Article Text

Abstract
The identification of the non-random chromosome rearrangements between the EWS gene on chromosome 22q12 and members of the ETS gene family in Ewing’s sarcoma, peripheral primitive neuroectodermal tumour, Askin tumour, and neuroepithelioma has been a key advance in understanding their common histogenesis and defining the Ewing’s sarcoma family of tumours (ESFT). In addition to improvements in diagnosis and potentially the stratification of patients for risk, biological investigations of these gene fusions may define targets for much needed therapeutic strategies to eliminate minimal residual disease or metastatic disease. Insight into their relation with other oncogenic events in ESFT will advance risk group analysis and ultimately may improve clinical management and survival for patients with this disease.
- Ewing’s sarcoma
- diagnosis
- prognosis
- treatment
- translocations
- ESFT, Ewing’s sarcoma family of tumours
- Euro EWING 99, European Ewing tumour working initiative of national groups 99
- FISH, fluorescent in situ hybridisation
- pPNET, peripheral primitive neuroectodermal tumour
- RT-PCR, reverse transcriptase polymerase chain reaction
Statistics from Altmetric.com
- ESFT, Ewing’s sarcoma family of tumours
- Euro EWING 99, European Ewing tumour working initiative of national groups 99
- FISH, fluorescent in situ hybridisation
- pPNET, peripheral primitive neuroectodermal tumour
- RT-PCR, reverse transcriptase polymerase chain reaction
Ewing’s sarcoma is the second most common malignant bone tumour occurring in children and young adults, and accounts for 10–15% of all primary bone tumours.1 The annual incidence is approximately 0.6/million total population, and it usually occurs between the ages of 10 and 20 years. It affects 13/million 0–24 year olds each year in the UK,2 and is slightly more common in males than females (ratio, 1.5 : 1). It has been described in siblings,3,4 although this is rare and the disease does not appear to be implicated in familial cancer syndromes. Genetic influences may play some role in its aetiology because black Afro-Caribbean and Chinese populations are less frequently affected than the white population.5,4
Ewing’s sarcoma can affect any bone but the most common sites are the lower extremity (45%), followed by the pelvis (20%), upper extremity (13%), axial skeleton and ribs (13%), and face (2%).6 The femur is the most frequently affected bone, with the tumour usually arising in the midshaft. Typically, by light microscopy, the tumour consists of small round cells with regular round nuclei containing finely dispersed chromatin and inconspicuous nucleoli, and a narrow rim of clear or pale cytoplasm. Ultrastructurally, the tumour contains primitive cells with a smooth nuclear surface, scanty organelles, and cytoplasmic glycogen in pools or aggregates.
“The femur is the most frequently affected bone, with the tumour usually arising in the midshaft”
Tumours with similar histology also arise in soft tissues. These include peripheral primitive neuroectodermal tumour (pPNET), neuroepithelioma, and Askin tumour. pPNET is the second most common soft tissue malignancy in childhood, accounting for 20% of sarcomas.7 It is frequently found in the chest wall (Askin tumour), paraspinal tissues, abdominal wall, head and neck, and extremities.7,8 However, soft tissue extension is common in osseous Ewing’s sarcoma and infiltration of adjacent bone is frequent in soft tissue pPNETs, which often makes it difficult to determine the primary site of tumour origin.
The identification of the non-random t(11;22)(q24;q12) chromosome rearrangement9,10 in these aggressive malignant tumours arising in bone and soft tissue is strong evidence for their common histogenesis, and provides a valuable characteristic for their differential diagnosis from other small round cell tumours of childhood. These tumours, including Ewing’s sarcoma, pPNET, Askin tumour, and neuroepithelioma, are now collectively recognised as the Ewing’s sarcoma family of tumours (ESFT). In this review, the diagnostic, prognostic, and therapeutic power of the t(11;22)(q24;q12) translocation and other molecular abnormalities in ESFT will be reviewed.
DIAGNOSIS
Accurate diagnosis of ESFT is crucial for the most appropriate clinical management of patients. Adequate clinical information and the recognition of the morphological, immunocytochemical, and sometimes ultrastructural features (table 1) of ESFT are all required for its differential diagnosis from other small round cell tumours of childhood, such as neuroblastoma, rhabdomyosarcoma, lymphoma (non-Hodgkin’s), other primitive neuroectodermal tumours, desmoplastic small round cell tumour, poorly differentiated synovial sarcoma, and small cell osteosarcoma.
EWS fusion types described in Ewing’s sarcoma family of tumours (ESFT) and other sarcomas
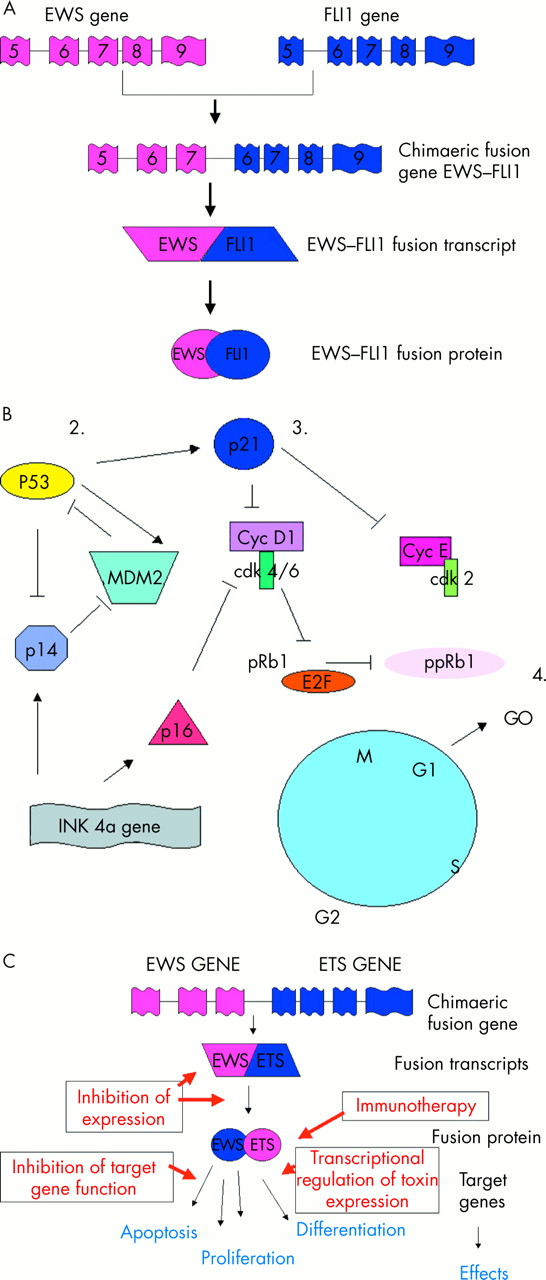
Increasingly, ESFT are being characterised by non-random gene rearrangements between the EWS gene on 22q12 and various members of the ETS gene family (table 1). The most frequent gene rearrangement is the t(11;22)(q24;q12) translocation, found in about 85% of these tumours (fig 1; table 1). Fusion of the EWS gene on 22q12 with the FLI1 gene on 11q24 results in a chimaeric fusion transcript EWS–FLI1 (fig 2A). Substitution of the EWS domain with a portion of the FLI1 transcriptional domain results in an EWS–FLI1 fusion transcript with increased transcriptional activity compared with that of normal FLI1. The chimaeric protein product is capable of transforming NIH3T3 cells in vitro,11–13 but neither EWS nor FLI1 alone have this capacity.
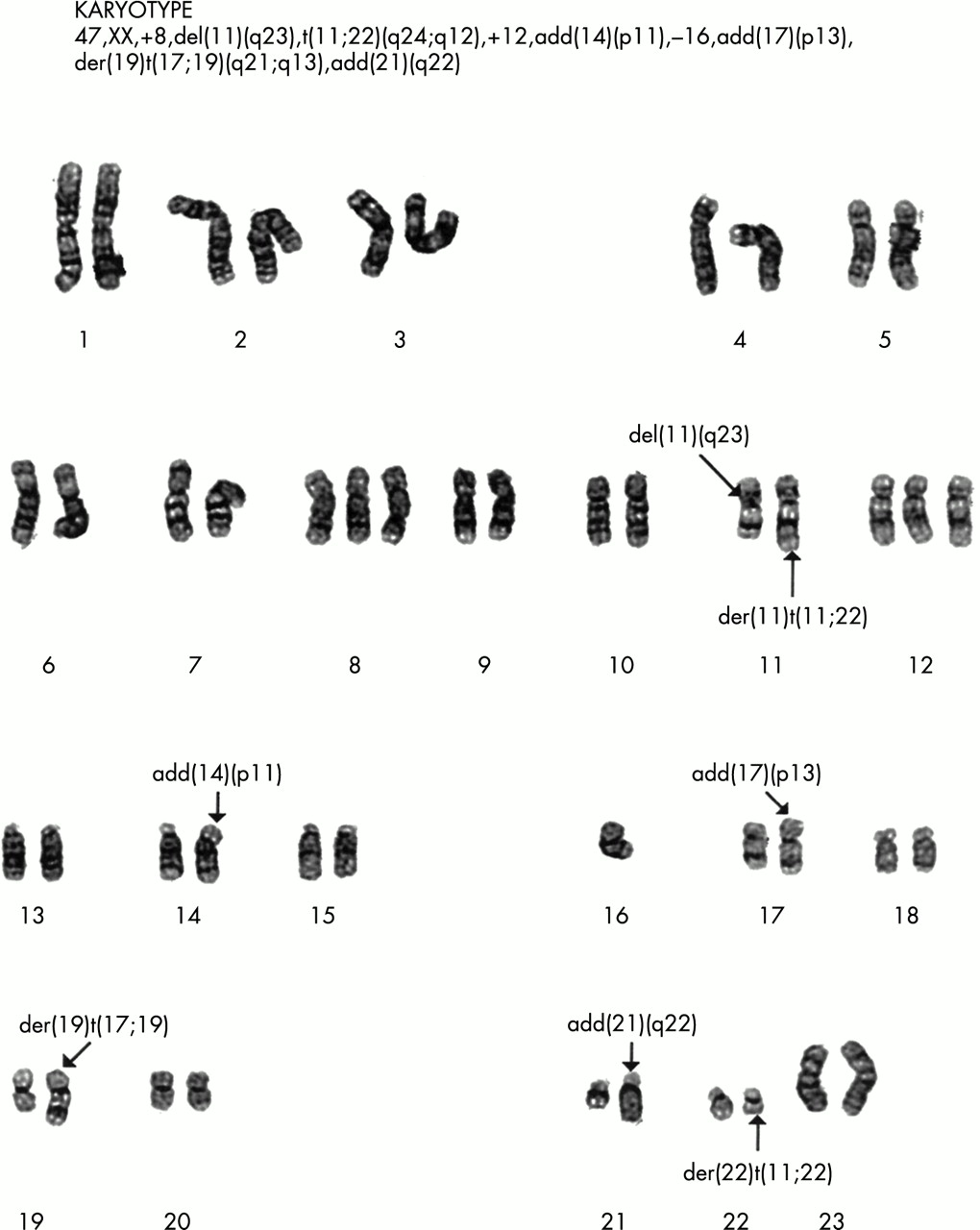
G banding of chromosomes from a Ewing’s sarcoma showing the t(11.22)(q12.24) translocation. This tumour is also characterised by trisomy of chromosome 8 and 12 and loss of chromosome 16, frequently described secondary aberrations in the Ewing’s sarcoma family of tumours. Reproduced with the permission of P Roberts, Department of Cytogenetics, St James’s University Hospital, Leeds, UK.
{kind=link}
{kind=link}
(A) Diagrammatical representation of the t(11;22) (q24;q12) translocation resulting in the generation of the EWS–FLI1 type 1 fusion transcript. (B) Cell cycle regulatory proteins implicated in regulation of the G1 checkpoint. 1. Despite the absence of cytogenetically detectable 9p21 chromosomal aberrations, p16 deletions are one of the most frequent secondary molecular aberrations identified to date in the Ewing’s sarcoma family of tumours (ESFT). 2. p53 is rarely mutated in primary ESFT, although mutation in this tumour suppressor gene has been described in up to 50% of ESFT cell lines. Mutation of p53 in ESFT is associated with a poor outcome for a small group of patients. 3. Mutation of the p21WAF gene and/or downregulation of its nuclear expression is a common aberration in ESFT. For most tumours, loss of p21WAF expression is independent of p53 status, suggesting gene silencing of WAF1 by mutation or hypermethylation. 4. Loss of pRb1 is rarely described in ESFT, but abrogation of the G1 checkpoint appears to be important in the progression and development of the ESFT clinical phenotype. Cdk, cyclin dependent kinase; Cyc, cyclin; pRb1, retinoblastoma protein 1; ppRb1, phosphorylated pRb1. (C) Potential strategies to exploit EWS–ETS gene rearrangements for therapeutic purposes.
Other EWS–ETS gene family rearrangements have been identified in the remaining 15% of tumours, with the t(21;22)(q22;q12) translocation resulting in fusion of EWS with the ERG gene on 21q22 being the second most common (table 1).14 The fusion protein derived from EWS–ERG is similar to that of the EWS–FLI1 gene product, with EWS replacing the transcriptional domain of ERG. Other variant translocations have been described where EWS is fused with ETV1 (t(7;22)(p22;q12)),14 E1AF (t(17;22)(q21;q12)),15 and FEV (t(2;22)(q33;q12)),16 in addition to more complex translocations involving multiple chromosomes such as t(11;14;22)(q24;q11;q12) and t(10;11;22)(p11.2;q24;q12). The biological importance of these more complex gene rearrangements is not yet clear, but they may be associated with more aggressive tumour behaviour (P Roberts, personal communication, 2002). There has also been at least one case of cryptic EWS–FLI1 fusion in a tumour with normal chromosomes 11 and 22.17
“Accurate diagnosis of the Ewing’s sarcoma family of tumours is crucial for the most appropriate clinical management of patients”
Therefore, cytogenetic evaluation of ESFT is an essential part of their diagnostic evaluation. Conventional G banding may identify EWS–ETS gene rearrangements in a proportion of tumours, although diagnostic accuracy can be enhanced using either the reverse transcriptase polymerase chain reaction (RT-PCR)18 and/or fluorescent in situ hybridisation (FISH).19–21 The presence of EWS–ETS gene fusion products detected by RT-PCR or FISH correlates well with conventional G banding studies, but perhaps more interestingly these techniques can be used to identify gene fusions in tumours where conventional G banding has been uninformative, either because of a very complex gene rearrangement or the lack of mitotic cells. Indeed, the ability of RT-PCR to detect fusion transcripts in small pieces of tumour, even in fine needle biopsies or bone marrow aspirations, makes this a very attractive tool to aid in the diagnosis of ESFT.18
The EWS gene is also rearranged in several other tumours, which are clinically and morphologically distinct from ESFT (table 1). A multiplex RT-PCR for these different gene rearrangements can be very useful to aid in the differential diagnosis of sarcomas.18 However, there are some reports of EWS–ETS gene arrangements in biphenotypic sarcomas with features of muscle and neural differentiation,22,23 and rarely in rhabdomyosarcomas,24 neuroblastomas,25 rhabdoid tumour,26 extraskeletal mesenchymal chondrosarcoma,27 and solid papillary tumour of the pancreas.28 Whether these examples represent extremes of the ESFT or rare pathological entities themselves is not clear. Consequently, the presence of an EWS–ETS gene rearrangement alone may not be sufficiently powerful to diagnose ESFT, but in conjunction with other clinical and pathological criteria it is pathognomonic of this tumour group.29
PROGNOSTIC VALUE
Improved outcome may be achieved by stratifying patients for treatment according to risk. A recent systematic review of prognostic tumour markers in ESFT has been undertaken (NHS health and technology assessment programme; grant number 97/15/03; for further details of these results see http://www.prw.le.ac.uk/epidemio/personal/rdr3/paed.html). However, very few prognostic markers are routinely used in ESFT, probably as a result of the small number of studies and patients evaluated in clinical outcome investigations. This identifies the need for multicentre, quality controlled, prospective clinical outcome studies in ESFT. Currently in ESFT the most useful prognostic indicators in clinical practice are clinical features, including the presence of metastatic disease at diagnosis (measured by imaging and histological examination of bone marrow), tumour volume, and primary tumour site (patients with a primary tumour of the pelvis have a worse prognosis).
Using RT-PCR to detect EWS–ETS fusion transcripts several alternative forms of EWS–FLI1 have been described, reflecting the different breakpoints in FLI1 and EWS. The most common type, designated EWS–FLI1 type 1, consists of the first seven exons of EWS joined to exons 6 to 9 of FLI1, and accounts for approximately 60% of cases (fig 2A). The type 2 EWS–FLI1 fusion also includes FLI1 exon 5 and is present in a further 25%.30 Recent studies have suggested that the fusion transcript type may be prognostically powerful,30,31 with the presence of EWS–FLI1 fusion transcript type 1 being associated with improved outcome compared with that in patients with other fusion transcript types.30,31 However, this may be limited to patients with localised disease.30 The EWS–FLI1 fusion transcript type 1 appears to encode a less active chimaeric transcription factor32 and to be associated with a lower proliferation index33 than that of tumours with other fusion transcript types; this might provide the biological basis for the correlation between fusion transcript type and clinical outcome. However, other studies have shown that the two gene fusion products EWS–FLI1 and EWS–ERG do not define distinct clinical phenotypes, suggesting that differences in the C-terminal partner of ESFT gene fusions are not associated with significant phenotypic differences.34 The clinical and biological relevance of different fusion transcript types is currently being evaluated through a European prospective, quality controlled, clinical outcome study, namely: the European Ewing tumour working initiative of national groups 99 (Euro EWING 99).
The presence of disseminated disease, detected by imaging and cytological and histological examination of bone marrow at diagnosis in ESFT, is one of the most powerful markers of poor prognosis. However, some patients with apparently localised disease at diagnosis rapidly develop metastases and aggressive disease, from which they subsequently die. This suggests that these patients have low level metastatic disease that is not detected by current routine methods, such as imaging and histological examination of bone marrow at diagnosis. Fusion products of specific EWS–ETS gene rearrangements have successfully been used as targets for RT-PCR to identify low numbers of circulating ESFT cells with improved sensitivity and specificity.35–37 Disease detected in the peripheral blood by RT-PCR appears not to be clinically informative, although in bone marrow it may predict a poor clinical outcome.35,36,38–41 This suggests that RT-PCR for the EWS–ETS fusion transcript type in bone marrow may be useful to improve the stratification of patients for treatment and could result in a redefinition of the term disease free. This hypothesis is currently being evaluated in Euro EWING 99.
Additional chromosomal abnormalities have also been described in ESFT, although these are less frequently described than rearrangements of the EWS–ETS genes and their clinical implication less well understood. They consist of both numerical and structural aberrations, including gains of chromosomes 842–46 and 12,43–46 the unbalanced translocation t(1;16),42,46–50 and deletions at the short arm of chromosome 1.46,50 Initial studies suggested that individuals with gain of chromosome 8 might have a poor clinical outcome,43,45 although this has recently been disputed.48 Deletion of chromosome 1 is reported to be associated with an unfavourable outcome in individuals with localised disease.48 Up to 15 different chromosomes, other than those relating to EWS–ETS gene rearrangements, have been implicated in ESFT, although much of this has been in the form of individual patient data (see http://www.prw.le.ac.uk/epidemio/personal/rdr3/paed.html). It is important to evaluate the frequency and clinical importance of these additional chromosomal aberrations in large clinical outcome studies, because a more complex karyotype with multiple chromosomal aberrations appears to be associated with poor outcome and may be prognostically powerful in ESFT.46,50
Loss of cell cycle control occurs in a variety of malignancies, often as secondary genetic changes during multistage progression. In ESFT, abrogation of the G1 checkpoint appears to be important in the progression and development of the clinical phenotype (fig 2B),50–52 consistent with the hypothesis of unchecked cell division. This hypothesis is supported by recent studies demonstrating changes in G1/S regulatory genes after downregulation and forced expression of the EWS–FLI1 fusion gene.53
Homozygous deletions and/or mutations of the INK4a gene on chromosome 9p21 have been reported in seven of 4154 and eight of 2750 primary ESFTs, although one study failed to identify mutations of this gene in all 70 primary ESFTs tested.55 Interestingly, loss of the INK4a gene product p16INK4a in four of 20 cases was associated with shortened survival and the presence of metastatic disease at diagnosis. Conversely, loss of the alternatively spliced INK4a variant p14ARF is rarely described even in tumours where p16INK4a is lost.52 This is consistent with the hypothesis that selective loss of p16INK4a function may save p14ARF,56 with epigenetic inactivation by promoter hypermethylation of the E1α exon resulting in the loss of p16INK4a expression but retaining expression of functional p14ARF.57
“Although the loss of p21WAF expression appears to be the most common aberration in primary Ewing’s sarcoma family of tumours, no correlation with clinical behaviour has yet been reported”
The loss of retinoblastoma gene expression is very rarely described in ESFT,50,52 and although mutations in the tumour suppressor gene p53 have been described in up to 50% of ESFT cell lines,58,59 this appears to be a rare event in primary tumours.51,60–62 However, in the small clinical subset of patients with ESFT in whom p53 is mutated this is associated with a very poor outcome,51,62 although the expression of p53 protein is reported by some to correlate with a poor prognosis.63 Amplification of the MDM2 gene, which inactivates the p53 protein, is also rare in ESFT,51,54 consistent with the hypothesis that p53 and regulators of its activity may not play a dominant role in ESFT transformation and progression.
One of the most frequent aberrations of a tumour suppressor in ESFT reported to date is loss of p21WAF expression (11 of 20,52 and 30 of 5062). Because p21WAF is transactivated by wild-type but not mutated p53, loss of its expression might reflect inactivation of p53. This is true for some ESFTs,64 but for most loss of p21WAF expression is independent of p53 status, suggesting that gene silencing of WAF1 by mutation or hypermethylation may be important in these tumours. Although the loss of p21WAF expression appears to be the most common aberration in primary ESFT, no correlation with clinical behaviour has yet been reported, although there are reports in other tumour types of its prognostic power.65–67
THERAPEUTIC VALUE
In human ESFT cells, knockout studies using anti-sense EWS–FLI1 oligonucleotides or cDNAs have shown reduced growth in in vitro68–70 and in vivo69,71 models. These observations imply that EWS–ETS gene rearrangements are important for the maintenance of the ESFT malignant phenotype. Coupled with the high frequency of these rearrangements in ESFT, this suggests that the products of these gene rearrangements play a role in the genesis of ESFT. As the genetics and biology of these gene fusions and their protein products are elucidated, their potential as targets for therapeutic intervention are being explored so they may be exploited for the benefit of adolescents and adults with these tumours (fig 2C).
In animal models, the chimaeric fusion transcripts are reported to act as dominant oncogenes, contributing to the development of ESFT by binding to the promoter region of specific genes, and altering their expression.11,12,72–74 Deletion analysis suggests that the EWS gene functions as the regulatory or modulating domain, with the ETS gene partners possibly acting to establish promoter activity and also to link with signal transduction pathways.75 Gene expression is a multistage process potentially providing several steps that may be targeted as a therapeutic strategy. Although general inhibitors of transcription and translation may effectively downregulate EWS–FLI1 protein expression and induce growth arrest in vitro,76 this is unlikely to be specific enough for therapeutic use. However, more specific targeting of oncogene expression, through the use of antisense reagents to interfere with ribosomal translation, degradation, or processing of pre-mRNA complexes may be more successful. Because EWS–ETS oncoproteins are thought to protect ESFT cells from apoptosis, antisense treatment may increase the efficacy of chemotherapeutic strategies,77 or conversely may have a direct effect by downregulating the role of EWS–FLI1 in maintaining cell proliferation.68,69,76 Recent studies have shown that the delivery of antisense oligonucleotides in polyalkylcyanoacrylate nanocapsules results in a greater inhibition of ESFT growth in nude mice than that seen with free antisense oligonucleotide injections,71 although the delivery of antisense reagents in vivo remains a challenge in therapeutic trials. Alternatively, the oncogenic function of the EWS–ETS proteins may be targeted by inhibition of the protein itself using—for example, a competitive dominant negative strategy for the selective disruption of gene translation and expression.68
“Recent studies have also shown that EWS–ETS may interfere with normal RNA processing, suggesting that the gene fusions may function both in transcriptional and post transcriptional processes to modulate growth”
The primary goal for much research that aims to exploit the therapeutic value of EWS–ETS gene fusions has been to identify the EWS–ETS fusion target genes responsible for its oncogenic activity. Because the transformed phenotype of these tumours is thought to be dependent on the deregulated expression of target genes, transcriptional repressors that downregulate the same transcriptional targets may be used to inhibit the function of these oncoproteins.78 This approach has been evaluated in alveolar rhabdomyosarcoma.79 Alternatively, fusion oncoprotein function may be inhibited by expressing antibody fragments intracellularly.80 Although the genes targeted by the EWS–ETS fusion proteins are not yet fully defined, analysis of RNAs differentially expressed by NIH3T3 cells expressing FLI1 or EWS–FLI1 have identified some interesting candidate genes including Manic Fringe (m-FNG),81 stromelysin,82 EAT-2,74 E2-C,83 transforming growth factor βII receptor (TGFβ2 receptor),84,85 and platelet derived growth factor C (PDGF-C).86 However, no single or group of gene(s) has yet been identified that reproduces the EWS–FLI1 phenotype, and thus far direct target genes have not been distinguished from indirect ones.
The function of these novel fusion proteins as potent transcription factors may also be exploited to increase the delivery of cytotoxin directed treatments. Because some of the fusion transcripts have been shown to be more potent transcriptional activators than their wild-type counterparts, the delivery of exogenous toxic genes under the control of these regulatory elements is a potentially specific mechanism to target these tumours. Although normal cells may express the wild-type gene, its reduced transcriptional activity is hypothesised to result in little or no activation of the exogenous toxin. This approach has yet to be evaluated in ESFT, but in alveolar rhabdomyosarcoma target expression of the diphtheria toxin A gene has resulted in expression and appropriately selective toxicity.87
Take home messages
-
EWS–ETS gene rearrangements are important for the differential diagnosis of the Ewing’s sarcoma family of tumours
-
EWS–ETS fusion transcript type may predict clinical behaviour
-
Elucidating the biology of these gene fusions and their protein products may provide targets for novel therapeutic intervention
-
Insight into other molecular abnormalities in ESFT is essential
Recent studies have also shown that EWS–ETS may interfere with normal RNA processing,88,89 suggesting that the gene fusions may function both in transcriptional and post transcriptional processes to modulate growth. Further evidence that EWS–ETS fusion transcripts may operate through mechanisms other than promoters of specific target genes comes from the observations that EWS–FLI1 binds a nuclear ribonucleoprotein involved in RNA splicing, which can result in inhibition of EWS–FLI1 transactivation,90 and that an artificial EWS–FLI1 DNA binding domain mutant that has no in vitro binding activity still retains some transforming activity.91
An alternative therapeutic strategy in ESFT may be to exploit the EWS–ETS gene fusions as novel tumour antigens for immunotherapy. The intracellular expression of translocation protein products protects them from immune surveillance. If the fusion transcripts are to be exploited as novel tumour antigens to increase immune recognition of tumour cells then the peptides derived from these fusion proteins (in conjunction with major histocompatability complex class I molecules) must be expressed on the tumour cell surface. This strategy is being explored in alveolar rhabdomyosarcoma,92 synovial sarcoma, clear cell sarcoma, desmoplastic small round cell tumour,93 and a variety of other sarcomas.94,95
CONCLUSIONS
In summary, as in many cancers, improved diagnostic and staging methodologies for patients with ESFT will lead to better risk group analysis and ultimately may improve clinical management and survival for patients with this disease. The identification and characterisation of the EWS gene rearrangements in ESFT has been the most important advance made in these tumours in the past two decades, leading to improvements in diagnosis and potentially stratification of patients for risk. Current biological investigations of these gene fusions may define targets for much needed therapeutic strategies to eliminate minimal residual or minimal metastatic disease, although delivery of these novel agents remains a major challenge. Understanding the role of these tumour specific fusion transcripts and protein products is essential, but insight into their relation with other oncogenic events in ESFT is crucial so that molecular abnormalities may be exploited for the maximum benefit of individuals afflicted with this disease.