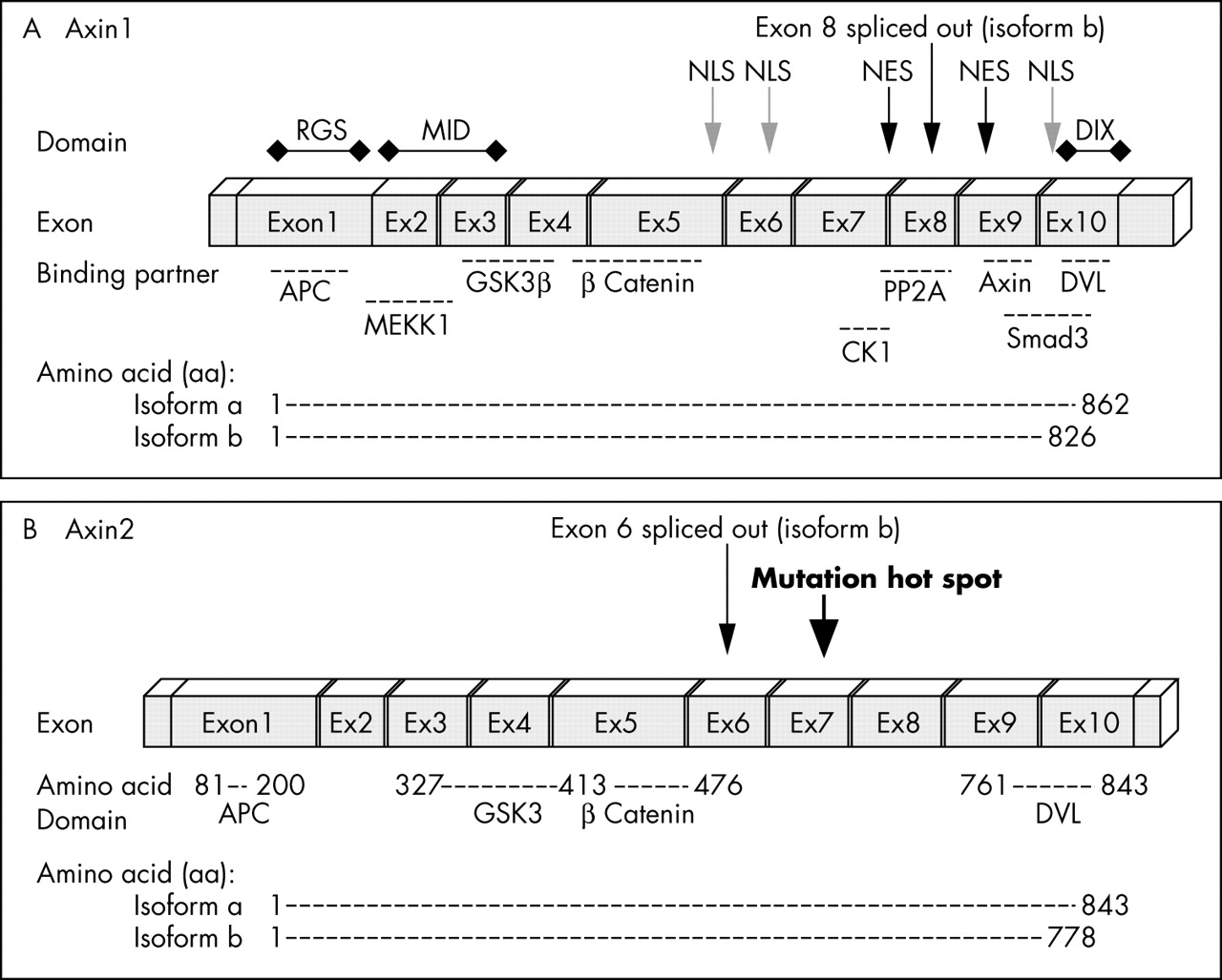
Genomic structure of Axin. (A) Axin1 is composed of 10 exons (encoding isoform a). Exon 8 is spliced out in isoform b. (B) Axin2 also consists of 10 exons that encode 843 amino acids (aa) (isoform a) or a 778 aa polypeptide (isoform b). Similar to Axin1, the binding partners of Axin2 are APC (aa 81–200), GSK3 (aa 372–413), β catenin (aa 414–476), and DVL (aa 761–843). So far, all mutations found in Axin2 are located in exon 7. APC, adenomatous polyposis coli; CKI, casein kinase I; DIX, Dishevelled and axin binding domain; DVL, Dishevelled; GSK3, glycogen synthase kinase 3; MEKK1, mitogen activated protein/extracellular regulated kinase kinase kinase 1; NES, nuclear export signal; NLS, nuclear localisation signal; PP2A, protein phosphatase.
“Depending on the cell type or the context of activation of other signalling pathways, SAPK/JNK can help mediate cell survival or apoptosis”
MEKK4, which is related to MEKK1, was recently identified as another axin binding protein that mediates SAPK/JNK activation.45 Interestingly, MEKK1 and MEKK4 compete for axin binding, even though the binding domain for MEKK4 is separate from the MEKK1 binding domain.46 Axin dimerisation and oligomerisation appear to be necessary for the activation of the SAPK/JNK pathway.37 Other components of Wnt signalling such as GSK3 and CKIε (casein kinase Iε) also compete for binding to axin, and thereby inhibit SAPK/JNK signalling, whereas the binding of APC and β catenin to axin does not interfere with axin mediated SAPK/JNK activation.44
AXIN GENE STRUCTURE, EXPRESSION, AND SUBCELLULAR LOCALISATION
Axin1 (also simply called Axin), which encodes isoforms a and b, and Axin2 (also called Axil or Conductin) have 45% identity at the nucleotide level and the proteins they encode appear to be functionally similar. However, whereas Axin1 is expressed ubiquitously during mouse embryogenesis, Axin2 is expressed in a restricted pattern.1 Axin1 is the constitutively expressed component of the β catenin degradation complex and is essential for the maintenance of low Wnt signalling activity in the basal state. In contrast, axin2 is upregulated in response to increased β catenin concentrations and thus serves to limit the duration and intensity of the Wnt signal.47,48 Axin is downregulated in a Wnt dependent manner and is dephosphorylated after Wnt stimulation, which leads to axin1 destabilisation over time. Cells that receive Wnt ligand signals have low concentrations of axin. Biochemical studies show that the intracellular concentrations of axin are approximately 1000 times lower than those of other destruction complex components, suggesting that axin is the limiting factor in this pathway.49
Axin1
Axin1 was first identified as the product of the mouse fused locus. Its human homologue was mapped to chromosome 16p13.3 and shows 87% similarity to the mouse protein.1 Axin1 “isoform a” encodes a 862 amino acid (aa) polypeptide (GenBank NP_003493), whereas “isoform b” is a shorter form of axin lacking 36 aa within the N-terminal domain encoded by exon 8 (GenBank NP_851393) (fig 3A). The function of the polypeptide region encoded by exon 8 is as yet unknown (fig 4A). This splice form is conserved between different species, suggesting a conserved role. The polypeptide encoded by exon 8 is located between the β catenin binding and Dishevelled and axin binding domain (DIX) domains (fig 3A). The region contains a predicted CKI phosphorylation site and is also close to the axin oligomerisation site, where axin binds to itself. It has been suggested that axin dimerisation is necessary for its stability and function in cells. The spliced exon is also located between two potential axin nuclear export signals.50

Axin expression pattern and subcellular localisation in tumour cell lines and tumour tissue. (A) Western blot analysis of protein extracted from MCF12A and SW480 cells confirmed the existence of two axin1 isoforms (a and b). (B) The subcellular localisation of axin1 was examined in a sporadic colorectal cancer case and axin1 was found to have a predominantly nuclear localisation.
Axin2
A homologue of axin, axin2 was identified by virtue of its interaction with β catenin.16,51,52 Mutation of the Axin2 gene leads to an increase in β catenin concentrations in colorectal cancers with defective mismatch repair systems.53 Molecular studies revealed that Axin2 contains 10 exons spanning more than 2.5 kb.54 Similar to Axin1, Axin2 encodes two isoforms (a and b). In isoform b, exon 6 is spliced out, leading to a transcript with 65 fewer aa (S Salahshor, unpublished data, 2004). Fluorescence in situ hybridisation analysis assigned it to human chromosome 17q24, a region that shows frequent loss of heterozygosity in breast cancer, neuroblastoma, and other tumours.54 Similar to Axin1, Axin2 has binding domains for APC, GSK3, and β catenin (fig 3B).16
Subcellular localisation
Based on the known function and proposed activities of axin, both axins are expected to be located in the cytoplasm of cells where they promote β catenin phosphorylation and degradation. Confocal microscopy of whole mount crypts from patients with colorectal cancer shows that staining for axin1 is diffuse within the nucleus and along lateral cell membranes where β catenin is located, and also in the cytoplasm where both GSK3 and the cytoplasmic pool of β catenin are found. Cell lysate fractionation of two human epithelial cell lines—HCT116 (colorectal cancer cells with β catenin mutation) and HEK293 (embryonic kidney cells)—reflects the axin localisation pattern seen in tissue. Although axin1 was found mainly in the nucleus of HCT116 cells, in HEK293 cells it was localised in both the nuclear and cytoplasmic compartments.55 Staining for axin1 is seen in the cytoplasm of normal epithelial cells, whereas it is located in the nucleus in adenocarcinomas and tumours (fig 4B). The subcellular localisation of axin1 appears to be cell type dependent (fig 5). In contrast, axin2 shows strong nuclear staining in normal tissue. Nuclear localisation of axin2 could also be detected in polyps and carcinomas with some cytoplasmic translocation.55 Despite the similarity between these two molecules, it appears that they are not found within the same compartment of the cell, and thus may have distinct functions. Axin1 can shuttle between the cytoplasm and the nucleus and act as a β catenin chaperone. Two nuclear export (between aa 532 and 667) and three nuclear import signals have been identified on axin1 (fig 3A). Axin1 translocation to the nucleus and its interaction with β catenin is required for axin induced cytoplasmic shifting of β catenin.50,56

Subcellular localisation of axin1 in MCF12A cells. Based on the known function of axin1, it is expected to be mainly localised in the cytoplasm. Confocal sections of MCF12A cells stained with (A and D) anti-axin1 and (B and E) anti-β catenin antibodies (C and F merged) show both nuclear and cytoplasmic axin1 staining, with a pronounced nuclear localisation, whereas β catenin is located mainly at the cell membrane (scale bars, 20 μm). The following antibodies were used: polyclonal anti-axin1 (1/500 dilution; Zymed, South San Francisco, California, USA), monoclonal anti-β catenin antibody (1/500 dilution; Signal Transduction Laboratories; Lexington, Kentucky, USA), secondary monoclonal and polyclonal antibodies (1/1000 dilution; Molecular Probes, Eugene, Oregon, USA). Other studies show a more cytoplasmic localisation for axin1. However, the subcellular localisation of axin1 appears to be cell type dependent.
AXIN BINDING PARTNERS
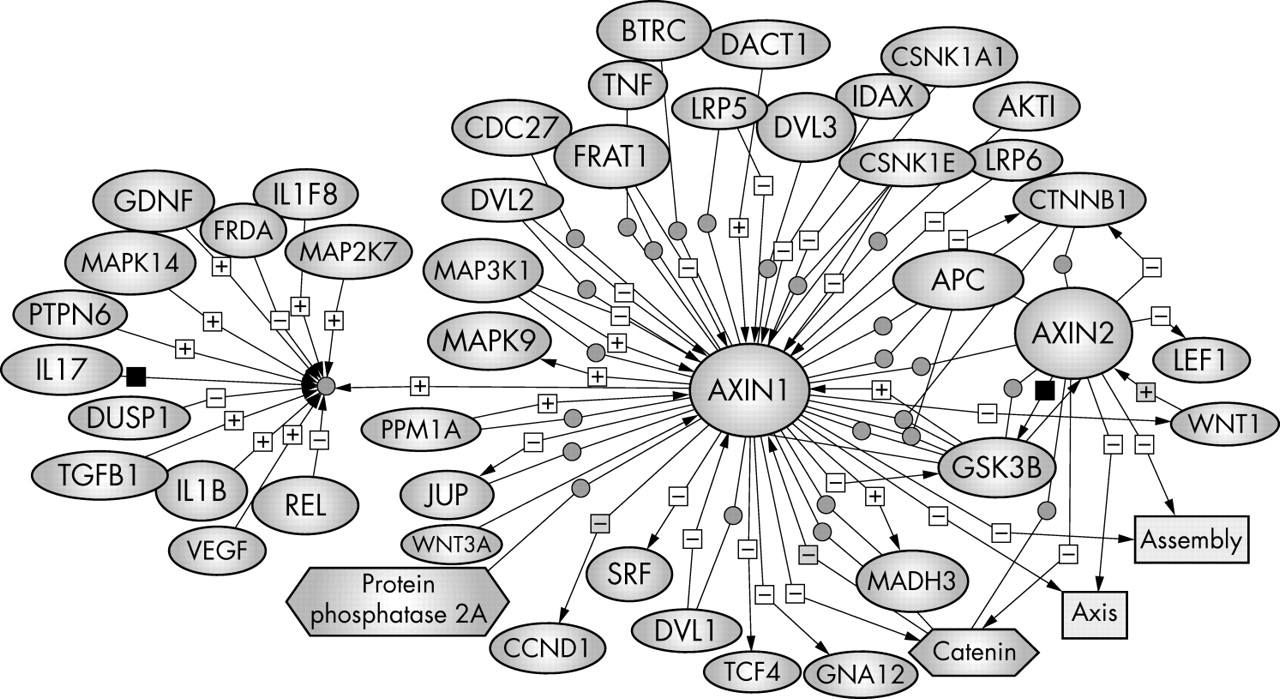
Both axins contain several domains that mediate direct binding to low density lipoprotein related protein receptor, the Frizzled coreceptor,57–59 APC (which appears to assist axin in recruiting β catenin to the axin complex),13 GSK3β/α,60 β catenin.61,62 DVL (DVL-1, DVL-2, and DVL-3),63 MEKK1, CKI,64 protein phosphatase 2A (PP2A),65 frequently rearranged in advanced T cell lymphomas (Frat1),66 and a homodimerisation domain (figs 3 and 6).67

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Axin regulators and binding partners. To bring all the relevant information about axin together, we have used PathwayAssist software (Stratagene) to generate a network of molecules that are either positively (+) or negatively (−) regulated by axin. Each interaction is annotated with the source references and contains hyperlinks to the source literature on PubMed. The biological data have been extracted from PubMed. The references that support the interaction or regulation for each molecule can be viewed by holding the mouse on the plus or minus sign. This pathway is an online navigation tool (http://kinase.uhnres.utoronto.ca/Sima/axins_pathway/).
APC
The region of axin involved in APC binding shows significant homology to members of the regulators of G protein signalling (RGS) family.1,12 The region of APC that interacts with axin consists of a conserved sequence of approximately 20 aa containing a Ser-Ala-Met-Pro (SAMP) motif.16 The APC gene contains three SAMP repeat sequences, all of which are located after its mutation cluster region, where most truncating mutations in cancer are found. These mutations eliminate all axin binding sites on APC, but preserve some of the β catenin binding domains, indicating the important role that the interaction between axin and APC plays in APC tumour suppressor activity.22,68 Thus, the tumorigenic potential of mutated APC correlates with loss of binding to axin1/axin2, rather than loss of β catenin binding. APC binding to axin via its RGS domain is required for the efficient downregulation of β catenin by APC.22,51 However, overexpression of axin in colorectal cancer cell lines bearing a mutated form of APC that lacks an axin binding domain can still promote β catenin downregulation.16,61,69 Similarly, axin mutants lacking the APC binding domain are still capable of β catenin downregulation when overexpressed.51,62 These results suggest that APC might be only a cofactor for axin1/axin2. However, multiple binding sites on APC for β catenin (10 binding sites) and axin1/axin2 (three binding sites) may be important for the efficient assembly of the β catenin degradation machinery in different cell types or various in vivo conditions. These results further indicate that the concentration of axin rather than its interaction with other components of Wnt signalling might control its activity in cells, and that axin is the rate limiting factor in these reactions. In contrast, in SW480 colorectal cancer cell lines, which carry a truncated form of APC, β catenin is stabilised. Expression of ectopic APC leads to β catenin degradation.22,70 Endogenous axin1 and axin2 are present in this cell line, but appear not be capable of downregulating β catenin in the absence of wild-type APC.
“Direct binding of APC to β catenin is not essential for APC to degrade β catenin, whereas the binding of axin to β catenin is necessary for it to degrade β catenin”
However, overexpression of exogenous axin2 can induce β catenin degradation in SW480 colon carcinoma cells. Overexpression of an axin2 mutant that lacks the β catenin binding domain does not induce β catenin degradation, whereas an axin2 mutant lacking the RGS domain (APC binding domain) can induce degradation.16 A β catenin mutant that does not interact with axin2 is resistant to degradation induced by the ectopic expression of axin2 in SW480 cells. However, these β catenin mutants were efficiently degraded when wild-type APC was introduced.71 These observations indicate that direct binding of APC to β catenin is not essential for APC to degrade β catenin, whereas the binding of axin2 to β catenin is necessary for it to degrade β catenin.
β Catenin
Axin1 has been shown to interact directly with β catenin at residues adjacent to the GSK3α/β binding domain,12 and β catenin binding to axin2 is abolished or strongly reduced by mutations in β catenin ARM (armadillo repeat) 3 and 4.71 A direct interaction between axin2 and β catenin in the same region as axin1 has been shown. This interaction promotes the phosphorylation of β catenin by CKIα and GSK3, which is required for the ubiquitination and subsequent degradation of β catenin. The degradation of β catenin can also occur in a GSK3/CKIα phosphorylation independent manner. Siab (mammalian homologue of drosophila gene sina) binds to the ubiquitin conjugating enzyme through its N-terminal region and forms a complex with Ebi. Ebi is an F-box protein that binds to β catenin and forms a complex that functions as a ubiquitin ligase and is able to downregulate β catenin in a p53 inducible manner.72,73 Recently, it was shown that activated p53 can downregulate β catenin. It was suggested that p53 induces the faster mobilisation of axin into the β catenin degradation complex, and thereby stimulates β catenin degradation.74 These observations imply that axin might be able to promote the degradation of β catenin, through both phosphorylation dependent and independent pathways.
GSK3
GSK3α and GSK3β are essential players in the β catenin destruction complex. GSK3α/β are serine/threonine kinases that phosphorylate three conserved serine and threonine residues in the N-terminal domain of β catenin.75,76 β Catenin is a poor substrate of GSK3 in vitro, and GSK3 does not bind β catenin directly, requiring axin and APC to facilitate its interaction with the β catenin target.1,77,78 Axin1 dramatically (> 20 000 times) facilitates the phosphorylation of APC and β catenin by GSK3 in vitro.61 Phosphorylation by GSK3α/β requires a priming phosphorylation by a third party kinase. In the case of β catenin, this priming enzyme is CKIα.79 Axin itself is phosphorylated by GSK3α/β and this modification increases its stability.80 Unphosphorylated axin has a lower affinity for β catenin, reducing its ability to promote the formation of the β catenin destruction complex.27,80,81 Axin1 contains several possible GSK3α/β phosphorylation sites, and mutation of these leads to reduced axin phosphorylation by GSK3.
“Axin1 dramatically facilitates the phosphorylation of APC and β catenin by glycogen synthase kinase 3 in vitro”
In drosophila, expression of a hypomorphic allele of Armadillo (the fly homologue of β catenin) on a background lacking Zeste-White3/Shaggy (the fly homologue of GSK3), revealed that the concentration of β catenin/Armadillo was still sensitive to expression of Wingless, the fly homologue of Wnts. This effect was mediated by changes in the concentration of axin.59,82 As mentioned previously, axin concentrations in cells are the lowest of all of the regulatory components and could therefore be rate limiting.49 However, Wnt signalling in mammalian cells does not appear to alter axin1 protein concentrations (B Doble and J Woodgett, unpublished observation, 2004). These data suggest that the Wnt pathway may use multiple mechanisms to regulate the concentration of β catenin, although the dominant mechanism appears to be via phosphorylation.
DVL
DVL binds to the C-terminal region of axin, which includes the DIX domain, and inhibits axin activation.83 The association of DVL with axin modulates the ability of axin to dimerise. DVL also binds CKI, which can promote Wnt3a mediated DVL phosphorylation. Phosphorylated DVL has a high affinity for Frat, which binds to and inhibits GSK3.63,84 In Wnt stimulated cells, Frat bound to DVL might be able to prevent GSK3 bound to axin from phosphorylating β catenin (fig 3A). Both DVL and axin have DIX domains that are necessary for the binding of DVL and axin to intracellular vesicles and actin filaments, which suggests that axin and DVL may regulate receptor mediated endocytosis of the Wnt signalling pathway.85 DVL also plays a role in relocating axin to the plasma membrane upon Wnt signalling.86 Studies on living embryos have confirmed previous findings that Wnt signals cause a relocalisation of axin from the cytoplasm to the plasma membrane and that this relocalisation is DVL dependent.59
CKI/II
CKI has recently been found to be an activator of Wnt signalling because microinjection of CKI into xenopus induces a secondary axis.87 The overexpression of CKI mimics Wnt signalling by stabilising β catenin and thereby inducing the expression of β catenin target genes. CKI has been detected in the axin–GSK3 complex and appears to phosphorylate APC and β catenin (at Ser45) in an axin dependent manner.64,79,88 CKI also competes for MEKK1 binding and can attenuate axin–SAPK/JNK signalling.89 In contrast, although CKII also interacts with axin, it does not have an inhibitory effect on SAPK/JNK signalling.89 CKII interacts and phosphorylates DVL, which is thought to inhibit the function of axin, thereby stimulating Wnt signal transduction.90
PP2A
PP2A is a broad specificity serine/threonine protein phosphatase that comprises a catalytic “C” subunit and various “A” and “B” regulatory subunits that confer selective binding to proteins and scaffolds within cells. PP2A physically interacts with axin and APC in the axin–GSK3–β catenin destruction complex and decreases the phosphorylation of axin, which suggests a positive role for PP2A in Wnt signal transduction.14 Phosphorylation of APC is also important for β catenin binding and its subsequent degradation. PP2A has been implicated as a positive effector for Wnt mediated signalling.91 However, the phosphatase has other, confounding effects, probably because of it action on both positive and negative effectors of the pathway, such that its role is clearly complex.14,65,92,93
AXIN VARIANTS IN CANCER
Alterations in both axin1 and axin2 have been detected in several different tumours (tables 1 and 2). Mutations are found in most axin domains including the APC (RGS) and β catenin binding domains. Axin sequence variants have also been found in colon, ovarian, endometrioid, adenocarcinoma, and HCC cell lines (table 3). Biochemical and functional studies have shown that these mutations interfere with the binding of GSK3 and that they also alter the interaction between axin and two upstream activators of TCF dependent transcription, Frat1 and DVL.94 Several studies have investigated the role of axin in several types of tumour.
Sequence variants of the Axin1 gene found in human cancer
Axin2 variants in human diseases
Axin1 variants identified in cancer cell lines
Medulloblastoma
Medulloblastoma (MB) is the most common malignant brain tumour in children.105 Most MBs are sporadic; however, patients with germline mutations in APC or PTCH (patched) genes carry a higher risk of this disease.106 In addition, mutations in β catenin, another component in the Wnt pathway, have been detected in MB tumours.107–109 Recently, several deletions and single somatic point mutations in Axin1 have been found in sporadic MB cases.20,21,110 These aberrations include somatic point mutations, deletions, and loss of heterozygosity. The C-terminal deletion in these cases corresponded to loss of exons 6–10, where the DIX and oligomerisation domains are located. Oligomerisation of Axin1 is crucial for the inhibition of TCF transcription,67 and C-terminal deleted axin1 fails to downregulate β catenin and cell growth in the SW480 colon cancer cell line.63 Another type of inframe deletion that removes exons 1–5, the APC binding domain, has also been found in MB. The truncated protein lacks binding sites for APC, GSK3, and β catenin (table 1). This mutant is also incapable of downregulating β catenin. In addition, some truncated forms of axin1 may act as dominant negatives and inactivate endogenous axin1.111
Colorectal cancer and familial tooth agenesis
Many components of the Wnt signalling system are mutated in colorectal cancer. Germline loss of function mutations in the APC gene are associated with an inherited form of colorectal cancer—familial adenomatous polyposis—with 90–95% penetrance. Somatic APC mutations are also found in most sporadic colorectal cancers.112 Alterations in other components of Wnt signalling, including β catenin, TCF, axin1, and axin2, found in colorectal cancer indicate the important role that this pathway plays in the aetiology of this disease.113 Most Axin1 mutations in colorectal cancer occur between exon 1 and 5, where the APC, GSK3, and β catenin binding domains are located (table 1). Mutations in axin2 have been found in approximately 20% of mismatch repair deficient colorectal tumours (table 2).6,53,94,101,103 In most cases, one base deletion or insertion occurs in the mononucleotide repeat sequences located in exon 7, leading to a frame shift and premature protein truncation.53 These mutations lead to elimination of the DIX domain, where DVL binds and negatively regulates axin activity. This domain is also essential for homo-oligomerisation of axin. The mutant form of axin2 appears to be more stable than the wild-type protein. Transfection of normal fibroblasts with axin2 mutants led to the accumulation of β catenin in the nuclei.
“Alterations in other components of Wnt signalling, including β catenin, TCF, axin1, and axin2, found in colorectal cancer indicate the important role that this pathway plays in the aetiology of this disease”
Immunohistochemical staining of β catenin in tumours with defects in axin2 also showed nuclear accumulation of β catenin in the cells. These results suggest a dominant negative effect of axin2 mutant proteins in cells. High levels of Axin2 gene expression have been detected in most human colon cancer cell lines (SW480, SW620, LoVo, SW620, Caco2, HT29, HCT116, T84, HCT15, and Alab), in addition to colorectal tumours. Axin2 appears to be a transcriptional target of the Wnt signalling pathway. Upregulation of Axin2 by Wnt–β catenin appears to constitute a negative feedback loop that acts to restrain or desensitise Wnt signalling. Axin2 gene expression increases in response to raised concentrations of β catenin, thereby modulating the duration and activity of the Wnt signal.47,48 In addition to colorectal cancer, several Axin2 mutations have been found in patients with oligodentia. Linkage analysis of a Finnish family with colorectal cancer in association with oligodentia revealed a high logarithm of the odds score in chromosome 17 between markers D17S949 and D17S1352, where more than 80 known or predicted genes (including Axin2) are located. Direct mutational analysis of the coding sequence of Axin2 identified a base pair transition in exon 7, leading to premature termination of translation of Axin2, whereas family members with normal dentition showed no sign of Axin2 mutation or neoplasia. Axin2 is expressed in developing dental tissues, and germline Axin2 mutations found in patients with oligodentia provided the first evidence for its function.104 In a form of familial colorectal cancer, Gardner syndrome, the occurrence of odontomas and supernumerary teeth in association with familial adenomatous polyposis has been reported.114–117 These results indicate that overactivation of Wnt signalling as a result of mutation of a component of the β catenin destruction complex, in addition to carcinogenesis, may lead to the failure of tooth development (tables 1–3).
Hepatocellular carcinomas
HCC and hepatoblastoma (HB) are primary liver cancers that occur predominantly in adults and children, respectively. Alteration of β catenin occurs in approximately 20% of HCCs and 40–89% of HBs.23,118–121 A fraction of HCCs (∼ 10%) with wild-type β catenin have mutations in Axin1 or Axin2 instead.2,122 These data suggest that alterations in the Wnt pathway are involved in the pathogenesis of a large proportion of both HBs and HCCs. The axin1 mutations identified in the previous reports include truncation mutations as a result of either small deletion/insertion or nonsense mutations, although Axin1 mutations found in liver cancer show a different spectrum of alterations. Most of the mutations found in these cases were predominantly missense. These Axin mutations apparently only affect one allele, so that their oncogenic effect may result from the dominant negative activity of the mutant Axin, which causes stabilisation of free β catenin.16 In addition, detection of loss of heterozygosity at the Axin1 locus suggests that axin1 functions as a tumour suppressor. However, in a subset of tumours mutations in Axin1 or Axin2 were found in addition to mutations in β catenin.97 The expression of wild-type Axin1 in HCC or colorectal cancer cells with APC, β catenin, or Axin1 mutations led to the induction of apoptosis.2 Even though axin1 and axin2 are both able to downregulate β catenin when overexpressed, and show similar biochemical characteristics, axin2 does not compensate for axin1 mutations in HCCs. Therefore, axin1 and axin2 expression is either cell type dependent or, more likely, these two proteins are not functionally equivalent.
Ovarian endometrial adenocarcinomas
Ovarian cancer is one of the most frequent gynaecological malignancies in women.123 A subtype of ovarian cancer, endometriosis, often shows defects in the components of the Wnt signalling pathway. Although mutations in APC are rare in this type of tumour, mutation of β catenin is found in 16–24% of cases.24,124,125 Recently, mutations in axin1 and axin2 have been detected in a subset of ovarian endometrial adenocarcinomas, where activation of TCF dependent transcription could be detected in a tumour with an Axin2 frameshift mutation (tables 1 and 2). As noted in cell lines, the mutated form of axin2 appears to be more stable than the wild-type axin2 protein.101
Oesophageal squamous cell carcinoma
Oesophageal cancer accounts for 7% of all gastrointestinal malignancies, but in some regions of Asia its incidence may be as high as 170/100 000 of the population. About half of the oesophageal cancers diagnosed are squamous cell carcinomas (SCCs). Several studies suggest a role for defects in Wnt signalling in the pathogenesis of SCC. Deletion of chromosome 17 (where Axin2 is located) has been found in 45% of SCCs.126 Immunohistochemical studies of axin1 have shown cytoplasmic localisation of the protein in normal stratified squamous epithelium of the oesophagus, whereas a mixed pattern of expression was detected in tumours. Protein expression analysis of seven oesophageal SSC cell lines revealed differential axin expression. Four cell lines (TE1, TE15, TT, and TTn) showed high expression, whereas another three cell lines (TE2, TE8 and TE13) showed very low axin1 expression. However, northern blot analysis of the same cells showed similar amounts of RNA in all cases. Several mutations and polymorphisms have been reported in SCC tumours and cell lines (tables 1 and 3), and a correlation between reduced axin1 expression and tumour progression has been suggested in oesophageal SCC.100,127
Roles of axin/GSK3 in Alzheimer’s disease
In addition to tumorigenesis, defects in the Wnt signalling pathway have also been postulated to contribute to the pathogenesis of Alzheimer’s disease (AD).128–130 The pathology of this form of dementia involves the deposition of amyloid plaques, which are made up of Aβ peptides derived from the abnormal cleavage of amyloid precursor protein, and neurofibrilliary tangles, which are composed of hyperphosphorylated forms of the microtubule associated protein, tau. These two lesions are associated with neuronal cell death. GSK3 is one of several protein kinases that phosphorylates tau at sites observed in the neurofibrilliary tangles.131–133 Axin negatively affects tau phosphorylation by GSK3.134 Tau that has been phosphorylated by GSK3 has a decreased capacity to bind and stabilise microtubules.135 Altered expression of PP2A, another protein implicated in Wnt signalling, has also been associated with AD pathology and appears to play a major role in regulating tau phosphorylation in the adult brain.136–139 These results have led to the hypothesis that deregulation of GSK3 might be involved in the pathogenesis of AD.140,141 Considering the central role that the scaffolding protein axin plays in regulating GSK3 mediated phosphorylation of β catenin, and the fact that axin negatively regulates GSK3 mediated tau phosphorylation, it has been suggested that deregulation of the phosphorylation status of GSK3 may contribute to tau pathology in AD. A direct role for Wnt signalling in AD pathology remains to be demonstrated. However, it can be speculated that events leading to a decrease in GSK3–axin complex formation, and ultimately axin concentrations, may allow the aberrant phosphorylation of tau by GSK3 at primed sites that are increased in the AD brain, and thus contribute to tau dysfunction and pathology.142
AXIN: A POTENTIAL TARGET FOR CANCER TREATMENT
Given the clear link between defects in Wnt signalling components and several human cancers, compounds that either positively or negatively regulate these molecules according to their role in the pathway will provide valuable reagents for the development of anticancer drugs. Because axin positively regulates the SAPK/JNK (apoptosis) pathway and negatively regulates the Wnt (survival) pathway, and is the limiting factor in these systems, a search for axin antagonists or agonists might lead to the discovery of compounds that have potential for the treatment of cancer. To ascertain whether the Axin mutations found in cancers were biologically important, normal Axin1 genes were inserted into the cancer cells and their effects on cell growth monitored. In the case of HCC cell lines, the introduction of Axin appeared to induce programmed cell death, suggesting a therapeutic potential. Although this opens up the possibility of using Axin as a cargo gene in gene therapy, small molecule approaches are also being pursued.
“A search for axin antagonists or agonists might lead to the discovery of compounds that have potential for the treatment of cancer”
The high frequency of mutations in the Wnt pathway in specific tumour types suggests that the activated transcriptional output of these pathways efficiently misregulates multiple growth regulatory functions. Because axin can degrade β catenin when overexpressed in cells that lack functional APC it is a good candidate for cancer treatment. One drawback is that although the overexpression of axin1 strongly promotes the downregulation of wild-type β catenin in colon cancer cells, mutant, oncogenic β catenin is unaffected.
Thus, Axin treatments would be selective for APC associated cancers. More studies of the mechanistic details of these molecules are required to elucidate the precise role and effect of axin stimulation in cancer treatment.
Take home messages
-
Axins (both Axin1 and its homologue Axin2) are tumour suppressor genes
-
Deregulation of axin has been found in different forms of cancer
-
Wnt signals modulate axin concentrations in the cell
-
Axin is a rate limiting component of different signalling pathways
-
The Wnt, stress activated protein kinase/Jun N-terminal kinase, and transforming growth factor β signalling pathways are regulated by axin
-
Upregulation of axin leads to the phosphorylation and degradation of β catenin
-
The overexpression of axin promotes apoptosis
CONCLUDING REMARKS
A large body of evidence indicates that the regulation of β catenin stability and concentration in cells by the axin–APC–GSK3 complex is crucial for both embryogenesis and carcinogenesis. Axins act as negative regulators for Wnt signalling, and positive regulators for SAPK/JNK signalling. Because axin itself is sensitive to Wnt signals, axin concentrations in cells fluctuate, which probably impacts upon its other functions. Despite being discovered in 1997, there is much to be learned about the physiological functions and roles of axin. In common with several other molecules in the Wnt signalling pathways, axin binds to different components and regulates opposite functions in the cell. The question is how axin operates to maintain the specificity of these different signalling pathways. Downregulation of axin can be just as catastrophic as too much activity. Axin2 overexpression in cells leads to apoptosis, yet upregulation of axin2 is found in most colorectal cancers. Deregulation of axin2 can be detected in adenomas where cells do not show β catenin nuclear accumulation, which indicates that axin2 might be an early marker of tumour initiation. Furthermore, axin2 is mostly located in the nucleus, and has access to the “active” form of its target molecule, β catenin. It remains to be determined how the basal concentrations of axins are controlled and how their nuclear and cytoplasmic localisation is accomplished. Currently, our understanding of axins suggests that these proteins play key assembly functions for promoting efficient protein–protein interaction and stand at the crossroads of several signalling paths—helping to integrate and coordinate the plethora of continuously changing signals.
APPENDIX 1 ONLINE LINKS AND DATABASES
-
http://kinase.uhnres.utoronto.ca/Sima/axins_pathway/ (Axin page)
-
http://us.expasy.org/prosite/ (Prosite database)
Acknowledgments
Supported by grants from the Canadian Institutes of Health Research and the National Institute of Cancer/Canadian Cancer Society.
REFERENCES
Supplementary materials
The third sentence in the abstract should read; �Overexpression of mutant axin results in axis duplication in mouse embryos�. Also, Figure 5; A and D (in green) are b-catenin and figures B and E (in red) are Axin1.
There are two errors in the abstract and figure 5 of this article. The third sentence in the abstract should read; �Overexpression of mutant axin results in axis duplication in mouse embryos�. Also, Figure 5; A and D (in green) are b-catenin and figures B and E (in red) are Axin1.
Linked Articles
- Correction