Article Text

Abstract
Colorectal cancer (CRC) that demonstrates microsatellite instability (MSI) is caused by either germline mismatch repair (MMR) gene mutations, or ‘sporadic’ somatic tumour MLH1 promoter methylation. MLH1 promoter methylation is reportedly correlated with tumour BRAF V600E mutation status. No systematic review has been undertaken to assess the value of BRAF V600E mutation and MLH1 promoter methylation tumour markers as negative predictors of germline MMR mutation status. A literature review of CRC cohorts tested for MMR mutations, and tumour BRAF V600E mutation and/or MLH1 promoter methylation was conducted using PubMed. Studies were assessed for tumour features, stratified by tumour MMR status based on immunohistochemistry or MSI where possible. Pooled frequencies and 95% CIs were calculated using a random effects model. BRAF V600E results for 4562 tumours from 35 studies, and MLH1 promoter methylation results for 2975 tumours from 43 studies, were assessed. In 550 MMR mutation carriers, the BRAF V600E mutation frequency was 1.40% (95% CI 0.06% to 3%). In MMR mutation-negative cases, the BRAF V600E mutation frequency was 5.00% (95% CI 4% to 7%) in 1623 microsatellite stable (MSS) cases and 63.50% (95% CI 47% to 79%) in 332 cases demonstrating MLH1 methylation or MLH1 expression loss. MLH1 promoter methylation of the ‘A region’ was reported more frequently than the ‘C region’ in MSS CRCs (17% vs 0.06%, p<0.0001) and in MLH1 mutation carriers (42% vs 6%, p<0.0001), but not in MMR mutation-negative MSI-H CRCs (40% vs 47%, p=0.12). Methylation of the ‘C region’ was a predictor of MMR mutation-negative status in MSI-H CRC cases (47% vs 6% in MLH1 mutation carriers, p<0.0001). This review demonstrates that tumour BRAF V600E mutation, and MLH1 promoter ‘C region’ methylation specifically, are strong predictors of negative MMR mutation status. It is important to incorporate these features in multifactorial models aimed at predicting MMR mutation status.
- Tumour features
- BRAF V600E mutation
- MLH1 methylation
- predictors of MMR mutation status
- genetic epidemiology
Statistics from Altmetric.com
- Tumour features
- BRAF V600E mutation
- MLH1 methylation
- predictors of MMR mutation status
- genetic epidemiology
Introduction
Lynch syndrome (LS) (formerly hereditary non-polyposis colorectal cancer (HNPCC)) is an autosomal dominantly inherited disorder caused by germline mutations in the DNA mismatch repair (MMR) genes, with ∼90% of the mutations identified occurring in the MLH1 and MSH2 genes, and the remainder in the MSH6 or PMS2 genes.1 2 A deleterious or pathogenic MMR gene mutation is associated with a significant increased risk of colorectal cancer (CRC), where approximately 53% of men and 33% of women who carry a mutation in MLH1 or MSH2 will be diagnosed with CRC by age 703 with corresponding risks for a mutation in MSH6 of 22% and 10% respectively,4 and for PMS2 mutation carriers the risks are 20% and 15%.5 LS is the most common hereditary CRC syndrome, accounting for 1–6% of all CRCs, depending on the population studied.6 7 Individuals with LS also have an elevated risk of developing cancers of the endometrium, stomach, small intestine, ovaries, urinary tract, brain, hepatobiliary tract, pancreas and skin.
Defects in MMR caused by a germline mutation in one of the MMR genes results in tumour development with a specific tumour phenotype. A functional defect in the MMR process results in genomic instability, exhibited as replication errors in tumour tissue DNA but not seen in the DNA from normal surrounding mucosal tissue. These replication errors are frequently observed in repetitive mononucleotide and dinucleotide microsatellite DNA markers such that high levels of microsatellite instability (MSI-H) are frequently observed in CRCs in LS when several of these microsatellite markers are assessed. Loss of tumour MMR protein expression assessed by immunohistochemistry (IHC) is reported to be evident in >90% of cases with clearly pathogenic MMR gene mutations and shows >90–95% correlation with the MSI-H phenotype.8 MMR IHC testing provides additional information to prioritise specific MMR genes for mutation testing, with the caveat that tumours with protein stable missense alterations that have reduced MMR function are not identified by IHC testing.9
However, CRCs that are MSI-H and/or demonstrate loss of MLH1 protein expression are not unique to LS and occur in up to 15% of all CRC without a germline mutation. MSI-H CRC that is not attributable to LS is commonly referred to as ‘sporadic MSI-H CRC’ and results from methylation of the CpG island in the MLH1 gene promoter, causing transcriptional silencing of the MLH1 gene. The distinction between LS related MSI-H CRC and sporadic MSI-H CRC is a clinically important one that is underpinned by a difference in tumourigenic pathways. The adenoma to carcinoma sequence has long been recognised to constitute the major pathway of colorectal carcinogenesis,10 and is the pathway that underlies the development of LS MSI-H CRC. An alternative non-overlapping pathway of tumourigenesis is the serrated neoplasia pathway where CRC develops from precursor polyps with serrated architecture.11 The serrated pathway is distinguished from the traditional adenoma-carcinoma pathway by highly correlated molecular characteristics, including CpG island methylator phenotype (CIMP), the BRAF V600E mutation, and methylation of the MLH1 gene promoter.12 It has been suggested that tumours with MLH1 promoter methylation and the BRAF V600E mutation should not be screened for MMR mutations,13–15 and testing for MLH1 methylation and the BRAF V600E mutation appears to have become increasingly utilised in molecular and genetic testing algorithms in clinical practice to discriminate sporadic MSI-H CRC from MSI-H CRC in LS.
Sequencing of MMR genes in suspected LS families often identifies rare sequence variants of unknown clinical significance, and figures as high as 20–50% have been reported for unique variants that may fall into this category.16 17 Such unclassified sequence variants include nucleotide changes predicted to cause missense substitutions, small in-frame insertions/deletions, or intronic sequence changes that may alter splicing. Currently, there are 7548 MMR gene variants that have been reported on the International Society for Gastrointestinal Hereditary Tumours (InSiGHT) database as having uncertain clinical significance (http://chromium.liacs.nl/LOVD2/colon_cancer/home.php?action=switch_db). Determining the pathogenicity of these variants has important implications for the clinical management of the families concerned, with respect to genetic counselling approaches, presymptomatic screening, and choice/timing of prophylactic surgery.
We are currently involved in a comprehensive research study to develop a multifactorial likelihood model for the clinical classification of MMR gene variants of uncertain significance.18 19 This statistical model draws on various commonly available, independent, data sources to assess the likelihood that a variant carrier demonstrates features expected for a carrier of a high risk pathogenic MMR gene mutation. For instance, tumour characteristics that are indicative of germline MMR gene mutation status may be used to derive a likelihood ratio for pathogenicity for use in a MMR gene multifactorial model. This review was undertaken to assess the utility of the BRAF V600E and MLH1 methylation tumour characteristics as predictive markers of MMR gene mutation status, specifically for future use in a multifactorial likelihood classification model for clinical classification of MLH1 gene variants of uncertain clinical significance. The appraisal summarises the frequency information currently published for BRAF V600E mutation and MLH1 methylation status in CRC according to MMR gene mutation status, and recommends appropriate approaches to determine the value of tumour markers as robust independent predictors in statistical prediction models. It also highlights limitations associated with use of these tumour features as clinical tools to triage patients for mutation testing.
Materials and methods
Literature review
The electronic database PubMed was searched to identify studies on cohorts of CRC patients where MMR gene mutation status, tumour BRAF V600E status and/or tumour MLH1 promoter methylation status had been investigated and published prior to July 2011. The keywords used to search this database were ‘HNPCC BRAF’, ‘HNPCC BRAF V600E’, ‘HNPCC MLH1 methylation’, ‘HNPCC MLH1 promoter methylation’, ‘Lynch BRAF’, ‘Lynch BRAF V600E’, ‘Lynch MLH1 methylation’, and ‘Lynch MLH1 promoter methylation’. Abstracts were read, and full copies were obtained for papers that had possible relevance to the topic. Inclusion criteria used for these papers were: (1) studies investigating a cohort of patients with colorectal tumours; (2) studies screening for the BRAF V600E mutation in tumours; (3) studies screening for MLH1 promoter methylation in tumours; (4) peer-reviewed articles in English. Studies were excluded if they described only individuals or families (case studies), or if data were presented with insufficient detail to determine actual counts of BRAF V600E positive/negative tumours and MLH1 promoter methylated tumours. There were 57 papers that were eligible and selected for review in this paper after application of these criteria.
Tumour counts were extracted from each study and the following information was noted: patient ascertainment (including clinical criteria for MMR testing), MMR gene mutation testing and methods, BRAF testing methods, MLH1 promoter methylation testing methods, MMR gene mutation status (positive, negative, unknown), and any additional notes considered of relevance to interpretation. Any patient with a MMR gene variant of uncertain clinical significance was excluded from analysis (n=16). When details of MMR gene sequence variant were not fully described, variants called a mutation were assumed to be a pathogenic variant.
Promoter regions tested were noted for the A, B, C and D regions proposed by Deng et al,20 shown in figure 1. Where primer sequences were given, these were checked against the sequence −1000 to −1, relative to the start codon of MLH1. This sequence was acquired through the UCSC Genome Bioinformatics web browser (http://genome.ucsc.edu/). Information abstracted from all eligible papers is shown in supplementary table 1 online (tumour BRAF V600E mutation status) and supplementary table 2 online (tumour MLH1 methylation status). Reported BRAF V600E status and methylation status were categorised (positive/negative) as described in the original reports, since few papers provided raw data that might permit re-assessment of status.
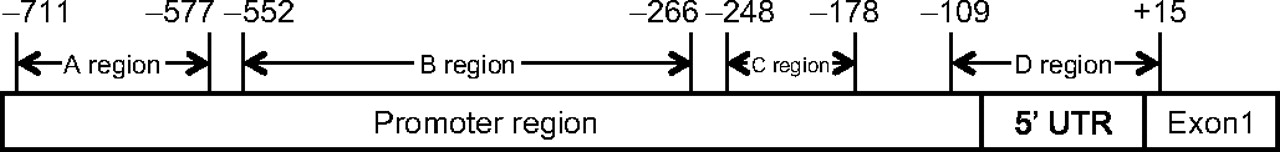
Schematic of MLH1 promoter regions. Four MLH1 promoter regions proposed by Deng et al.20 The base pair numbering is in relation to the start codon (ATG) of MLH1. 5′ UTR, 5′ untranslated region.
Statistical analyses
Meta-analysis was performed using the statistical package, R (http://www.r-project.org). Due to heterogeneity between studies, proportions were combined via an inverse variance random effects model. Variance estimates were calculated using a Freeman-Tukey double arcsine transformation. Frequencies in different groups were compared using a χ2 test.
Results
Tumour BRAF V600E mutation status and germline MMR gene mutation status
From the literature review, 35 studies assessed the BRAF V600E mutation status in CRC tumours, and 32 studies provided information confirming the carrier status of a pathogenic mutation in the MMR genes MLH1, MSH2, MSH6 or PMS2 (see supplementary table 1 online for details). The summary frequency of the BRAF V600E mutation was determined in individuals with CRC stratified by their MMR mutation status, and by MSI status within individuals without a MMR mutation (table 1).
Summary frequencies of tumour BRAF V600E mutation status
A total of 550 tumours were from MMR gene mutation carriers. As expected, the lowest frequency of the BRAF V600E mutation was observed in this sample group (1.40%, 95% CI 0.0006 to 0.0252), with four BRAF-positive CRCs reported (1.40%, 95% CI 0.06% to 2.52%).21 22 Two patients with a tumour BRAF V600E mutation had the same truncating PMS2 mutation c.736 del6 ins11, and the other two patients carried a truncating mutation in MLH1 or MSH2. In contrast to MMR gene mutation carriers, the BRAF mutation frequency was higher in the 331 MMR non-mutation carriers with MSI-H CRC with no information provided regarding MLH1 tumour expression (36.1%, 95% CI 20.95% to 52.84%). BRAF mutation frequency was further increased in the 332 tumours known to display MLH1 methylation or MLH1/PMS2 protein loss (63.5%, 95% CI 46.98% to 78.53%), with frequency >70% in 6/10 reports.13 22–26 The frequency of the BRAF V600E mutation in the 1623 microsatellite stable (MSS) CRC investigated was only 5.0% (95% CI 3.55% to 6.88%). There was evidence for heterogeneity in frequency estimates (p<0.0001) across the studies of non-MMR mutation carriers between the MSI-H group and the MLH1 deficient group.
MLH1 promoter methylation and MMR gene mutation status
Information on 2934 tumours from 43 papers was available (see supplementary table 2 online). For 33 of these papers, adequate information was provided to determine the promoter region tested (see methods). The most commonly tested regions were A and C. MMR mutation status was recorded in 29 papers, with sufficient mutation details provided in 22 papers to allow assessment of methylation frequency for the subset of known MLH1 mutation carriers. The summary frequency of methylation was determined in individuals with CRC stratified by their MMR mutation status (including MLH1 mutation status specifically), and MSI status within individuals without a MMR mutation (table 2). Frequencies were also determined according to promoter region tested.
Summary frequencies of tumour MLH1 promoter methylation
Across the multiple studies, MLH1 promoter methylation testing was performed on a total of 446 tumours from patients with a confirmed MMR gene mutation, including 289 known to carry a mutation in MLH1. Of these, 109 were tested for the A region, and 191 were tested for the C region, with some overlap in the studies. Results reported for MSI-H tumours from known mutation-negative colorectal cancer patients included 143 tested for A region methylation, and 680 tested for C region methylation, with some overlap in the studies. Results for MSS tumours from mutation-negative patients included 312 tested for A region methylation, and 469 tested for C region methylation, again with some overlap in the studies.
From the summary frequencies in table 2, it is evident that some MLH1 mutation carriers do exhibit MLH1 promoter methylation. Considering the region tested, the A promoter region was much more commonly methylated than the C region in MSI-H tumours from MLH1 mutation carriers (42% vs 6%; p<0.00001). The combined data confirm preliminary findings by three studies comparing A and C region methylation in the same MLH1 mutation carrier cases.27–29 Similarly, methylation within the A region was greater than within the C region for MSS tumours from MMR non-mutation carriers (17% vs 0.06%; p<0.00001). However, MSI-H tumours from known mutation-negative cases demonstrated a non-significantly lower frequency of methylation for the A region compared to the C region (40% vs 47%; p=0.1163). Similar trends were seen for the subset of MMR mutation-negative cases demonstrating tumour MLH1 protein loss (32% vs 40%; p=0.1352).
Based on observations in table 2, only the C region of the MLH1 promoter was considered for further analyses when assessing methylation as a predictor of MMR mutation status. Tumour methylation in the C region of the MLH1 promoter was significantly decreased in known MLH1 mutation carriers compared to MSI-H tumours from MMR mutation-negative cases overall (6% vs 47%; p<0.00001), and also compared to the subset of MSI-H tumours from MMR mutation-negative cases demonstrating tumour MLH1 protein loss (6% vs 40%; p<0.00001).
Interestingly, there was evidence of heterogeneity in MLH1 methylation frequency within the subset of MMR mutation-negative MSI-H cases with known MLH1 expression loss. A graphic representation of frequencies by study (figure 2) demonstrates heterogeneity in a proportion of MLH1 methylated cases, where five case series reported in four studies investigating suspected LS cases reported low frequencies of MLH1 methylation in their sample set.29–32 The frequency in these reported mutation-negative familial cases was 0–13%, compared to ≥75% for the other four studies investigating mutation-negative cases not selected for family history (p<0.00001).33–36

{kind=link}
{kind=link}
Forest plot of MLH1 C region methylation frequencies in MLH1 expression-negative tumours from mismatch repair (MMR) mutation-negative cases. The two cohorts in the Gylling study were obtained using different ascertainment criteria.
Discussion
This systematic review provides the first meta-analysis of the BRAF V600E mutation status and MLH1 promoter methylation in CRC cases, stratified by MMR mutation status, MSI status and/or MLH1 expression loss.
Tumour BRAF mutation status
A total of 4562 tumours that have had BRAF testing are summarised in this review, including 550 from mutation carriers, and 663 from mutation-negative cases with known or assumed MSI-H status. The largest individual sample set of confirmed MMR mutation carriers was 111 by Domingo et al,37 followed by Schofield et al26 with only 36 carriers. Similarly, the two largest sample sets of mutation-negative cases included only 111 tumours38 and 63 tumours.35 Moreover, some data could not be included in the summary counts of BRAF mutation status by MMR mutation status because of missing information, particularly mutation status26 39 but also BRAF status of known mutation carriers.21 Of note, Lagerstedt-Robinson et al21 excluded 37 tumours from BRAF testing specifically because these tumours were from cases with germline MMR gene mutations.
Nevertheless, current data confirm that BRAF tumour mutations are most commonly found in mutation-negative cases with MSI-H tumours demonstrating MLH1 expression loss. However, the data also show that BRAF mutations may occasionally be identified in MMR gene mutation carriers (summary frequency of 1% across all studies). The four tumours identified to have a BRAF V600E mutation and a germline MMR gene mutation were from carriers of mutations in MLH1 or MSH2 (mutation nomeclature not detailed),22 or PMS2 (two tumours, both PMS2 c.736 del6 ins11).21 Interestingly, the patients with the truncating PMS2 mutations were not classified as LS in the original report because of the presence of BRAF V600E.21 Another study, excluded from this review because of insufficient detail, also described three PMS2 mutation carriers (two with the PMS2 c.736 del6 ins11 mutation, another with a PMS2 exon 10 deletion) that presented tumours with the BRAF V600E mutation.5 Given the rarity with which a BRAF V600E positive CRC has been reported in a MMR mutation carrier, the possibility that these results are false positives resulting from technical artefacts or in suboptimal mutation detection techniques cannot be discounted. Similarly, the presence of a sporadic MSI-H phenocopy tumour harbouring a BRAF V600E mutation in a mutation carrier is also plausible. In support of this, a case report excluded from this review discussed a mixed lineage family containing both an MLH1 mutation (c.350C>T p.T117M) and serrated neoplasia.40 Until this issue is further resolved, the presence of a BRAF V600E mutation in a CRC, while a strong negative predictor of carrying a MMR mutation, should not be assumed to exclude positive mutation status for any of the four MMR genes, particularly in the context of a family history suggestive of LS.
It has been suggested that using BRAF status as a selection criteria for referring patients to germline genetic testing, particularly for tumours with an MLH1 negative IHC test, could be a strategy for reducing costs and effort.14 38 While this may be relevant for population health screening initiatives,26 the information summarised in this review indicates that use of tumour BRAF V600E status alone to triage cases for MMR mutation screening in the clinical setting will fail to identify some MMR mutation-positive individuals. The use of BRAF testing for clinical test triaging may be even further complicated when testing for MMR epimutations in LS becomes routine, given the recent proposal that tumours from patients carrying a constitutional epimutation may mimic MSI-H sporadic tumours, based on the observation of a somatic BRAF mutation in a carrier of a constitutional MLH1 epimutation in a LS-suspected patient.41
MLH1 promoter methylation
A major observation from the data reviewed is that the proportion of individuals exhibiting tumour MLH1 methylation varies greatly depending on the promoter region analysed. Although not enough data were available to assess methylation in the promoter B and D regions, the evidence suggests that methylation of the A region is not predictive of MSI status: it was observed in 16% of MSS cases. By inference, region A methylation is thus not associated with MSI-H status or MLH1 protein expression loss. In contrast, methylation of the C region was rare in MSS cases (0.06%) but common in MSI-H mutation-negative cases, including those shown to have loss of MLH1 protein expression loss. These cumulative data support previous findings that methylation of the C region is associated with tumour MLH1 expression loss,2 and indicates that targeted screening of the C region of the MLH1 promoter demonstrates the best correlation with somatic changes in MLH1 protein expression. Methylation of the MLH1 promoter C region was reported in 5% of the tumours from MLH1 mutation carriers. Akin to the explanation for the rare occurrences seen with the BRAF V600E mutation, these eight mutation carriers demonstrating MLH1 methylation from the literature could be attributed to false positive findings. However, the possibility that MLH1 promoter methylation is the ‘second hit’ in these carriers is also feasible.42 Further, there is increasing evidence for a role of constitutional epimutations in LS,41 43 44 but the relationship between such epimutations and methylation of the MLH1 C region has yet to be comprehensively investigated. In any case, a proportion of MLH1 mutation carriers may be undiagnosed when relying on MLH1 methylation alone for triaging LS.
It is possible that the predictive power of MLH1 methylation for somatic MLH1 protein expression loss is even greater than that implied by the current dataset, since it is likely that some MMR gene mutations were missed due to limitations in screening methods. As noted in the results section, the frequency of methylation in mutation-negative cases with MLH1 tumour expression loss was significantly lower in studies investigating suspected LS cases,29–32 compared to those that were not selected for family history. The extent of mutation testing in the suspected LS cases ranged from multiplex ligation-dependent probe amplification (MLPA) and denaturing high performance liquid chromatography followed by confirmatory sequencing of MLH1 only, to MLPA of MLH1, MSH2 and MSH6, followed by direct sequencing of MLH1 and MSH2 only. Furthermore, even for those cases subjected to direct sequencing, there are several examples in the literature of hereditary mutations that would escape detection using ‘standard’ screening methods. These include variants resulting in constitutional epigenetic silencing as mentioned above,41 43 44 a deep intronic mutation in MSH2,45 and transcriptional silencing as a result of 3′ large genomic rearrangements.46 Thus we cannot exclude the possibility that some of these suspected LS cases may yet be identified to carry a germline alteration, including those leading to tumour MLH1 expression loss.
Combined BRAF-MLH1 methylation status
There were only three studies that investigated both BRAF V600E mutation status and methylation status of the MLH1 promoter C region, in the same tumours. In the 72 MMR mutation-negative cases identified, methylation was more common than BRAF V600E mutation (54 vs 26), and all 26 of the BRAF-positive cases were C region methylated. Of the 35 MLH1 mutation carriers identified, none showed BRAF mutations but three exhibited tumour methylation. These data suggest that tumour BRAF status is more sensitive than tumour methylation status as a predictor of MMR gene mutation status, as inferred by overall results for BRAF alone and methylation alone. However, it would be worthwhile to assess the combined value of these markers in a much larger dataset.
Use of BRAF and MLH1 methylation as predictors of MMR gene mutation status
While tumour BRAF mutation status and MLH1 promoter methylation are not a conclusive test for MMR mutation status, these tumour characteristics are clearly strong negative predictors of mutation status and thus have potential to be included into statistical models that predict the pathogenicity of variants of uncertain clinical significance. A likelihood model has been developed for quantitative classification of BRCA1 and BRCA2 variants into five classes that are linked to patient management recommendations.47–49 This model is currently being adapted for use with MMR gene variants.18 19 47 The MMR prediction model proposes to assess whether a variant of uncertain clinical significance is displaying the characteristics of a pathogenic mutation by estimating the likelihood of pathogenicity based on multiple features: bioinformatic analysis of sequence conservation and physicochemical properties of amino acid substitutions; co-segregation with disease in families; reported family history; co-occurrence of a variant with a pathogenic mutation in the same gene; and tumour characteristics.18 Colorectal tumour MSI-H status is clearly recognised as a marker for loss of MMR function and thus a marker to predict MMR gene mutation status. However, it would be helpful to include additional tumour features that distinguish MSI-H sporadic tumours from MSI-H tumours from MMR mutation carriers, and data from this comprehensive review indicate that separation of these two tumour groups would be assisted by the inclusion of BRAF V600E status and MLH1 promoter methylation as negative predictors of MMR mutation status in prediction models.
However, this review has detected heterogeneity in frequency across studies, likely resulting from a combination of random error due to small study sizes, as well as differences in patient ascertainment, sensitivity and extent of MMR gene mutation testing, and tumour testing methodologies. Moreover, it would be optimal to estimate likelihood ratios for all tumour features simultaneously, to account for non-independent association of these features with MMR gene mutation status. In order to derive the most robust likelihood ratio estimates for inclusion of tumour MSI status, BRAF V600E mutation status and MLH1 methylation into a MMR variant classification model, we thus advocate simultaneous analysis of these tumour features in large reference sample sets with extensive and sensitive MMR gene mutation testing. Of importance, these reference sample sets need to be appropriately matched by ascertainment to the population from which unclassified variants will be identified, and all molecular testing (germline mutations and tumour characteristics) have to be undertaken in an unbiased manner.
In addition, the baseline tumour information derived for use in the MMR variant classification should also be used to supplement prediction models that are used clinically to assess the probability that a colorectal cancer patient is a MMR mutation carrier.50 Currently, the MMRpro, MMRPredict and PREMM (1,2,6) models base their predictions on only proband diagnosis age and family history. Inclusion of tumour characteristics will improve the performance of such models, and importantly, facilitate use of multiple features rather than a single tumour datapoint to predict LS diagnosis and select patients for MMR gene mutation testing.
Conclusion
Colorectal tumour BRAF V600E mutation status and MLH1 C region promoter methylation status are strong predictors of MMR mutation-negative status although not definitive, and caution should thus be exercised in excluding cases with strong family history from MMR gene mutation screening. However, these two tumour features will have application in models predicting MMR mutation status and clinical significance of MMR gene variants. Further assessment of the combined predictive power of tumour MSI-H status, BRAF status and MLH1 methylation requires simultaneous analysis of these tumour features in large and appropriate reference sample sets with extensive and sensitive MMR gene mutation testing.
Acknowledgments
We thank Leesa Wockner for statistical support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online tables
Footnotes
Funding This research has been supported by funding from NHMRC and Cancer Australia. BT is supported by a TCCQ PhD scholarship, JY is a TCCQ Senior Research Fellow, and ABS is an NHMRC Senior Research Fellow.
Competing interests None.
Patient consent Being a review article, all data included were from other publications.
Provenance and peer review Not commissioned; externally peer reviewed.