


Abstract
Endogenous cardiotonic steroids (CTS), also called digitalis-like factors, have been postulated to play important roles in health and disease for nearly half a century. Recent discoveries, which include the specific identification of endogenous cardenolide (endogenous ouabain) and bufadienolide (marinobufagenin) CTS in humans along with the delineation of an alternative mechanism by which CTS can signal through the Na+/K+-ATPase, have increased the interest in this field substantially. Although CTS were first considered important in the regulation of renal sodium transport and arterial pressure, more recent work implicates these hormones in the regulation of cell growth, differentiation, apoptosis, and fibrosis, the modulation of immunity and of carbohydrate metabolism, and the control of various central nervous functions and even behavior. This review focuses on the physiological interactions between CTS and other regulatory systems that may be important in the pathophysiology of essential hypertension, preeclampsia, end-stage renal disease, congestive heart failure, and diabetes mellitus. Based on our increasing understanding of the regulation of CTS as well as the molecular mechanisms of these hormone increases, we also discuss potential therapeutic strategies.
I. Introduction
The topic of this review is the digitalis-like factors, which are also referred to as inhibitors of the Na+/K+-ATPase (de Wardener and Clarkson, 1985; Goto et al., 1992; Schoner, 1992) or endogenous cardiotonic steroids (CTS1). As we will discuss, these CTS link dietary NaCl and cardiovascular and renal disease. Although the importance (and the very existence) of such factors has been a matter of controversy (Kelly and Smith, 1992; Hansen, 2003), remarkable progress has been achieved during the past 15 years. These breakthroughs are illustrated in a series of articles and include 1) positive identification of specific CTS in experimental animals and humans (Hamlyn et al., 1991; Lichtstein et al., 1993; Bagrov et al., 1998; Komiyama et al., 2005), 2) establishment of alterations in concentrations as well as the role(s) of CTS in animal models and human disease states (Ferrandi et al., 2005; Haddy, 2006; Huang et al., 2006; Schoner and Scheiner-Bobis, 2007), and, in parallel, 3) the discovery of cell signaling functions of the Na+/K+-ATPase and its involvement in many aspects of basic cell biology (Xie and Askari, 2002; Wasserstrom and Aistrup, 2005; Orlov and Hamet, 2006; Nesher et al., 2007; Schoner and Scheiner-Bobis, 2007). The main goals of the present review are to emphasize the clinical implications of CTS in human health and disease and to demonstrate potential targets for new therapies.
II. Na+/K+-ATPase
A. Structure and Function of the Na+/K+-ATPase
The discovery of the sodium pump was a critical step in the 300-year study of the cell as a basic unit of animal life. More importantly, the sodium pump gave substance to the concept of the cell membrane, which isolates the milieu interior from the external environment and/or the environment of other cells. Based on the asymmetrical distribution of sodium and potassium ions, the scientific community was ready to accept the existence of “submicroscopic pumps, installed across the cell membrane,” which could actively participate in “fine-tuning” of the transmembrane ion gradients in accordance with changes in the physiological needs of cells (Ling, 2007). The discovery of the sodium pump is generally credited to Skou (1957) for his experiments with crab nerve homogenate that clearly demonstrated the existence of a protein-based structure, incorporated in the cell membrane, which pumped sodium ions outside and potassium ions inside living cells, and in so doing, converted chemical energy into work. It is noteworthy that this discovery was possible because of the existence of ouabain, a specific sodium pump inhibitor of steroidal nature and plant origin, which later was found to be identical to one of the endogenous mammalian inhibitors of activity of sodium pump (Hamlyn et al., 1982, 1991).
The sodium pump, or Na+/K+-ATPase [(Na+ + K+)-stimulated adenosine triphosphatase; EC 3.6.3.9], is an active transport system of sodium and potassium ions that is highly conserved in all eucaryote cells. It is a member of the P-type ATPase family of membrane-incorporated proteins, and it is directly responsible for the maintenance of the low intracellular Na+/K+ ratio by the active transport of these ions across the plasma membrane using the hydrolysis of ATP to provide the necessary energy (Skou and Esmann, 1992). The Na+/K+-ATPase controls multiple essential cellular functions. Specifically, it maintains the electrical membrane potential, which is necessary for nerve transmission and muscle contraction, excitability, and many other cellular functions, which depend on the necessary sodium-potassium gradients (Lingrel, 1992). The sodium pump can also drive secondary active co-/countertransporters, which are coupled to the gradient of extracellular to intracellular [Na+], such as the Na+/Ca2+-exchanger (Blaustein, 1993). It logically follows that the Na+/K+-ATPase, acting via ATP hydrolysis, determines a very substantial fraction of the cellular metabolic rate of most tissues (Blanco and Mercer, 1998).
The Na+/K+-ATPase consists of two polypeptides in equimolar ratios. The α catalytic subunit has a molecular mass (Mr) of approximately 110 kDa and the glycosylated β glycoprotein subunit has a Mr of 31.5 kDa. The α subunit has 10 transmembrane segments. It contains the binding sites for Na+ and CTS on the extracellular segments and the binding sites for K+ and ATP on the intracellular loops. Asp369, in particular, is critical for the binding of a phosphate group and is an essential site for ATP hydrolysis (Lingrel, 1992). Two more residues, Lys501 and Asp586, may also participate in the binding of ATP (Farley et al., 1997) (Fig. 1). The regulatory β subunit is a single-transmembrane protein with a glycosylation site controlling the activity of Na+/K+-ATPase (Blanco and Mercer, 1998). At present, the αβ complex is believed to form the functional dimeric units of the sodium pump. These dimers may also be organized in tetramers or possibly stack into ribbons (Söderholm et al., 1988). According to the Albers-Post model, the Na+/K+-ATPase “pumps” sodium ions from the inside of the cell to the outside while moving potassium ions in the opposite direction, both against existing concentration gradients in an energy-dependent process. The α subunit, in the presence of Na+ and Mg2+, is phosphorylated by ATP, followed by occlusion of three cytosolic Na+ ions. This high-energy E1P form of the enzyme, loaded with Na+ ions, undergoes a conformational change to the low-energy E2P form. When the sodium cation sites are exposed to the extracellular medium, Na+ ions are released, and in the presence of potassium ions the E2P form is dephosphorylated. Dephosphorylation is followed by occlusion of two K+ ions to specific potassium binding sites. Uptake of K+ ions leads to the transition of the E2 form to the E1 form, which is accelerated by ATP. This transition is followed by the release of K+ ions to the intracellular medium. Then the E1 form with bound ATP undergoes the cycle again (Albers, 1967; Post et al., 1972; Jørgensen, 1986).

Structure of Na+/K+-ATPase. Na+/K+-ATPase consists of two α and β polypeptides in equimolar ratios. The α catalytic subunit has 10 transmembrane segments, schematically presented in an “unfolded” disposition; in reality, there is a bungle around M4, M5, and M6 transmembrane segments. The extracellular segments of α subunit form a binding site for CTS (shown in red), which include TM1–TM2, TM5–TM6, and TM7–TM8 loops and several amino acids from the transmembrane regions M4, M6, and M10 (see the explanation in the text). The binding site for ATP is located on the intracellular loop TM4–TM5 (shown in blue), which forms the “pocket” for this nucleotide. The phosphorylation domain (P; shown in orange) located on the proximal and distal parts of intracellular loop TM4–TM5; phosphate of ATP is transiently transferred on the aspartyl residue 376 of DKTGT motif. The actuator domain, specifically its TGES motif, is responsible for the dephosphorylation step; it is constituted by the cytoplasmic NH2-terminal and TM2–TM3 intracellular loop (shown in green). The regulatory β glycoprotein subunit is a single-transmembrane protein with a several glycosylation sites (two are shown). The extracellular part of β subunit interacts with a conserved motif SYGQ on the extracellular loop TM7–TM8 of α subunit. The αβ-subunit complex of Na+/K+-ATPase associates with third subunit, which contains the conserved motif FXYD identical for all seven proteins from this family. FXYD2 protein is known as the earlier described γ subunit of Na+/K+-ATPase. These proteins, including the γ subunit, are not an integral part of sodium pump, but they are associated with specific domains of αβ-subunit complex and modulate catalytic properties of the Na+/K+-ATPase.
Seven additional single-transmembrane proteins, which colocalize with the αβ subunit complex have been identified and named after the sequence of their conserved motif FXYD (Sweadner and Rael, 2000). One of these proteins, FXYD2, is also known as the γ subunit of the Na+/K+-ATPase; it has an Mr of 7.3 kDa and exhibits a high degree of interspecies homology (Blanco and Mercer, 1998) (Fig. 1). These proteins, which associate with the Na+/K+-ATPase, including the γ subunit, are not integral parts of sodium pump per se. However, they are associated with specific domains of the αβ subunit complex and modulate the catalytic properties of the Na+/K+-ATPase (Cornelius and Mahmmoud, 2003; Crambert and Geering, 2003; Zouzoulas et al., 2003).
There are four α (α1, α2, α3, and α4), and three β (β1, β2, and β3) isoforms of Na+/K+-ATPase, thus allowing numerous combinations of αβ complexes among the tissues with different characteristics including differential sensitivities to different cardiotonic steroids (Adams et al., 1982). Evidence for the existence of different Na+/K+-ATPase isozymes emerged from analysis of the digitalis sensitivity of the rodent sodium pump. Thus, it has been demonstrated that ouabain induced heterogenous inhibition curves of Na+/K+-ATPase with Ki values ranging from 0.1 μM in brain to 100 μM in renal tissues (Marks and Seeds, 1978). The structural basis for the functional heterogeneity of the sodium pump was found in brine shrimp (Peterson et al., 1978) and later in mammals (Sweadner, 1979). Specifically, the α subunit can be resolved into distinct forms by Western blotting. In the latter article, one of the isoforms expressed in brain tissue was found to be identical to the renal α-subunit, which displayed lesser sensitivity to ouabain (α1 isoform). The brain α+ isoform, which displayed high sensitivity to ouabain, was later renamed α3. Structural dissimilarities at the NH2 terminus of the α1 and α3 isoforms suggested that the existence of multiple isozymes of Na+/K+-ATPase has a genetic basis (Lytton et al., 1985; Blanco and Mercer, 1998). Indeed, at present, four genes in humans have been found to control four α isoforms, three genes to control the β isoforms, and seven genes for the FXYD peptides (Shull et al., 1985, 1986; Sverdlov et al., 1987; Shamraj and Lingrel, 1994; Franzin et al., 2005).
Both α and β isoforms are expressed in a tissue-specific fashion. The distribution of the α1 isoform is ubiquitous (Blanco and Mercer, 1998); it is also the main isozyme expressed in the kidney (Jørgensen, 1990; Skou and Esmann, 1992; Glynn, 1993). The α1/β1 complex is found in nearly every tissue and is often the major form of the enzyme (Fambrough, 1988; Sweadner, 1989; Levenson, 1994). The α2 isoform is expressed in adult heart (Zahler et al., 1992; Shamraj et al., 1993), vascular smooth muscle (Zhang et al., 2005), skeletal muscle (Hundal et al., 1992), brain (Urayama et al., 1989; McGrail et al., 1991), adipocytes (Lytton et al., 1985), and cartilage and bone (Mobasheri et al., 2000). The expression of the α2 isoform is also insulin-dependent (Russo and Sweadner, 1993). The α3 isozyme is expressed mostly in excitable tissues (Urayama et al., 1989), being most abundant in the central and peripheral nervous tissues (Hieber et al., 1991; McGrail et al., 1991) and in the conductive system of the heart (Zahler et al., 1992). It is noteworthy that the α4 isoform seems to be testis-specific (Shamraj and Lingrel, 1994). The expression of β1 is also ubiquitous. The β2 and β3 isoforms are expressed in brain, cartilage, and erythrocytes, whereas β2 can also be found in cardiac tissues and β3 can be observed in lung tissues. Studies of the artificial expression of different combinations of α and β isoforms in heterologous cells have revealed that the α subunit isozymes are responsible for substrate and CTS affinities of human Na+/K+-ATPase (Müller-Ehmsen et al., 2001) and also demonstrated an important function of the β subunit (Blanco et al., 1995; Blanco and Mercer, 1998; Pierre et al., 2008).
Physiological functions of the Na+/K+-ATPase have been deduced from its role as an ion pump. In particular, the ability of the sodium pump to establish and maintain ion gradients makes it essential for the generation and maintenance of membrane potentials, which are required for neuronal excitability (Gillis and Quest, 1979). In renal tubular epithelial cells, the expression and function of α1/β1 isoforms of the Na+/K+-ATPase are key in determining sodium ion reabsorption from the glomerular filtrate. As we will discuss in detail in section VII, variable levels of endogenous sodium pump inhibitors seem to regulate this reabsorption, both by inhibiting enzymatic function and through altering plasmalemmal expression, and, thus, regulate renal sodium handling. The sodium pump also participates in the maintenance of intracellular sodium concentrations necessary for smooth muscle cell and cardiac myocyte functions (Blanco and Mercer, 1998). Again, cardiotonic steroids are essential participants in this regulation. In the classic or “ionic” signaling schema for sodium pump function, it is quite clear how the inhibited sodium pump, coupled to the Na+/Ca2+-exchanger, may cause an increase in intracellular sodium, which in turn may lead to an increase in cytosolic calcium, the latter being a key second messenger for a variety of cell functions (Blaustein, 1993; Juhaszova and Blaustein, 1997a,b).
B. Na+/K+-ATPase as a Specific Receptor for Cardiotonic Steroids
Although digitalis-like substances were almost certainly a part of herbal remedies administered as long ago as the Roman empire, most authorities agree that serious scientific study and medicinal application began with William Withering's observation of a salutary effect of foxglove (Digitalis purpurea). Withering, a British physician and herbalist, apparently obtained foxglove initially from “an old woman in Shropshire,” who had apparently administered a variety of herbal remedies to local patrons. In his 1785 monograph, “An Account of the Foxglove and Some of Its Medical Uses; With Practical Remarks on Dropsy and Other Diseases,” Withering detailed nearly a decade of studying both the beneficial and toxic effects of foxglove. In 1869, Claude Adolphe Nativelle, a French pharmacist isolated a material he called “digitalin” from the foxglove plant. Six years later, the German chemist, Oswald Schmiedeberg, isolated the first pure glycoside in crystal form from foxglove, which he called “digitoxin” (Lesney, 2002). CTS belonging to the class of bufadienolides were also introduced into routine clinical practice more than 1000 years ago. The dried skin of toads, which contains various bufadienolide derivatives, comprises one of the main active principles of traditional Chinese medicine Sen-So (or Ch'an Su), and has been used in the treatment of cardiac dysfunction (Chen and Kovaríková, 1967). The pharmacological effects of digitalis and related compounds were reviewed in many excellent articles (Braunwald and Klocke, 1965; Langer, 1972; Repke, 1972; Smith and Haber, 1973; Lindenmayer, 1976; Akera and Brody, 1977; Gillis and Quest, 1979; McDonough et al., 1995).
Digitalis-like inhibitors, or CTS, have a specific binding site on the extracellular loops (TM1–TM2, TM5–TM6, and TM7–TM8) of the α subunit of the Na+/K+-ATPase. Some amino acids from the transmembrane regions (M4, M6, and M10) also interact with ouabain; this suggests that the hydrophobic regions of CTS may actually be inserted in the membrane inside which they interact with the α subunit of sodium pump (Burns et al., 1996; Croyle et al., 1997; Vasilets et al., 1998). Amino acids from 111 to 122 sequences (extracellular TM1–TM2 loop) form the most important part of the putative CTS binding site (Fig. 1). It is noteworthy that the dissimilarities in the amino acid sequence between different species and different isoforms underlie the different sensitivity of the sodium pump to a variety of cardiotonic steroids (Blanco et al., 1999; Mobasheri et al., 2000; Geering, 2005, 2006), which include cardenolides and bufadienolides (this issue is reviewed in detail in sections IV.A and IV.B) (Fig. 2). For example, rodent renal epithelial Na+/K+-ATPase, consisting nearly exclusively of the α1 isoform, may be inhibited by a bufadienolide marinobufagenin (MBG) at nanomolar concentration range, whereas ouabain is 3 orders of concentration less active with renal Na+/K+-ATPase (Fedorova et al., 2000, 2001b).
The sensitivity of the sodium pump to cardiotonic steroids is controlled by multiple mechanisms in addition to the tissue specificity of α and β isoforms distributions. Thus, it is known that protein kinases phosphorylate the Na+/K+-ATPase in a tissue- and isoform-specific fashion (Bertorello and Katz, 1993; Blanco and Mercer, 1998; Therien and Blostein, 2000). The α subunit is phosphorylated by protein kinase A and protein kinase C (PKC) (Bertorello et al., 1991); PKC directly phosphorylates Ser18 on the intracellular domain (Feschenko and Sweadner, 1997; Feschenko et al., 1997) of the α1 isoform of Na+/K+-ATPase in vascular smooth muscle from human mesenteric artery, which in turn causes increased sensitivity of sodium pump to MBG but not to ouabain (Fedorova et al., 2002a). Some studies indicate that dopamine-induced phosphorylation of the α subunit at Ser18 results in a clathrin-dependent internalization of the sodium pump (Chibalin et al., 1999). Another mechanism of modulation of sodium pump activity involves the cGMP-sensitive phosphorylation of Na+/K+-ATPase; this may be initiated by atrial natriuretic peptide (ANP). It is noteworthy that whereas cGMP induces Na+/K+-ATPase phosphorylation in renal epithelial cells, it actually reduces levels of Na+/K+-ATPase phosphorylation in smooth muscle cells (Fedorova et al., 2006).
The three classic features of Na+/K+-ATPase (the pump, the enzyme, and the receptor to cardiotonic steroids) are being understood in substantially greater detail, and, in some aspects, have undergone a paradigm shift in understanding. In particular, there is evidence that extremely low concentrations of cardiotonic steroids, which are unlikely to inhibit the enzymatic function of the sodium pump (Akimova et al., 2005, 2008a; Orlov and Hamet, 2006), are able to initiate several signaling pathways, which may be extremely important for a variety of cell functions. This process is discussed in detail in section V. Szent-Gyorgyi (1953) was one of the first to hypothesize that digitalis drugs, are, in fact, a substitute of endogenous compounds participating in the regulation of cardiac muscle contractility. Thus, the further discovery of Na+/K+-ATPase with a receptor site for ouabain became a landmark in the discovery of the endogenous CTS.
III. Endogenous Digitalis
A. Early Evidence
For many years, increased dietary NaCl intake has been suspected to increase the risk of cardiovascular and renal diseases (Ritz, 1996; Meneton et al., 2005). However, during the several last decades, the importance of dietary NaCl for development of hypertension has shifted from a topic of debate to a well established phenomenon (De Wardener and MacGregor, 2002; Ritz et al., 2006; Weinberger, 2006). The impact of dietary NaCl on cardiovascular disease is indicated by data from several large-scale studies, such as the International Study of Salt and Blood Pressure (INTERSALT) (Stamler et al., 1991) and the Dietary Approaches to Stop Hypertension (DASH) (Appel et al., 2006). On this background, understanding of the specific mechanisms underlying the deleterious effects of NaCl becomes critically important. Our understanding of blood pressure and blood volume regulation has been significantly enhanced by our detailed understanding of the renin-angiotensin-aldosterone, vasopressin, and sympathetic nervous systems (Schrier, 1974). However, it became clear in the 1960s that although these systems can be invoked to explain the physiology and pathophysiology associated with acute or chronic volume depletion, they are inadequate to explain responses to acute or chronic expansion of blood volume. The history of the “Third Factor,” so called because it was a factor in addition to aldosterone and glomerular filtration rate (GFR) in determining renal sodium handling, has been elegantly reviewed (Ritz, 1994; Meneton et al., 2005). This existence of this Third Factor was demonstrated in 1961 by De Wardener et al. (1961). In this classic article, these workers demonstrated that natriuresis induced by saline infusion was maintained even if renal perfusion pressure and glomerular filtration rate were not allowed to increase. After this landmark study, the relationship between volume and this Third Factor was elucidated as described in the following section.

Chemical structures of cardiotonic steroids: cardenolides (ouabain and digoxin) and bufadienolides (marinobufagenin and proscillaridin A). Rostafuroxin, a digitoxin derivative, is an antihypertension compound that opposes the prohypertensive effects of endogenous ouabain.
B. Regulation of Volume and the Third Factor
The Third Factor was a topic of great interest in the 1960s and 1970s (Guyton et al., 1980). Cort and Lichardus (1963) demonstrated that a circulating substance in cats subjected to carotid artery occlusion induced natriuresis in rats and inhibited sodium transport in frog skin. Buckalew et al. (1970) demonstrated that an ultrafiltrate of volume-expanded dogs inhibited sodium transport in toad bladders and postulated that this ultrafiltrate contained an inhibitor of the Na+/K+-ATPase. Extremely important contributions were made by Schrier and de Wardener in the measurement and implications of this factor (De Wardener et al., 1961; de Wardener et al., 1968, 1971; Schrier et al., 1968a,b; Schrier, 1974). Kramer and Gonick (1974) were later able to demonstrate that volume expansion in rats produced a substance that could inhibit ATPase activity in rat kidney. Bricker and coworkers incorporated the concept of a circulating Na+/K+-ATPase inhibitor in a “trade-off” schema for renal failure progression and the pathogenesis of the uremic syndrome (Bourgoignie et al., 1970; Kaplan et al., 1974; Schmidt et al., 1974; Bricker et al., 1975). Although some of the effects attributed to a Third Factor are, in fact, caused by several factors in addition to aldosterone and GFR (e.g., atrial and brain natriuretic factors), this work initiated the search for these factors, of which cardiotonic steroids feature prominently.
Haddy and Overbeck (1976) and Overbeck et al. (1976) discovered that the Na+/K+ pump activity (ouabain-sensitive 86Rb uptake) in arteries and veins of dogs with low renin hypertension is decreased and that Na+/K+-ATPase activity in cardiac microsomes from rats with low renin hypertension is inhibited. Gruber et al. (1980) demonstrated that plasma volume expansion is associated with elevated levels of circulating digoxin-like immunoreactive material in dogs. Shortly thereafter, Hamlyn et al. (1982) demonstrated that plasma Na+/K+-ATPase inhibitory correlated positively with blood pressure in patients with essential hypertension. Kojima et al. (1982) showed that anti-digoxin antibody lowers blood pressure in rats with deoxycorticosterone-salt-induced hypertension. Taken together, we argue that these studies strongly implicated CTS in the pathogenesis of hypertension.
C. Hypertension, Dietary NaCl Intake, and Concept of Natriuretic Hormone
Despite the excitement of the 1960s and 1970s, enthusiasm for the concept of Na+/K+-ATPase inhibitors as third factors decreased during the 1980s and 1990s in part because of inconsistencies with the experimental data. Most assays were based on cross-reactivity to the medication digoxin, and such cross-reactivity to other CTS varied dramatically (Sheiner et al., 1974; Bergdahl et al., 1979; Gruber et al., 1980; Bergdahl and Molin, 1981; Kelly, 1986; Pleasants et al., 1986). However, the most important inconsistency was that digitalis, the prototypical inhibitor of the Na+/K+-ATPase was not natriuretic in normal subjects (Hauptman and Kelly, 1999). In addition, atrial (and brain) natriuretic peptide(s) were discovered; these hormones clearly were natriuretic and could be easily measured, and their circulating concentrations were obviously increased by volume expansion (de Bold et al., 1981; Agnoletti et al., 1987; Bruneau et al., 1997; Lee and Burnett, 2007).
Interest in the study of CTS has steadily increased in the past decade for several reasons. First and perhaps foremost, multiple endogenous digitalis-like factors have been isolated and chemically characterized in experimental animals and humans (Hamlyn et al., 1991; Lichtstein et al., 1993; Bagrov et al., 1998; Komiyama et al., 2005). Specifically, ouabain, MBG, telocinobufagin, and bufalin have each been unequivocally identified in human plasma and/or urine. Also quite importantly, a signal cascade that does not seem to depend on enzymatic inhibition of the Na+/K+-ATPase but rather occurs when CTS bind caveolar Na+/K+-ATPase in the company of Src and EGFR has been identified (Xie and Askari, 2002; Liu et al., 2003; Wang et al., 2004; Pierre and Xie, 2006; Liang et al., 2007). The measurement of CTS has evolved from bioassays and cross-reaction with digitalis because of the definite identification of CTS and the development of high-quality immunoassays. At present, there is a commercially available assay for ouabain, whereas measurements of MBG (Fedorova et al., 2002b, 2008), telocinobufagin, and bufalin still require research assays. Plasma levels of ouabain and MBG in humans seem to be in the range of 200 to 1500 pM in health and disease.
IV. Salt-Sensitive Hypertension, Concept of Natriuretic Hormone, and Elucidation of Nature and Roles of Endogenous Cardiotonic Steroids
A. Endogenous Cardenolides
1. Endogenous Ouabain.
Hamlyn and coworkers reported that human plasma contains CTS indistinguishable from ouabain (Hamlyn et al., 1991; Ludens et al., 1991) (Fig. 2). Since that time, endogenous ouabain (EO) has also been isolated from bovine adrenal gland (Schneider et al., 1998), bovine hypothalamus (Tymiak et al., 1993), and rat adrenomedullary cells (Komiyama et al., 2001). Using NMR and mass spectrometry techniques, mammalian EO was found to be identical to the plant-derived ouabain (Hamlyn et al., 1991; Tymiak et al., 1993; Schneider et al., 1998). The adrenal cortex and hypothalamus are considered to be the sites of EO production (Komiyama et al., 2001; el-Masri et al., 2002; Murrell et al., 2005). ACTH, angiotensin II, vasopressin, and phenylephrine were shown to facilitate the in vitro release of EO from adrenal cortex (Laredo et al., 1997; Shah et al., 1999).
Evidence indicates that EO does not fulfill the criteria for a putative natriuretic hormone but does play a role in the adaptation to both sodium depletion and sodium loading. Although some studies of salt loading in normotensive rats demonstrate increases in plasma [EO] (Butt et al., 1997; Ho et al., 1997), results of many other experiments performed in dogs (Ludens et al., 1993; Bagrov et al., 1996b), rats (Fedorova et al., 2001a,b), and humans (Manunta et al., 2001; Balzan et al., 2005) are contradictory. In hypertensive subjects, results are similarly inconsistent. In 180 untreated hypertensive patients, Manunta et al. (2001) demonstrated that plasma [EO] did not change during volume expansion but rather increased after 2 weeks of sodium depletion. In a more recent study, in 13 healthy individuals, Manunta et al. (2006) demonstrated that NaCl depletion produced a 4-fold elevation in plasma [EO], but in the same study, 3 days of NaCl loading (171 mEq of NaCl/day) caused a transient 13-fold elevation in plasma [EO]. A similar pattern of EO response (i.e., elevation followed by decrease) has been observed in Dahl salt-sensitive (Dahl-S) rats after acute and chronic NaCl loading (Fedorova et al., 2000, 2002b) and in normotensive human subjects during 6 days of high NaCl intake (4 mmol/kg/day) studied by other workers (Anderson et al., 2008). Suffice it to say that plasma [EO] cannot be used as an indicator of plasma volume in either normal or hypertensive subjects.
Despite lack of evidence for the natriuretic activity of EO, several lines of experimental evidence clearly demonstrate the prohypertensive role of EO, including induction of hypertension in ouabain-treated rodents, elevation of EO levels in hypertensive rats, and observations of the central prohypertensive action of this hormone. In rats, chronic peripheral administration of low doses of ouabain (10–50 μg/kg/day) has been reported to increase arterial pressure and to induce cardiac hypertrophy (Rossoni et al., 2002, 2006; Ferrandi et al., 2004; Briones et al., 2006; Cheung et al., 2006; Dostanic-Larson et al., 2006). Padilha et al. (2008) have shown in rats that chronic administration of a low dose of ouabain is associated with an increase in the expression of cyclooxygenase-2 and with impairment of the release of endothelium-derived hyperpolarizing factor.

Prohypertensive effects of endogenous ouabain. In the kidney, in the presence of adducin polymorphism, EO increases sodium reabsorption via interaction with a pool of ouabain-sensitive α1 Na+/K+-ATPase located in the caveolae of renotubular cells. The resultant activation Src-EGFR-dependent tyrosine phosphorylation pathway reduces internalization of the Na+/K+-ATPase and increases the net sodium pump activity (left column). EO may also increase vascular tone via inhibition of ouabain-sensitive α2 Na+/K+-ATPase in vascular sarcolemma and resultant activation of Na+/Ca+2 exchange (right column) and, in addition, directly induces hypertrophic signaling in cardiovascular tissues. Rostafuroxin exhibits its antihypertensive effects via antagonism of the interaction of EO and adducin on vascular and renal Na+/K+-ATPase. MAPK, mitogen-activated protein kinase.
Three mechanisms, the “adducin paradigm,” the existence of highly sensitive ouabain-binding sites in vascular smooth muscle, and central effects, were proposed to link EO to vasoconstriction in hypertension (Fig. 3). A mechanism for the prohypertensive effect of EO has been suggested by experiments performed in Milan hypertensive rats, which carry a mutation of a gene of a cytoskeletal protein, adducin (Bianchi et al., 1994). In this strain of rats, both increased levels of EO and adducin mutation are associated with heightened expression and Na+/K+-ATPase activity in the renotubular epithelium. This heightened expression is apparently due to an increase in the resident time of the sodium pump in the cellular membrane (Efendiev et al., 2004; Ferrandi et al., 2004). In this strain of animals, ouabain apparently leads to further up-regulation of the renal Na+/K+-ATPase expression, renal sodium retention, and hypertension (Ferrandi et al., 2004). Administration of an ouabain antagonist, rostafuroxin (PST2238), a digitoxin derivative (Figs. 2 and 3), attenuates the hypertension in this strain (Ferrari et al., 1999).
Elevated levels of EO can also elevate blood pressure via inhibition of α2 Na+/K+-ATPase and promotion of Ca2+ entry via the Na+/Ca2+ exchanger in vascular smooth muscle (Zhang et al., 2005; Dostanic-Larson et al., 2006). Thus, the genetically engineered mice that express ouabain-resistant α2 Na+/K+-ATPase, unlike control mice with more ouabain-sensitive sodium pumps, do not manifest an increase in BP after chronic administration of ouabain (Dostanic-Larson et al., 2006). Accordingly, vascular smooth muscle from these mice with ouabain-insensitive α2 sodium pumps is insensitive to the pressor effect of ouabain (Dostanic-Larson et al., 2005). Furthermore, genetically engineered mice with reduced expression of α2 (but not of α1) Na+/K+-ATPase become hypertensive, and their arteries exhibit enhanced vascular tone in vitro (Zhang et al., 2005).
Alternatively, other workers have not found major differences in ouabain binding to the caveolar compared with noncaveolar Na+/K+-ATPase (Liang et al., 2007). Liu et al. (2004, 2005) have observed that both signal transduction through the Na+/K+-ATPase-Src-EGFR cascade and clathrin-mediated endocytosis of the Na+/K+-ATPase in renal proximal tubular epithelial cells are limited to the Na+/K+-ATPase residing in caveolae. It is possible that different levels of expression of the γ subunit of the Na+/K+-ATPase in the preparations used explain some of these differences as suggested by Nguyen et al. (2007). In accord with either of these scenarios, patients with hypertension and mutations in the adducin gene also exhibit altered renal sodium reabsoption (Wang et al., 2003). Manunta et al. (2008) demonstrated that saline loading produces renal sodium retention in hypertensive patients with elevated plasma EO levels and mutant adducin. In Milan hypertensive rats, administration of the novel antihypertensive compound, PST2238 (Ferrari et al., 1999), antagonizes the interaction of EO and adducin on the renal sodium pump, lowers the BP, and inhibits the activity of Na+/K+-ATPase in the renal medulla (Ferrandi et al., 2004).
In rats, centrally administered ouabain elicits pressor and natriuretic responses that depend on the activation of the renin-angiotensin system (RAS) (Takahashi et al., 1984; Yamada et al., 1994; Huang and Leenen, 1996a,b). Likewise, there is substantial evidence to indicate that brain EO contributes to the pathogenesis of NaCl-sensitive hypertension and is stimulated by acute and chronic NaCl loading (Goto et al., 1992; Huang and Leenen, 1996a,b, 1998; Fedorova et al., 2005a, 2007). Increases in brain EO in Dahl-S rats are caused by central administration of NaCl or by NaCl loading; these increases are mediated by the brain RAS and by sympathoactivation (Huang and Leenen, 1996b). Leenen and coworkers demonstrated in rats that an increase in cerebrospinal fluid NaCl precedes the development of hypertension (Huang et al., 2004) and that sodium ions in the brain enter the intracellular space via epithelial sodium channels (Wang and Leenen, 2003). Moreover, they also demonstrated that this process is modulated by central mineralocorticoid receptors (Amin et al., 2005). Accordingly, Gabor and Leenen (2009) demonstrated in Wistar rats that immunoneutralization of brain EO prevented the potentiating effect of exogenously administered aldosterone on pressor response induced by central administration of sodium chloride. Some data, however, suggest that brain-specific rather than epithelial sodium channels are involved in this process (Orlov and Mongin, 2007).
Haupert and coworkers have described a specific pathway for the biosynthesis of EO in the hypothalamus of Milan hypertensive rats (Murrell et al., 2005). However, no such pathway could be identified in the adrenal cortex. Therefore, it seems that in NaCl-sensitive hypertension, EO is likely to act as a central regulator rather than as a peripheral effector. This concept is integral to the schema we propose in Fig. 4.
2. Endogenous Digoxin.
There is some evidence that one of the endogenous CTS represents digoxin. Thus, Goto et al. (1990) purified a substance indistinguishable from digoxin from human urine. Later, another group (Qazzaz et al., 1996) reported that bovine adrenal glands contain deglycosylated analogs of digoxin. It is noteworthy that digoxin (200 μg/kg/day) has been previously reported to reverse hypertension induced by chronic ouabain administration (Huang et al., 1999). Although the concept of endogenous digoxin as an antagonist of EO in humans is quite appealing, the extensive variability of commercial digoxin immunoassays to detect endogenous CTS (Sheiner et al., 1974; Bergdahl et al., 1979; Bergdahl and Molin, 1981; Kelly, 1986; Goto et al., 1991a, 1992) argues strongly against this possibility.
B. Endogenous Bufadienolides
Amphibians are known to produce CTS of bufadienolide nature, which are different from cardenolides in having a doubly unsaturated six-membered lactone ring (Fig. 2) (Meyer and Linde, 1971). Bufadienolide-containing preparations from frog and toad skin have been (and still are) used for treatment of congestive heart failure (CHF) in the traditional medicine of the Far East (Chen and Kovaríková, 1967). It is noteworthy that the highest levels of bufadienolides are detected in the skin of those amphibian species, which migrate from a dry to an aquatic environment (Flier et al., 1980). Because in Amphibia the skin regulates water and electrolyte homeostasis, it has been hypothesized that the sodium pump and bufadienolides in the skin of Amphibia are integral to this process (Flier et al., 1980; Lichtstein et al., 1991). Supporting this concept, brain and skin levels of bufadienolides in toads have been found to respond appropriately to changes in environmental salinity (Lichtstein et al., 1991).

Interaction between brain endogenous ouabain, the renin-angiotensin system, and marinobufagenin in pathogenesis of salt-sensitive hypertension. In NaCl-loaded Dahl salt-sensitive rats, the impairment of renal sodium transport causes sodium retention, which stimulates brain endogenous ouabain in hippocampus, hypothalamus, and pituitary gland. Brain endogenous ouabain stimulates the brain RAS in the hypothalamus and pituitary and activates sympathetic nervous system (SNS), which activates adrenocortical RAS. Angiotensin II activates adrenocortical production of MBG with a primary adaptive aim to induce natriuresis via inhibition of renotubular Na+/K+-ATPase. Excessive MBG production, however, induces a maladaptive effect by inhibiting the Na+/K+-ATPase in vascular smooth muscle cells and by potentiating vasoconstriction.
These observations triggered a search for mammalian bufadienolides. At first, Kieval et al. (1988) and Goto et al. (1991a) detected bufalin-immunoreactive material in human bile and plasma. Subsequently, the presence of bufalin-like immunoreactivity in human plasma was reported by two other groups (Numazawa et al., 1995; Oda et al., 2001). Lichtstein et al. (1993) used mass spectroscopy to detect bufalin derivatives in the eye lenses of several mammalian species. Sich et al. (1996) reported that human plasma and bovine adrenal glands contain material cross-reacting with an antibody to proscillaridin A. Hilton and coworkers (1996) identified a bufadienolide compound in human placentae and plasma using mass spectroscopy. Other workers, using specific immunoassays, mass spectrometry, and, subsequently, NMR spectrometry, demonstrated that mammalian plasma and urine contain MBG (Bagrov et al., 1998; Komiyama et al., 2005). MBG emerged as a candidate CTS largely because of studies characterizing the pharmacological properties of bufadienolides (Bagrov et al., 1993a, 1995b, 1996a,b). It has been shown that the venom from the Bufo marinus toad contains digoxin-like immunoreactive material with vasoconstrictor, sodium pump-inhibitory, and positive inotropic effects (Bagrov et al., 1993a). Subsequently, this substance was identified as MBG, a steroid described previously in toads (Fig. 2) (Bagrov et al., 1995a). Various anti-MBG antibodies were found to cross-react with material present in the urine and plasma of humans, dogs, and rats (Bagrov et al., 1995a,b, 1998; Gonick et al., 1998; Fedorova et al., 2001a,b). In normotensive rats, plasma MBG increased in response to acute plasma volume expansion and after chronic administration of a high NaCl diet (Fedorova et al., 1998, 2001a; Periyasamy et al., 2005). Enhanced production of MBG has been demonstrated in humans with volume expansion (Bagrov et al., 1995a) and in patients with preeclampsia (Lopatin et al., 1999), essential hypertension, primary aldosteronism, and end-stage renal disease (Gonick et al., 1998). At concentrations comparable with in vivo plasma levels of this hormone, MBG produces vasoconstriction in isolated human pulmonary and mesenteric arteries (Bagrov et al., 1996a; Bagrov and Fedorova, 1998) and substantial inhibition of the ouabain-resistant α1 Na+/K+-ATPase isolated from rat aorta and kidney (Fedorova and Bagrov, 1997; Fedorova et al., 2000). In addition, it has been observed that immunoneutralization of MBG with a specific antibody administered in vivo reduced blood pressure and renal sodium excretion in NaCl-loaded Dahl-S rats (Fedorova et al., 2002b, 2007). This is, in fact, the same experimental model of hypertension in which Dahl et al. (1969) predicted the existence of an endogenous vasoconstrictor and natriuretic substance. The model of NaCl-induced hypertension in these Dahl-S rats strongly suggests an important interaction between brain and peripheral CTS. It has been observed in both acute and chronic NaCl loading in Dahl-S rats that a transient increase in circulating EO precedes a sustained increase in circulating MBG (Fedorova et al., 2000, 2002b). This finding has led Fedorova et al. (2005a) to postulate that EO, acting as a neurohormone, triggers MBG production and secretion, which in turn effectuates the increases in cardiac contractility, peripheral vasoconstrictor, and natriuresis observed in this model (Fig. 4). Subsequently, Fedorova et al. (2007) demonstrated that in salt-loaded Dahl-S rats, the brain EO exhibits peak transient responses in the amygdala, hippocampus, and supraoptic nucleus of hypothalamus. From this study, brain EO seems to activate the central RAS, which, possibly via sympathoactivation, in turn activates the RAS in the adrenal cortex. Activation of the adrenocortical RAS then seems to facilitate MBG production and secretion, which results in elevated plasma [MBG] and urinary MBG excretion. This sequence of events (Fig. 4) was fully mimicked by intrahippocampal administration of a very low (60 pg) dose of plant-derived ouabain (Fedorova et al., 2007).
Komiyama et al. (2005), using tandem mass and NMR spectrometry, demonstrated that uremic plasma contains increased levels of another bufadienolide, telocinobufagin, in addition to MBG. Telocinobufagin is different from MBG in that it has a hydroxyl at the 14-position of the lactone ring versus an epoxy group in the 14,15-position. The authors hypothesized that telocinobufagin may be a natural precursor of MBG (Komiyama et al., 2005).
Bufalin is a very potent Na+/K+-ATPase inhibitor, and bufalin derivatives were the first bufadienolides positively identified in the mammals (Lichtstein et al., 1993). Bufalin-like immunoreactive material has been identified in human and rat tissues (Kieval et al., 1988; Goto et al., 1991a; Oda et al., 2001). Although levels of bufalin-like compound in plasma from hypertensive Dahl-S rats were found to be moderately elevated, little is known at present about the physiological significance of this CTS in mammals.
C. Biosynthesis of Cardiotonic Steroids
Despite substantial progress in the elucidation of the structure and mechanisms of action of CTS in mammals, the biosynthesis of these endogenous CTS is still poorly understood. Based on the hypothesis that CTS have a steroidal structure, Schreiber et al. (1981b), demonstrated that rat adrenal extracts contain a digitalis-like immunoreactive material. Since that time, evidence for the adrenocortical origin of CTS has grown. First, the de novo biosynthesis of EO was demonstrated in cultured adrenocortical cells (Perrin et al., 1997; Lichtstein et al., 1998; Qazzaz et al., 2004). el-Masri et al. (2002) found that human adrenocortical cells also produce dihydro-ouabain in addition to EO. In agreement with the classic scheme of adrenocortical steroidogenesis, Lichtstein et al. (1998) using radiolabeled hydroxycholesterol as a possible CTS precursor, demonstrated that the side chain cleavage of cholesterol is essential for synthesis of EO. The pharmacological inhibition of the next step of classic steroidogenesis (i.e., conversion of pregnenolone to progesterone) was shown to reduce the synthesis of EO by adrenocortical cells (Perrin et al., 1997). Unfortunately, the enzymes controlling the conversion of progesterone into CTS via inversion of the configuration at carbons 5 and 14 to form the cis-trans-cis configuration so far remain unknown (Hamlyn, 2004). Because we do not know the enzymes responsible for the synthesis of specific CTS, our ability to develop knockout and/or overexpression models has also been rather limited so far.
Murrell et al. (2005) found that the genes coding P450scc (an enzyme controlling the side chain cleavage of cholesterol) and Δ5–3β-hydroxysteroid dehydrogenase isomerase (an enzyme that converts pregnenolone to progesterone) are up-regulated in the hypothalamus of Milan hypertensive rats but not in the hypothalamus of their normotensive counterparts (Milan normotensive rats). Accordingly, the knockdown of Δ5–3β-hydroxysteroid dehydrogenase in rat brain led to a marked reduction in the production of sodium pump-inhibitory material (Murrell et al., 2005). Surprisingly, in the same study, no such mechanism of CTS biosynthesis was identified in the adrenal cortex of Milan hypertensive rats. Evidence for neuroregulatory roles of EO and for the neuroendocrine source of EO has accumulated (Bagrov et al., 2002; Huang et al., 2006). Komiyama et al. (2001) specifically hypothesized that EO may be produced by and secreted from the adrenal medulla. These researchers purified a substance from the PC12 pheochromocytoma cell line that originated from progesterone and that, by its chromatographic and mass spectrometric properties, was not different from authentic ouabain (Komiyama et al., 2001). The above findings agree with a clinical observation of a marked reduction in the levels of circulating EO in a patient after the surgical removal of a pheochromocytoma (Komiyama et al., 1999).
In toads, bufadienolides are synthesized from cholesterol, as demonstrated by experiments with [14C]cholesterol (Siperstein et al., 1957). However, in several toad species, it has been demonstrated that pregnenolone is not incorporated into bufadienolides. This result indicates that cholesterol side chain cleavage is not involved in the biosynthesis of the predominant bufodienelide CTS in these species (Siperstein et al., 1957; Porto and Gros, 1970, 1971; Garraffo and Gros, 1986). Chen and Osuch (1969) demonstrated in toads that bufadienolide CTS may be synthesized from cholesterol via a “bile acid” pathway (i.e., from cholanates). A series of studies performed by Doris and coworkers confirmed that the biosynthesis of bufadienolide CTS does not involve the pathway implicated in synthesis of “classic” steroid hormones and EO. First, this group established that the production of digitalis-like immunoreactive material by adrenocortical cells did not depend on cholesterol side chain cleavage (Doris et al., 1989, 1994). Later, they demonstrated in murine Y1 adrenocortical cells that the de novo biosynthesis of MBG does require cholesterol as a precursor but does not involve the conversion of cholesterol to pregnenolone via side chain cleavage by P450scc as EO does (Dmitrieva et al., 2000, 2005). Lichtstein et al. (1998), using rat primary cultured adrenal cells and labeled precursors, demonstrated that production of bufalin-like immunoreactive material derives from pregnenolone and does require cholesterol side chain cleavage. Because in this study (Lichtstein et al., 1998) the detection of bufalin was based on an assay using antibody with very high (19.5%) cross-reactivity to the cardenolide strophanthidin, it is difficult to exclude the possibility that these results may, in fact, refer to the production of EO. Murine Y1 adrenocortical cells have been demonstrated to produce marinobufotoxin, an MBG-suberylarginine conjugate previously isolated from various amphibian tissues (Yoshika et al., 2007). At this juncture, it seems that although both bufadienelide and cardenolide CTS derive from cholesterol, only cardenolide CTS synthesis requires cholesterol side chain cleavage.
The release of endogenous CTS is controlled by various humoral stimuli including ACTH, ATII, vasopressin, and catecholamines. Release of EO from bovine adrenocortical cells is sensitive to ACTH, stimulation of α1-adrenergic receptors agonists, and ATII (Laredo et al., 1994, 1995, 1997). In human adrenocortical cells, however, the release of EO is insensitive to ACTH and ATII, whereas it is stimulated by phenylephrine and vasopressin (Laredo et al., 1997). It is noteworthy that in bovine adrenocortical cells ATII acting via AT1 receptors stimulates the release of aldosterone, whereas the release of EO is regulated by AT2 receptors (Laredo et al., 1997). In rat adrenocortical cells, EO secretion was shown to increase after stimulation of nicotinic cholinoceptors (Göôz et al., 2004). el-Masri et al. (2002) demonstrated that, unlike for EO, production of endogenous dihydroouabain by human adrenocortical cells is stimulated by cAMP. In bovine adrenocortical cells, Shah et al. (1998) showed that AT2 receptor antagonists blocked the effects of ATII on release of EO but not aldosterone. Treatment of these cells with dibutyryl cAMP, on the contrary, stimulated secretion of aldosterone with no effect on release of EO. These mechanisms may explain in vivo rapid and dramatic peak responses of EO to stimuli such as exercise (Bauer et al., 2005) and acute NaCl loading (Fedorova et al., 2000). Fedorova et al. (2005), in primary cultures of rat adrenocortical cells, demonstrated that ATII via AT1 receptors stimulates the production of MBG.
Both clinical and experimental animal studies have shown that plasma levels of CTS, including EO and MBG, decrease after adrenalectomy and rise after administration of ACTH (Gault et al., 1988; Fedorova et al., 1998; Sophocleous et al., 2003; Lorenz et al., 2008). It should be noted, however, that in a few clinical and experimental studies (Takahashi et al., 1988; Naruse et al., 1994; Bernini et al., 1998) levels of EO were found to be independent of adrenalectomy and ACTH administration. One explanation for these discrepancies is that, similar to other hormones (Miller, 2008), the biosynthesis of EO may occur both in the adrenal cortex and in extraadrenal tissues.
Because all classic steroid hormones circulate in a protein-bound form, it is not surprising that this also seems to be the case for mammalian CTS. Lichtstein et al. (1993) and Butler et al. (1996) addressed this issue. They found that a substantial portion of bufadienolides are protein-bound in toads; in these animals, plasma levels of bufadienolides reach an enormously high (micromolar) level. Subsequently, Antolovic et al. (1998) isolated from mammalian plasma and kidneys a specific cardiac glycoside-binding globulin with higher- and lower-affinity binding sites for ouabain and possibly for endogenous CTS. Later, CTS-binding protein was identified as a fragment of IgG (Komiyama et al., 1998; Parhami-Seren et al., 2002). The importance of CTS-binding protein(s) on the function of CTS has not been studied consistently yet; however, Antolovic et al. (2000) suggested that binding of CTS to and unbinding of CTS from this transport protein may be involved in a rapid and transient 36-fold rise of EO in response to exercise.
D. Differential Sensitivity of Na+/K+-ATPase Isoforms to Cardiotonic Steroids
A separate line of evidence indicates that various endogenous sodium pump ligands exhibit selectivity toward different isoforms of the Na+/K+-ATPase. Ferrandi et al. (1993) demonstrated that a Na+/K+-ATPase inhibitor purified from rat and bovine hypothalamus inhibits the Na+/K+-ATPase from rat brain (α3 isoform) similar to ouabain and surpasses ouabain in its ability to inhibit the renal sodium pump (α1 isoform). Another sodium pump inhibitor, a labile factor derived from the peritoneal dialysate of patients with chronic renal failure that is clearly dissimilar from ouabain in its elution profile, exhibited greater affinity toward the α1 and α2 rather than the α3 isoform of Na+/K+-ATPase (Tao et al., 1996, Graves et al., 2000). Subsequently, this material was shown to potently inhibit the Na+/K+-ATPase from rabbit kidney (α1 isoform) and by its vasoconstrictor activity exceeded ouabain by more than 2 orders of magnitude (Graves et al., 2000). Crambert et al. (1998) purified digitalis-like immunoreactive material from adult and neonatal plasma. This material exhibited greater affinity toward the α1 and α2 versus α3 Na+/K+-ATPase isoforms.
Fedorova and Bagrov (1997) have studied mechanisms of the vasoconstrictor effects of ouabain and MBG in isolated rat aorta. In this preparation MBG preferentially inhibited the Na+/K+-ATPase from vascular smooth muscle sarcolemma (α1 isoform), whereas ouabain exhibited higher affinity toward Na+/K+-ATPase from vascular nerve endings (α3 isoform). Subsequently, amphibian MBG and MBG-like immunoreactive material purified from hypertensive Dahl-S rats was found to be a potent inhibitor of ouabain-resistant Na+/K+-ATPase from rat kidney (Fedorova et al., 2000, 2001a).
Pierre et el. (2008), using ERK phosphorylation as an indicator of ouabain-induced signaling, demonstrated that α3/β1 and α4/β1 but not α2/β1 Na+/K+-ATPase responded to ouabain treatment, indicating the importance of isoform-specific differences in Na+/K+-ATPase signaling.
E. Hypertension and Cardiotonic Steroids
It seems that two scenarios involving different patterns of CTS responses are involved in pathogenesis of hypertension. One of them, the adducin paradigm, has been shown to be relevant to human hypertension. The levels of EO clearly become elevated in hypertensive individuals, who possess the appropriate mutation(s) of the adducin gene (Wang et al., 2003; Manunta et al., 2008). Moreover, although the clinical relevance of the other scenario, the interplay between brain EO and MBG, still remains to be established, it is certainly likely that these CTS are important modulators of blood pressure outside of the adducin paradigm. It is noteworthy that in a study of chronically NaCl-loaded female subjects, the levels of MBG negatively correlated with the height of blood pressure during high NaCl intake (Anderson et al., 2008). Thus, not only excessive production of MBG but also its failure to adequately increase after NaCl loading may be a factor triggering salt-sensitive hypertension.
V. Endogenous Cardiotonic Steroids and Cell Signaling
The first or classic model to explain interactions of endogenous CTS with the plasmalemmal Na+/K+-ATPase was based on the observation that many CTS inhibit the enzymatic and transport functions of the Na+/K+-ATPase (Klein et al., 1971). The model, which has been enumerated in a large number of reviews, basically proposes that exposure of cells to CTS results in some degree of inhibition of the Na+/K+-ATPase pumping function and consequently an increase in cytosolic sodium, at least within some domain of the cell. This increase, in turn, alters the transport function of the Na+/Ca2+ exchanger, which then results in increases in cytosolic calcium, again, at least in some domain of the cell. With this connection to cytosolic [Ca2+], it is easy to see how innumerable cellular functions might be influenced (Blaustein, 1993). This model is illustrated in Fig. 5.
Several lines of experimental evidence derived from the laboratory of Xie suggest that this classic or ionic mechanism might not be sufficient. First, circulating levels of the CTS that have been identified in humans (see section IV), do not typically have significant effects on the enzymatic function of purified Na+/K+-ATPase (Liu et al., 2002). Second, it is extremely difficult to detect changes in cytosolic Na+ in mammalian cells exposed to those concentrations of endogenous CTS (Liu et al., 2000; Tian et al., 2003). Third, increases in cytosolic Na+ induced with Na+ ionophores do not reproduce effects of CTS in most cell systems (Liu et al., 2000). Fourth, and perhaps most important, effects of CTS on the phosphorylation of key signaling proteins such as the EGFR can be observed in cell-free systems, in which changes in [Na+] are essentially excluded (Wang et al., 2004). This pattern of protein phosphorylation and stimulation of other intracellular messages such as reactive oxygen species can be clearly dissociated from any changes in cytosolic Na+ in multiple experimental systems and can actually be demonstrated to explain many of the changes in cytosolic Ca2+ originally attributed to the inhibition of the pumping function of the Na+/K+-ATPase (Liu et al., 2000). That said, the positive evidence supporting this alternative or “signaling” function for the Na+/K+-ATPase is derived from the aforementioned studies and others and can be summarized as follows. There is a “pool” of plasmalemmal Na+/K+-ATPases that reside in the caveolae of cells, do not seem to actively “pump” sodium, and are closely associated with other key signaling proteins, namely Src and the EGFR (Liang et al., 2007). Exposure of several different cell types in culture to several CTS including ouabain and/or MBG induce rapid phosphorylation of the EGFR in a Src-dependent manner. The phosphorylation pattern for the EGFR is distinctly different from that observed when EGFR induces autophosphorylation of this protein (Haas et al., 2000, 2002). In addition to the EGFR, other signaling proteins seem to be recruited including phospholipase C, TRP proteins, PI(3)K, and several isoforms of PKC (Kometiani et al., 1998; Liu et al., 2000, 2003, 2004, 2005; Tian et al., 2003; Wang et al., 2004). Studies using sophisticated fluorescence transfer methods in intact cells suggest that under basal circumstances, the caveolar Na+/K+-ATPase binds closely with Src and maintains Src in an inactive form. The binding of CTS to the Na+/K+-ATPase induces a conformation change that, in turn, alters the relationship between the Na+/K+-ATPase and Src and allows Src to become activated. This active Src is then able to phosphorylate other proteins (Tian et al., 2006). It is noteworthy that the binding of CTS to the plasmalemmal Na+/K+-ATPase induces the endocytosis of the CTS-Na+/K+-ATPase-Src-EGFR complex in a manner analogous to that for classic receptor tyrosine kinases (Liu et el, 2002, 2004, 2005; Periyasamy et al., 2005). One might speculate that the Na+/K+-ATPase and Src together form an effective receptor tyrosine kinase (Pierre and Xie, 2006).

Schematic diagram contrasting “classic” (“ionic”) versus “signaling” pathways for CTS effects. In the classic pathway, any signaling through the Na+/K+-ATPase requires inhibition of the Na+/K+-ATPase pumping activity, which in turn is accompanied by changes in cytosolic [Na+] and [K+]. As discussed in the text, some authors feel that the caveolar Na+/K+-ATPase may be more sensitive to CTS in terms of enzymatic function. The increases in [Na+] and decreases in [K+] then induce an increase in cytosolic [Ca2+], which, in turn, activates a variety of pathways that have combinations of genomic and nongenomic effects. In contrast, the signaling pathway involves the association of Src with the Na+/K+-ATPase in a caveolar domain. Binding of the CTS to the Na+/K+-ATPase activates Src, which, in turn, transactivates the EGFR and phospholipase C (PLC). This leads to a cascade that involves generation of ROS, activation of mitogen-activated protein kinase (ERK) through activation of its mitogen-activated protein kinase kinase (MEK), activation of PI(3)K, stimulation of endocytosis and activation of Akt as well as activation of protein kinase C. This signaling construct proposes that these steps induce increases in cytosolic [Ca2+] and induce the combinations of genomic and nongenomic effects. Note that whereas both the classic and signaling pathways allow for intervention at the level of CTS binding to the Na+/K+-ATPase [(a), immunoneutralization or pharmacological neutralization], the signaling pathway presents a number of potential targets such as (b) interference with Src activation and EGFR transactivation, (c) PLC activation, (d) MEK activation, (e) ROS generation or scavenging, or (f) PI(3)K activation. Modulation of the signaling pathways at the level of PKC (g), ERK, and Akt might also be possible.
Once CTS bind to the Na+/K+-ATPase, increases in cellular ROS can also been observed, and these increases in ROS can be easily dissociated by changes in the cytosolic Ca2+ concentration (Liu et al., 2000). These increases in ROS seem to depend on the function of the RAS as well as on the participation of mitochondrial K channels, but the precise mechanisms by which CTS effect an increase in cellular ROS are still unclear (Tian et al., 2003). Many of the downstream biochemical (e.g., activation of ERK) and physiological (e.g., increases in cytosolic Ca2+) consequences of CTS binding to the Na+/K+-ATPase can be prevented by ROS scavenging (Xie et al., 1999; Liu et al., 2000; Priyadarshi et al., 2003; Kennedy et al., 2006b). This model is summarized in Fig. 5.
Fascinating inquiries have been made into the relationship between signaling through the Na+/K+-ATPase and cell death (Masuda et al., 1995; Watabe et al., 1996; Orlov and Hamet, 2004; Akimova et al., 2008b; Mijatovic et al., 2008; Stoklosa et al., 2008; Uddin et al., 2008b). In particular, Orlov et al. (2004) demonstrated that high concentrations of ouabain seems to induce a “death signal” in endothelial cells that can be readily dissociated from the effects of Na+/K+-ATPase inhibition by extracellular potassium depletion. The interactions between signaling through the Na+/K+-ATPase and cell death seem to be both complex and variable, depending on cell type and on treatment conditions (Winnicka et al., 2007; Newman et al., 2008). It should also be noted that beyond death signals, the Na+/K+-ATPase signal cascade seems to be involved in a variety of cell biological functions including adhesion and differentiation (Zhang et al., 1991; Numazawa et al., 1994; Kawazoe et al., 1999; Ghoumari et al., 2006; Larre et al., 2006; Rodrigues-Mascarenhas et al., 2006).
The debate between the classic (ionic) and alternative (signaling) pathways may ultimately end in a tie. Although it seems clear that the alternative pathway can explain many of the effects of CTS, it does not seem to explain all of them. For example, it is well known that red blood cells of patients with end-stage renal disease have decreases in ouabain-sensitive rubidium uptake and enzymatic Na+/K+-ATPase function that can be reversed by incubation, in vitro, with antibodies to digitalis and/or other CTS (Periyasamy et al., 2001; Bagrov et al., 2005). Another point is that among Na+/K+-ATPase isoforms, predominantly the α1 isoform seems to reside in caveolae in close physical proximity to Src. However, it is very clear from the elegant studies of Lingrel and colleagues that changes in the CTS sensitivity of the α2 isoform produce profound physiological effects (Dostanic et al., 2003, 2004, 2005). In addition, Feldmann et al. (2007) have demonstrated that even the endocytosed Na+/K+-ATPase may still have a pumping function; these investigators have been able to distinguish the pH within early endosomes, depending on whether ouabain still interacted with the endocytosed Na+/K+-ATPase. From these and other data that have been summarized in several reviews, we propose that the classic and alternative pathways may work both in parallel and synergistically to effect physiological consequences of CTS binding to the Na+/K+-ATPase.
VI. Effects of Endogenous Cardiotonic Steroids on Kidney and Sodium Metabolism
As discussed in section III, a natriuretic substance is defined as one that increases urinary Na+ excretion; however, the prototypal CTS, digitalis or digoxin, is not natriuretic when administered in typical clinical doses in vivo to experimental animals or normal human subjects, although suprapharmacological doses of digoxin do alter sodium transport in the isolated kidney and impair transcellular transport of virtually all renal epithelial cells (Hauptman and Kelly, 1999). In contrast, administration of the atrial or brain natriuretic peptides induces prompt natriuresis in vivo and alters distal nephron sodium reabsorption in a well defined cGMP-mediated process (Itabashi et al., 1987; Nakamoto et al., 1987; Rubattu and Volpe, 2001; Lee and Burnett, 2007).
In distinction from digoxin or ouabain, the endogenous CTS described by Hillyard et al. (1976) did have an appreciable effect on renal Na+/K+-ATPase activity. Work from our laboratories has demonstrated that MBG has potent effects on rat renal Na+/K+-ATPase activity, which can be reversed in vivo by administration of anti-MBG antibodies. In addition to the direct inhibition of the Na+/K+-ATPase by MBG, other studies from our laboratories have shed new light on this subject. We observed that the administration of CTS induced endocytosis of the plasmalemmal Na+/K+-ATPase in LLC-PK1 cells, a cell line maintaining features of proximal tubule cells (Liu et al., 2002). In fact, CTS actually induced decreases in plasmalemmal Na+/K+-ATPase density in a dose- and time-dependent manner in these cells, whereas in MDCK cells, a cell line resembling distal tubular cells, no such depletion of the plasmalemmal Na+/K+-ATPase was observed. Among the CTS that were studied in LLC-PK1 cells, digoxin, MBG, and ouabain differed in their ability to induce this endocytosis; specifically, digoxin seemed to be the least effective whereas other CTS (e.g., MBG) were substantially more effective, even when results were normalized for the ability of the CTS to inhibit the Na+/K+-ATPase enzymatic function. Further studies demonstrated that CTS could actually inhibit transcellular Na+ transport in LLC-PK1 cells (Liu et al., 2002). Endocytosis of the Na+/K+-ATPase was subsequently shown to proceed through clathrin-coated pits and require PI(3)K activation and the plasmalemmal pump being in the context of caveola as well as signaling through the Src-EGFR pathway (Liu et al., 2004, 2005).
Extending these studies to an in vivo rat model, we observed evidence for CTS (specifically MBG)-induced endocytosis participating in the altered sodium reabsorption seen with increases in dietary sodium. Specifically, we observed that increases in dietary sodium induced increased urine excretion of MBG along with decreased proximal tubular Na+/K+-ATPase expression along with increased presence of the Na+/K+-ATPase in both early and late endosomes. Administration of an antibody to MBG blocked both the endocytosis of the Na+/K+-ATPase and blunted the increase in urinary sodium excretion seen with the high-salt diet (Periyasamy et al., 2005). Further work demonstrated that CTS can induce decreases in the apical expression of one of the plasma membrane Na+/H+ exchangers, NHE3 (Oweis et al., 2006; Liu and Shapiro, 2007). Some of the long-term decrease in NHE3 expression is related to decreases in NHE3 transcription, whereas, acutely, binding of ouabain to the basolateral Na+/K+-ATPase seems to rapidly induce endocytosis of the apical NHE3 (Cai et al., 2008). Taken together, these data suggest that increases in the circulating levels of MBG accompany salt loading, which may in turn induce decreases in both basolateral and apical sodium transport in the proximal tubule through both the classic (ionic) or signaling mechanism and the more recently described Na+/K+-ATPase-Src-EGFR pathway. This decrease in renal sodium reabsorption would then result in increases in urinary sodium excretion (Fig. 5). Therefore, through direct effects on renal epithelium, some CTS do seem to function as natriuretic substances in vitro and in vivo (Liu and Shapiro, 2007).
In addition to the effects of bufadienolides on the heart and vascular system, these hormones have renal effects that are important physiologically. First, increases in MBG accompany decreases in renal function; this has been established both in experimental animals and humans (Gonick et al., 1998; Priyadarshi et al., 2003; Komiyama et al., 2005; Kennedy et al., 2006a, 2007). More importantly, it has been demonstrated that MBG induces endocytosis of the proximal tubular Na+/K+-ATPase and decreases renal sodium reabsorption, both in vitro and in vivo; in particular, our group has demonstrated that administration of antibodies to MBG alters the endocytosis of the proximal tubular Na+/K+-ATPase and decreases urinary sodium excretion in Sprague-Dawley rats (Liu et al., 2002; Periyasamy et al., 2005). In agreement with the notion that inhibition of renal α1 Na+/K+-ATPase by MBG represents one of the mechanisms regulating renal sodium excretion, genetically engineered mice that express ouabain-sensitive α1 Na+/K+-ATPase have been shown to exhibit greater natriuretic responses to saline loading than mice with ouabain-insensitive α1 Na+/K+-ATPase (Loreaux et al., 2008).
VII. Chronic Renal Failure and Uremic Cardiomyopathy
The modern concept of uremic cardiomyopathy is that patients with renal disease develop a progressive loss of diastolic function and ventricular hypertrophy, which ultimately over time may progress to eccentric hypertrophy and, rarely, systolic dysfunction (Mohmand et al., 2005). Based on echocardiographic studies, diastolic dysfunction and ventricular hypertrophy are extremely common whereas systolic dysfunction occurs in less than 20% of patients with end-stage renal disease (ESRD); neither the diastolic dysfunction nor the ventricular hypertrophy can be explained only by the hypertension and anemia that usually complicate ESRD (Middleton et al., 2001). As mentioned in section IV.B, plasma levels of telocinobufagin and MBG have been noted to be substantially elevated in patients with ESRD (Komiyama et al., 2005). To examine this important issue, our laboratory has established a model of chronic renal failure using partial nephrectomy in the rat (Kennedy et al., 2003) and the mouse (Kennedy et al., 2007). Animals subjected to partial nephrectomy develop increases in MBG similar to that seen clinically in subjects with renal failure (Gonick et al., 1998; Kennedy et al., 2006a); these animals also develop other features similar to human uremic cardiomyopathy including diastolic dysfunction and ventricular hypertrophy as well as evidence for signaling through the Na+/K+-ATPase-Src-EGFR cascade and ERK activation (Kennedy et al., 2003, 2007). In this rodent model, cardiac fibrosis is especially prominent (Kennedy et al., 2006a, 2007; Elkareh et al., 2007), although to be fair, it also seems to complicate human uremic cardiomyopathy (Harnett and Parfrey, 1994; London and Parfrey, 1997). In the rat, infusion of MBG to achieve elevations in plasma [MBG] similar to those seen with partial nephrectomy results in activation of the Na+/K+-ATPase-Src-EGFR cascade and ERK and many of the phenotypical features of experimental uremic cardiomyopathy, whereas active immunization against MBG attenuates most of the biochemical, physiological, and morphological features of uremic cardiomyopathy in animals subjected to partial nephrectomy (Kennedy et al., 2006a; Elkareh et al., 2007).
Additional work in this area has demonstrated that in the rodent very small amounts of MBG (and other CTS), virtually identical to the circulating plasma concentrations seen in experimental and clinical renal failure, directly stimulate the production of collagen in the primary culture of cardiac fibroblasts (Elkareh et al., 2007). Again, this direct effect seems to require signaling through the Na+/K+-ATPase-Src-EGFR cascade. Although some activity of the transforming growth factor-β-Smad pathway is necessary for this stimulation, no changes in the amount of transforming growth factor-β or the Smads were identified. The stimulation of collagen production was associated with increased translational procollagen as well as increases in mRNA for procollagen, whereas no change in procollagen or collagen stability could be identified (Elkareh et al., 2007). Low concentrations of CTS have been shown to induce collagen synthesis by dermal fibroblasts, which could potentially be exploited to accelerate wound healing (El-Okdi et al., 2008).
VIII. Congestive Heart Failure
CHF is associated with fluid retention and plasma volume expansion; therefore, one would expect CTS to be stimulated under these circumstances (Schrier and Abraham, 1999). As early as 1981, Schreiber et al. (1981a) hypothesized that CTS might be involved in the regulation of tissue growth and myocardial hypertrophy. Morise et al. (1988) demonstrated that development of CHF in rats is associated with increased plasma Na+/K+-ATPase inhibitory activity. Liu et al. (1990) showed in a group of 50 patients that the severity of CHF is positively associated with plasma levels of digoxin-like immunoreactivity as well as erythrocyte sodium concentrations. The relationship between CTS, cardiac geometry, and central hemodynamic parameters has been analyzed in several studies. Gottlieb et al. (1992) found that although plasma EO did not exhibit graded increases with the progression of cardiac failure, EO levels were elevated in patients with severely impaired left ventricular (LV) performance (LV ejection fraction less than 21%). Manunta et al. (1999) demonstrated that plasma EO positively correlated with systolic and diastolic blood pressure in a group of 110 normotensive subjects and 100 hypertensive subjects; these workers found that EO levels positively correlated with LV mass index and LV end diastolic volume in only the hypertensive subjects. Later, Pierdomenico et al. (2001) found that circulating EO levels in 92 hypertensive patients were positively correlated with mean BP and total peripheral resistance index, whereas LV end-diastolic volume index, stroke index, and cardiac index exhibited inverse correlations with the plasma EO. Plasma EO was found to independently predict total peripheral resistance index, cardiac index, and relative wall thickness. Moreover, the plasma EO was substantially higher in patients with eccentric remodeling compared with those subjects with normal LV geometry or concentric hypertrophy (Pierdomenico et al., 2001). In another study performed in patients with LV dysfunction (Balzan et al., 2001), the plasma [EO] was found to be elevated compared with that in normal subjects but did not correlate with the LV ejection fraction. Fridman et al. (2002) studied 23 consecutive hypertensive male patients with CHF; in these patients, the plasma MBG exhibited progressive increases that paralleled the progression of CHF and increases in plasma α-human atrial natriuretic peptide (α-hANP). Although in the same study, the plasma EO was not found to be elevated compared with that in normal subjects, the levels of EO were found to be substantially elevated in a subset of patients with CHF who had ejection fractions less than 30% (A. Y. Bagrov and V. I. Novikov, unpublished observations), similar to the observation of Gottlieb et al. (1992).
Experimental data also indicate an association between elevated plasma CTS and cardiovascular remodeling. Normotensive rats consuming a 4% NaCl diet demonstrate increases in plasma MBG and correlated increases in cardiac weight in the absence of hypertension (Fedorova et al., 2001a). Sustained ouabain infusion, which causes a 2-fold elevation of plasma ouabain immunoreactivity, is also sufficient to induce LV hypertrophy in normotensive rats (Ferrandi et al., 2004).
Studies performed in Dahl-S rats subjected to a high-salt diet coordinated shifts between the physiological function of the left ventricle (first compensated LV hypertrophy develops, which later transitions into a dilated cardiomyopathy), plasma levels of CTS, and the amounts of α1 and α2 Na+/K+-ATPase isoforms within the LV myocardium (Fedorova et al., 2004). Specifically, the advancing stages of hypertrophy were associated with elevated plasma MBG, increased levels of α1 (compared with α2) Na+/K+-ATPase in the LV myocardium, and a heightened sensitivity of the cardiac sodium pump to MBG. The transition to CHF was associated with a decline in MBG levels and decreased levels of α1 Na+/K+-ATPase protein in the left ventricle. Levels of EO in Dahl-S rats with CHF rose substantially, and the enhanced EO production was associated with increased levels of ouabain-sensitive α3 Na+/K+-ATPase in LV myocardium, along with an increase in the sensitivity of the cardiac Na+/K+-ATPase to ouabain (Fedorova et al., 2004).
It has been demonstrated that ouabain and MBG in vitro in cultured renal tubular cells induce apoptosis and growth-promoting signaling, respectively (Akimova et al., 2005). The existence of endogenous CTS with distinct effects on cell survival makes teleological “sense,” because different in vivo scenarios may require modulation of sodium pump-dependent functions with or without cytotoxic actions. Thus, the transition from compensated LV hypertrophy to CHF is accompanied by a decrease in the plasma MBG and by a 3-fold increase in plasma EO when the induction of cell death may be desirable (Neuss et al., 2001).
Involvement of CTS in pathogenesis of experimental CHF is not limited to hypertensive models. In rats with CHF complicating experimental myocardial infarction, the chronic blockade of brain EO with Digibind (Fab fragments of polyclonal anti-digoxin antibody developed for treatment of digoxin intoxication) substantially inhibited the development of LV dilation and dysfunction (Leenen et al., 1999).
In humans, the development of CHF is associated with decreased Na+/K+-ATPase activity in LV myocardium along with a decline in the expression of both α1 and α3 Na+/K+-ATPase isoforms (Schwinger et al., 1999). In animal studies, changes in Na+/K+-ATPase isoform composition observed in the context of LV remodeling are complex and seem to be specific to both the species and experimental models. Thus, similar to those observed in hypertensive Dahl-S rats (Fedorova et al., 2004), significantly elevated LV levels of α1 Na+/K+-ATPase have been reported in rats with diabetic cardiomyopathy (Gerbi et al., 1997). In other studies of LV hypertrophy occurring in hypertensive rats, levels of α1 Na+/K+-ATPase in LV myocardium were unchanged, whereas those of α2 Na+/K+-ATPase were decreased (Book et al., 1994, Charlemagne et al., 1994).
In short, clinical and experimental studies demonstrate that in CHF levels of CTS change and accompany changes in the cardiac expression of their molecular target, the Na+/K+-ATPase. Considering growing evidence for an important role of CTS in cell signaling, growth, and apoptosis, it seems clear that these molecules represent potential biomarkers for CHF and cardiovascular remodeling as well as potential therapeutic targets.
IX. Preeclampsia
Despite the fact that preeclampsia occurs in between 3 and 5% of all pregnant women and represents the leading cause of maternal and fetal mortality, its pathogenesis remains poorly understood. Moreover, treatment for preeclampsia has not changed significantly in many years (Funai et al., 2005; Sibai et al., 2005). That said, one theory of the pathogenesis of preeclampsia attributes an important role to CTS.
Because pregnancy is associated with plasma volume expansion involving renal sodium and fluid retention (Gallery et al., 1979; Masilamani et al., 1994), it is logical to examine the role of CTS in pregnancy and pregnancy-associated diseases. Buckalew and coworkers (Gusdon et al., 1984) were the first to demonstrate increased circulating levels of CTS in pregnancy and to hypothesize that CTS are involved in the pathogenesis of pregnancy-induced hypertension and preeclampsia. Subsequently, several groups of investigators demonstrated elevated levels of digitalis-like immunoreactivity and elevated Na+/K+-ATPase inhibitory activity in preeclamptic plasma (Graves et al., 1984; Graves, 1987; Shrivastav et al., 1988; Poston et al., 1989; Kaminski and Rechberger, 1991; Gilson et al., 1997). After establishment of specific assays for cardenolide and bufadienolide CTS, levels of both EO and MBG were found to be moderately increased in normotensive pregnant women. In contrast, MBG but not EO was markedly elevated in patients with severe preeclampsia (average blood pressure 160/104 mm Hg) (Lopatin et al., 1999). In subjects with milder preeclampsia (blood pressure 149/93 mm Hg), elevated levels of MBG but, again, not of EO were observed and accompanied the observed inhibition of Na+/K+-ATPase in the erythrocytes. Ex vivo treatment of erythrocytes derived from these preeclamptic women with anti-MBG, but not anti-ouabain antibody, reversed the preeclampsia-associated Na+/K+-ATPase inhibition (Averina et al., 2006).
Although the exact role(s) of CTS in pregnancy is not yet understood, Digibind lowered the blood pressure in 15 patients with preeclampsia (Goodlin, 1988; Adair et al., 1996, 2009). The mechanism underlying this effect is believed to be cross-reactivity with endogenous CTS. These clinical observations are in agreement with the results of an experimental study, which demonstrated a vasorelaxant action of Digibind in isolated perfused preeclamptic placentae (Di Grande et al., 1993). Accordingly, the Na+/K+-ATPase from preeclamptic placentae was shown to be up-regulated and to exhibit heightened sensitivity to digitalis glycosides (Amler et al., 1994). This concept has been explored further in experimental animals. In pregnant Sprague-Dawley rats, levels of both MBG and EO doubled by the end of gestation. Sodium chloride (1.8%) supplementation of rats during days 14 to 20 of gestation was associated with a 33 mm Hg increase in systolic blood pressure and proteinuria and decreases in fetal weight, size, and number of off-spring (Fedorova et al., 2005b). Compared with normotensive pregnant rats, the development of hypertension in NaCl-supplemented pregnant rats was accompanied by increased plasma [MBG] but not [EO] (Fedorova et al., 2005b). In the same study, administration of anti-MBG antibody to NaCl-supplemented pregnant rats resulted in a 28 mm Hg decrease in blood pressure and caused a substantial increase in the activity of the sodium pump in thoracic aortae. A similar pattern of CTS levels has been observed in another model of preeclampsia, specifically, pregnant rats treated with deoxycorticosterone acetate and given 0.9% saline as drinking water for the duration of their pregnancy (Vu et al., 2005). Uterine arteries from these rats exhibited enhanced sensitivity to the vasoconstrictor effects of MBG. Moreover, anti-MBG antibody substantially reduced the blood pressure in these animals. It has been demonstrated that in patients with preeclampsia, a monoclonal antibody against MBG detects elevated levels of MBG in plasma and placenta and ex vivo restores activity of the Na+/K+-ATPase in erythrocytes (Fedorova et al., 2008).
In preeclampsia, besides exhibiting direct vasoconstrictor effects, heightened CTS levels can alter the function of cytotrophoblasts and thus affect placentation. In cultured human cytotrophoblast cells, MBG, via activation of c-Jun NH2-terminal kinase, p38, and Src and possibly via induction of apoptosis, has been shown to impair cell proliferation, migration, and invasion (Uddin et al., 2008a,b).
Thus, it seems that CTS contribute to pathogenesis of preeclampsia and that increases in [MBG] rather than changes in [EO] are responsible for preeclampsia-induced Na+/K+-ATPase inhibition. This notion is consistent with the bufadienolide nature of endogenous CTS purified from human placentae (Hilton et al., 1996).
X. Other Conditions
Considering the fact that the CTS are one of the factors regulating transport and signaling functions of a key membrane enzyme, Na+/K+-ATPase, it is not a surprise that the list of disorders in which CTS are implicated, is not limited to essential hypertension, ESRD, preeclampsia, and cardiac failure.
A. Myocardial Ischemia/Infarction
Arrhythmogenicity is one of the most serious side effects of digitalis glycosides, and acute myocardial ischemia is known to be associated with heightened sensitivity of LV myocardium to the arrhythmogenic action of digitalis (Akera and Brody, 1977). Maixent and Lelièvre (1987) demonstrated that acute myocardial ischemia is associated with a loss in the number of high-affinity ouabain binding sites and with inhibition of the Na+/K+-ATPase in canine LV myocardium. Bagrov et al. (1989) hypothesized that endogenous CTS may be responsible for myocardial ischemia-induced inhibition of LV Na+/K+-ATPase and that heightened CTS levels may contribute to ischemia-induced ventricular arrhythmias. In agreement with this notion, Delva et al. (1990) observed a rapid increase in plasma Na+/K+-ATPase inhibitory activity in patients with coronary artery disease associated with decreased cardiac output during a transient period of myocardial ischemia while undergoing percutaneous coronary angioplasty. Subsequently, it was demonstrated in patients with acute myocardial infarction that an increase in plasma digoxin-like immunoreactivity accompanies the inhibition of erythrocyte Na+/K+-ATPase (Bagrov et al., 1991, 1994). Accordingly, pretreatment with anti-digoxin antibody was shown to prevent myocardial ischemia-induced inhibition of cardiac Na+/K+-ATPase and to decrease the incidence of ventricular arrhythmias in coronary artery-ligated rats (Bagrov et al., 1993b; Ke et al., 2004). Subsequently, Bagrov et al. (1998) demonstrated that an elevation in plasma CTS could be attributed to increases in plasma MBG and purified MBG from urine of patients with myocardial infarction.
B. Diabetes Mellitus
Elevated levels of CTS, along with perturbed functions of the Na+/K+-ATPase, were found in patients and experimental animals with diabetes mellitus (Clerico and Giampietro, 1990; Chen et al., 1993; Straub et al., 1996). Because insulin resistance often accompanies NaCl-sensitive hypertension, it has been hypothesized that in type 2 diabetes, CTS rise as a result of insulin-dependent renal sodium retention and that excessive CTS elaboration contributes to hypertension (Chen et al., 1993; Martinka et al., 1997). Additional evidence indicates that rats with type 1 diabetes exhibit greater levels of MBG and more profound sodium pump inhibition than rats with type 2 diabetes (Bagrov et al., 2005). Along with clinical data demonstrating that CTS become stimulated during an oral glucose challenge (Carroll et al., 2001), this evidence indicated that CTS may also be implicated in determining insulin sensitivity. Accordingly, our laboratory demonstrated that in vivo administration of anti-MBG antibodies to rats after an oral glucose challenge produced greater elevations of plasma glucose and insulin compared with that in the vehicle-treated animals (Bagrov et al., 2007). Kotova et al. (2006) demonstrated that ouabain and MBG increase glycogen synthesis in skeletal muscle. This effect was additive to insulin and was mediated by activation of a Src-, ERK1/2-, p90rsk-, and glycogen synthase kinase 3-dependent signaling pathway. In skeletal muscle cells, MBG exceeded ouabain both with respect to sodium pump inhibition and induction of cell signaling. Thus, physiological mechanisms of regulation of carbohydrate metabolism and of glucose tolerance may involve CTS.
C. Obstructive Sleep Apnea
Obstructive sleep apnea (OSA) is frequently associated with the metabolic syndrome, a condition that, in turn, is characterized by associated volume-dependent hypertension (Ehlenz et al., 1991) and impaired glucose tolerance (Levinson et al., 1993; Coughlin et al., 2004). In OSA, repeated episodes of apnea/hypopnea lead to an increase in the arterial tension of carbon dioxide (PaCO2) (Fuse et al., 1999). Elevated PaCO2 levels decrease plasma pH and produce renal sodium retention, a major stimulus for CTS production (Anderson et al., 1995; Fedorova et al., 1996). Bagrov et al. (1995) observed that in healthy humans, the pressor response to voluntary hypoventilation is associated with a substantial inhibition of Na+/K+-ATPase in the erythrocytes and with a simultaneous increase in plasma levels of MBG but not of EO. In agreement with this observation, Paci et al. (2000) demonstrated that hypertensive subjects with OSA exhibit elevated plasma levels of an unidentified endogenous Na+/K+-ATPase inhibitor, which, by its chromatographic behavior, is different from EO and resembles MBG. In 52 patients with OSA, Zvartau et al. (2006) demonstrated that levels of MBG were elevated in proportion to the severity of the OSA. In the same study, in patients with OSA 24-h MBG excretion exhibited positive correlations with diastolic blood pressure and plasma levels of leptin and insulin. This finding suggests that in these patients, MBG may be one of the factors linking mechanisms of the pathogenesis of hypertension and insulin resistance (Zvartau et al., 2006).
D. Exercise
Early evidence indicated that prolonged strenuous exercise increased the circulating levels of CTS (Clerico et al., 1988), whereas decreased levels of digoxin-like immunoreactive CTS were implicated in the blood pressure-lowering effects of the exercise (Koga et al., 1992; Komiyama et al., 1997). Bauer et al. (2005) reported intriguing data demonstrating that within 15 min of ergometry, athletes demonstrated 30-fold increases in the plasma levels of EO that rapidly decreased (within 5 min) with rest. A similar observation was made in beagles exposed to moderate exercise (Bauer et al., 2005). These observations demonstrate that very dramatic effects of EO may be transient. This finding seems to be in agreement with reports demonstrating that EO can be implicated in behavioral stress (Goto et al., 1995, Weidemann et al., 2004) and via activation of hypothalamic-pituitary-adrenocortical axis contributes to the onset of NaCl-sensitive hypertension, which was discussed previously (Fedorova et al., 2005a).
E. Behavioral Stress
Several observations demonstrate possible involvement of endogenous ouabain-like compound in behavioral stress. Goto et al. (1995) demonstrated in rats that swimming stress is associated with enhanced levels of ouabain-like immunoreactivity in plasma and adrenal glands but not in the pituitary gland and hypothalamus. Fedorova et al. (2001a) have shown that social isolation of rats is associated with a transient increase in the renal excretion of ouabain-like material.
F. Mood Disorders
In many studies both mania and bipolar depression have been associated with decrements in red blood cell Na+/K+-ATPase activity (Naylor et al., 1973; Choi et al., 1977; Hesketh et al., 1977; Rybakowski and Lehmann, 1994). Subsequently, Grider et al. (1999) studied plasma levels of digoxin-like immunoreactive factor in manic bipolar patients. In this study, normal subjects exhibited seasonal variations in plasma digoxin-like factor; levels of this compound were significantly lower in winter than those in spring, summer, and autumn. Manic subjects did not exhibit such seasonal variations, the level of digoxin-like immunoreactivity in winter was comparable with that in healthy control subjects, and no changes were observed in ill subjects during spring, winter, and autumn (Grider et al., 1999). Goldstein et al. (2006) demonstrated that levels of ouabain-like immunoreactivity in the parietal cortex of bipolar patients are significantly higher than those in normal individuals and depressed patients. These observations suggest that the defect in the Na+/K+-ATPase could be an important factor in pathogenesis of mood disorders (el-Mallakh and Wyatt, 1995).
Experimental findings support this contention. Acute intracerebroventricular administration of 5 μl of 1 mM ouabain solution in rats produced a “mania-like hyperactivation” of the animals studied with automated activity monitors (el-Mallakh et al., 1995). Later, the same group reported that persistent hyperreactivity induced by central administration of ouabain was associated with substantial increases in ouabain binding and Na+/K+-ATPase activity in the hippocampus (Ruktanonchai et al., 1998). Pretreatment of animals with lithium prevented the above effects of ouabain (Li et al., 1997). There is additional evidence that Na+/K+-ATPase inhibitors may affect mood and behavior; several amphibian-derived preparations containing bufadienolides have been used as recreational drugs with apparently addictive consequences (e.g., “toad licking psychosis”) (Howard and Foerstl, 1990; Lyttle et al., 1996).
G. Ethanol Addiction
Neurochemical events associated with the effects of ethanol and with the development of ethanol addiction include changes in Na+/K+-ATPase activity. The effects of ethanol may be associated with both inhibition and activation of the sodium pump, depending on the dose, route of administration, and length of exposure (Nhamburo et al., 1986, 1987; Foster et al., 1989; Foley and Linnoila, 1995). Foley and Rhoads (1994) demonstrated a stimulatory effect of ethanol on the Na+/K+-ATPase in rat cerebral cortex synaptosomes; this is apparently related to its action on ouabain-sensitive (α2 and α3) rather than relatively ouabain-resistant (α1) Na+/K+-ATPase isoforms. These authors concluded that ethanol induces sodium pump activation possibly via antagonizing the effect of an unidentified Na+/K+-ATPase endogenous inhibitory factor (Foley and Rhoads, 1994). Considering the involvement of Na+/K+-ATPase in the effects of ethanol and the existence of multiple endogenous CTS in the mammalian brain, Bagrov et al. (1996c) hypothesized that CTS may be implicated in the development of alcohol addiction. In rats, administration of digoxin (chosen to mimic the action of CTS) caused a decrease in voluntary alcohol consumption whereas immunization of the animals against digoxin, ouabain, and MBG markedly potentiated voluntary ethanol consumption (Bagrov et al., 1996c, 1999). In Wistar rats, ethanol administration stimulated cerebrospinal EO and peripheral MBG (Bagrov et al., 1999). In addition, MBG apparently mediates the pressor response to ethanol withdrawal in rats, which is associated with renal sodium retention (Kashkin et al., 2008). Therefore, in rats, ethanol administration stimulates CTS and suppresses the free choice of alcohol, whereas immunization against MBG and ouabain is associated with alcohol-seeking behavior. Based on the similarities in the neurobiology of depression and drug/alcohol dependence, Markou et al. (1998) suggested that the development of dependence may reflect self-medication of substances of abuse (i.e., alcohol) to normalize altered neurobiological mechanisms. Thus, the deficit of endogenous CTS might be expected to cause depression-like disorders and facilitate self-medicating behaviors, and CTS may be one of the factors linking the pathogenesis of mood disorders and ethanol dependence.
H. Cancer
One of the most novel areas is the current CTS research examining the pathogenesis and treatment of cancer. Here, the initial evidence came from the clinical studies demonstrating that patients with breast cancer who were taking digitalis exhibited less recurrence and better survival than subjects not receiving digitalis treatment (Stenkvist et al., 1982; Mijatovic et al., 2007). This rather staggering observation, taken together with a mounting body of evidence of the growth-promoting but anti-cancer effects of CTS in vitro, suggests the possibility of using digitalis drugs to treat cancer (Mijatovic et al., 2007). It is noteworthy that CTS may also be involved in cancer development. In a group of 84 patients with breast cancer, 73.6% had plasma levels of digitalis-immunoreactive CTS that were 3 times lower than those seen in control subjects, whereas 10.8% of these patients with cancer exhibited extremely high CTS levels (Weidemann et al., 2004; Weidemann, 2005). Therapeutic strategies involving either addition of or antagonism of CTS are currently under development (Mijatovic et al., 2007).
XI. Possible Strategies for Pharmacological Antagonism of Endogenous Cardiotonic Steroids
A. Blockade of Receptor Site on the Na+/K+-ATPase
The potential roles of spironolactone and canrenone (the major metabolite of spironolactone) as CTS antagonists (Semplicini et al., 1995) may be very interesting. Excellent experimental studies demonstrate that both spironolactone and canrenone, which acts as a partial agonist of the digitalis receptor site on the Na+/K+-ATPase, antagonize ouabain binding as well as shift Na+/K+-ATPase inhibition by ouabain (Finotti and Palatini, 1981; Garay et al., 1985). Canrenone has even been proposed as a treatment for digitalis toxicity (Selye et al., 1969; Yeh and Lucchesi, 1974; Waldorff and Buch, 1979). Canrenone has also been shown to reduce BP and to restore Na+/K+-ATPase activity in rats with experimental volume-dependent hypertension (de Mendonça et al., 1988; Pamnani et al., 1990). Clinical data also demonstrate that canrenone is capable of antagonizing the effects of CTS. Boero et al. (1989) have demonstrated in hypertensive subjects that canrenone can prevent inhibition of red blood cell Na+/K+-ATPase induced by intravenous saline infusion. Later, Semplicini et al. (1993) demonstrated that a 4-week administration of canrenone (100 mg daily) abolished vasoconstriction induced by a bolus infusion of ouabain in 50% of untreated hypertensive patients. In those patients in whom ouabain-induced vasoconstriction was inhibited, canrenone significantly lowered blood pressure. In the other patients the blood pressure reduction was not statistically significant.
The next obvious approach, which also does not discriminate between the classic and “signaling” schemas of the Na+/K+-ATPase, would be the use of receptor “antagonists.” Extremely interesting data exist for PST2238 (rostafuroxin) (Fig. 3). This is a steroid compound designed to serve as an EO antagonist. Rostafuroxin has been demonstrated to have beneficial effects in a rodent ouabain infusion model (Ferrari et al., 1999, 2006; Ferrandi et al., 2004). The efficacy of rostafuroxin in hypertensive subjects and the possible dependence of its efficacy on adducin cytoskeleton proteins is being assessed in the ongoing Ouabain and Adducin for Specific Intervention on Sodium in HyperTension (OASIS-HT) (http://clinicaltrials.gov/ct2/show/NCT00415038) phase II multicenter study (Staessen et al., 2005). Vu et al. (2006) reported that resibufagenin, a bufadienolide sodium pump inhibitor, in pregnant rats reverses hypertension induced by deoxycorticosterone acetate-salt and by chronic marinobufagenin administration.
B. Immunoneutralization
Although at first glance the in vivo immunoneutralization of CTS may appear to be an eccentric approach to the treatment of hypertension and blockade of volume-sensitive hormones in a volume-contracted state may seem counterintuitive, clinical evidence for the efficacy of Digibind in preeclampsia comprises one of the most convincing arguments in favor of the prohypertensive effects of CTS. Goodlin (1988) reported a decrease in blood pressure in a 25.5-week preeclamptic patient after two intravenous infusions of Digibind (0.087 mg/kg each). Later, Adair et al. (1996) reported another case of successful use of Digibind in preeclampsia. Subsequently, the same group, in a placebo-controlled double-blind study, demonstrated that Digibind (0.76 mg/kg) lowered the blood pressure in 13 patients with postpartum preeclampsia (Adair et al., 1996, 2009). It is noteworthy that Digibind did not exert adverse effects in these studies. It is hoped that the results of a multicenter, double-blind, placebo-controlled study of the efficacy of Digibind in preeclampsia (http://clinicaltrials.gov/show/NCT00158743) will elucidate the potential utility of CTS immunoneutralization in preeclampsia. The preliminary results of this study indicate that administration of Digibind to patients with severe preeclampsia is associated with an improvement in renal function manifested by an increase in creatinine clearance and by a reduction of plasma Na+/K+-ATPase inhibitory activity (Hopate et al., 2008; Lam et al., 2008).
The clinical impact of CTS immunoneutralization may not be limited to hypertensive emergencies. In fact, CTS antagonism may also be effective in patients with cerebral salt-wasting syndrome, a condition frequently accompanying cerebral injury and associated with a life-threatening natriuresis. Digibind was shown to reduce renal sodium excretion in a patient after brain tumor removal (Menezes et al., 2003). Likewise, the magnitude of the pressor response after intracerebroventricular infusion of blood correlated with plasma sodium pump-inhibitory activity, and Digibind in vivo reduced natriuresis in rats.
C. Modulation of Interaction of Cardiotonic Steroids with Na+/K+-ATPase
1. Protein Kinase C.
Protein kinases are among the factors that regulate Na+/K+-ATPase phosphorylation, activity, and sensitivity to CTS (Blanco et al., 1998). In particular, PKC and PKG can affect the Na+/K+-ATPase activity and sensitivity to CTS via altering its phosphorylation state (Feschenko and Sweadner, 1997; Vasilets et al., 1999). The modulatory effect of PKC is specific to the Na+/K+-ATPase α1 isoform (Blanco et al., 1998; Fedorova et al., 2002a). In human arterial sarcolemma, PKC induces the phosphorylation of α1Na+/K+-ATPase and enhances the sensitivity of Na+/K+-ATPase to nanomolar concentrations of MBG (Fedorova et al., 2002a). Accordingly, in isolated human mesenteric arteries, nonselective activation of PKC by phorbol diesters markedly potentiates vasoconstrictor activity of MBG (Fedorova et al., 2002a). This synergistic interaction of MBG and PKC on vascular Na+/K+-ATPase is likely to participate in fine-tuning of the sodium pump activity and represent a potential therapeutic target. Bagrov et al. (2000) have demonstrated that cicletanine, a furopyridine antihypertensive compound with natriuretic and vasorelaxant actions, directly inhibits PKC activity and antagonizes the vasoconstrictor and Na+/K+-ATPase inhibitory effects of MBG via a PKC-sensitive mechanism. In a subsequent study, in Dahl-S rats with NaCl-induced hypertension, in which levels of MBG are elevated and contribute to vasospasm, chronic administration of cicletanine prevented development of hypertension, reduced the MBG sensitivity of the Na+/K+-ATPase in LV myocardium, reduced the level of phosphorylation of the myocardial α1 Na+/K+-ATPase, and prevented up-regulation of PKC β2 and δ-isoforms in LV sarcolemma (Fedorova et al., 2003).
2. Protein Kinase G.
ANP is capable of modulating CTS sensitivity of the Na+/K+-ATPase via PKG-dependent phosphorylation. cGMP regulates activity of the Na+/K+-ATPase by modulation of its phosphorylation (Blanco et al., 1998; Fotis et al., 1999) and activation of the guanylyl cyclase-cGMP-PKG pathway underlies both renal and vascular effects of ANPs (Vesely et al., 1987; Hedge et al., 1989; Scavone et al., 1995). Unlike PKC, which directly phosphorylates the Na+/K+-ATPase, the effect of PKG on sodium pump phosphorylation is more complex and involves the recruitment and regulation of additional phosphatases (Aperia et al., 1994; Fotis et al., 1999). Fedorova et al. (2006) have shown that α-hANP and prepro-ANP, via a cGMP-dependent mechanism, dephosphorylate Na+/K+-ATPase from vascular sarcolemma and markedly reduce its sensitivity to MBG. In the same study, ANP exhibited an opposite effect; i.e., it induced Na+/K+-ATPase phosphorylation and sensitized the sodium pump to the inhibitory effect of MBG in renal medulla. Because aortic sarcolemma and renal medulla express PKG1 and PKG2 isoforms, respectively, these two PKG isoforms are likely to mediate the opposing effects of ANP on Na+/K+-ATPase phosphorylation and MBG sensitivity (Fedorova et al., 2006). Thus, ANP may be capable of antagonizing undesirable effects of CTS while potentiating desired effects in some settings. Accordingly, plasma levels of MBG and α-hANP exhibit a strong positive correlation in hypertensive patients with heart failure (Fridman et al., 2002). As our understanding of this signaling pathway expands, additional targets for clinical intervention will become apparent.
D. Antagonism of Cardiotonic Steroid-Induced Cell Signaling
If we accept the Na+/K+-ATPase-Src-EGFR paradigm for CTS signaling, several rather obvious targets present themselves for consideration. A number of EGFR antagonists, both at an antibody (immunoneutralization) and a tyrosine kinase level, have been developed because of the importance of the EGFR for cancer. Application of these agents may be surprisingly effective in the management of cardiovascular disease. The role of reactive oxygen species in Na+/K+-ATPase signaling is another obvious target. Neutralization of ROS may be effective in several experimental models of cardiovascular disease although the clinical benefits of this strategy are still debated.
Another possible approach to interfere with the effect of CTS is inhibition of Na+/Ca2+ exchanger, NCX1. CTS inhibit Na+/K+-ATPase in vascular smooth muscle cells; the elevation of local Na+ facilitates Ca2+ entry through NCX1, resulting in vasoconstriction (Iwamoto, 2006). The Na+/Ca2+ exchange inhibitors having a benzyloxyphenyl structure interact with a specific receptor site in the NCX1 molecule and block its reverse (Ca2+ influx mode) mode more effectively than the forward Ca2+ efflux mode (Iwamoto et al., 2004). Thus, the benzyloxyphenyl inhibitors exhibit their effect under pathological conditions (high Na+i) rather than under normal (low Na+i) conditions. In consideration of such a profile, benzyloxyphenyl inhibitors appear promising as therapeutic agents for the treatment of conditions associated with intracellular Na+i, such as salt-dependent hypertension, in which levels of CTS are elevated. Indeed, the administration of NCX1 inhibitors, SEA0400 and KB-R7942, lowered arterial pressure in several rat models of salt-sensitive hypertension, in which levels of CTS are elevated and contribute to enhanced vascular tone in deoxycorticosterone acetate-salt hypertensive rats, salt-loaded Dahl-S rats, and rats with ACTH-induced hypertension. Remarkably, NSX1 inhibitors exhibited minimal vasodepressor effects in normal animals and in salt-independent forms of hypertension (Iwamoto et al., 2004; Iwamoto, 2006).
E. Biosynthesis and Release of Cardiotonic Steroids
One potential opportunity is to decrease the circulating concentrations of CTS. Unfortunately, we are somewhat limited in achieving this end because we do not fully understand the biosynthesis of the CTS. That said, it will be interesting to note how much of the therapeutic “accomplishments” of inhibiting the RAS are related, in fact, to the ATII dependence of the EO-MBG axis (Fig. 4). The benefits of inhibiting ATII synthesis or action have been well established in hypertension, renal failure progression, and CHF. Unfortunately, although chronic angiotensin-converting enzyme inhibition was shown to reduce levels of circulating CTS in young spontaneously hypertensive rats (Kähönen et al., 1995) and blockade of ATII receptors by losartan prevented NaCl-induced stimulation of MBG in the adrenal cortex (Fedorova et al., 2005a), we do not have robust clinical data about the long-term effects of RAS inhibition on the EO-MBG axis.
F. Conclusions and Perspectives
Our understanding of the role of endogenous cardiotonic steroids has evolved considerably. More than 50 years ago, it was hypothesized that digitalis glycosides represented the exogenous counterparts of endogenous factor(s) involved in the regulation of cardiovascular system (Rein, 1949; Szent-Gyorgyi, 1953). When it became clear that the Na+/K+-ATPase was the specific receptor for digitalis, the concept of the Third Factor emerged (de Wardener and Clarkson, 1985). Within a few years, many studies demonstrated that these endogenous CTS did exist and implicated them in the regulation of the cardiovascular system, fluid-electrolyte homeostasis, and the pathogenesis of essential hypertension (Goto et al., 1992; Schoner, 1992).
The discovery of EO was a modern breakthrough in these studies of CTS (Hamlyn et al., 1991), and the discovery of the cell-signaling functions of the Na+/K+-ATPase, along with the definitive chemical characterization of both cardenolide and bufadienolide CTS, has largely erased skepticism and reawakened interest in this topic. At present, it is clear that multiple endogenous Na+/K+ ATPase inhibitors exist in mammals.
One of the factors underlying skepticism concerning endogenous CTS is the marked differences from their pharmacological prototype, digitalis, that endogenous CTS display. First, the classic pharmacology of endogenous CTS was based on the presumption that the effects of these compounds were mediated via inhibition of transmembrane sodium transport (Braunwald and Klocke, 1965; Akera and Brody, 1977). Data reviewed elsewhere (Xie and Askari, 2002; Schoner and Scheiner-Bobis, 2007; Tian and Xie, 2008) and briefly in this article, clearly demonstrate that many signaling effects of CTS may be independent of this mechanism. Second, many classic pharmacological studies of digitalis used micromolar concentrations of these agents, whereas circulating concentrations of endogenous CTS, at least in mammals, seem to be in the subnanomolar range. Very different effects of these lower concentrations of CTS on cell signaling have been observed (Nesher et al., 2007; Schoner and Scheiner-Bobis, 2007). Thus, it seems that an enormous number of studies of cardiac glycosides not only did not facilitate the progress of our understanding of endogenous CTS but also became rather an obstacle in the studies of endogenous sodium pump ligands.
Despite controversies and periods of skepticism (Manunta et al., 2009; Nicholls et al., 2009), it is clear that endogenous CTS represent an important class of hormones with profound consequences in health and disease, related but not limited to their roles as natriuretic hormones. These hormones are synthesized in the adrenal cortex, and, similar to neurosteroids, in the brain. Endogenous CTS circulate bound to transport proteins, and levels of CTS in plasma vary from subnanomolar to nanomolar concentrations in different physiological conditions. These circulating levels of CTS seem to be both sufficient to alter transmembrane sodium transport in some cell types as well as to induce a variety of cellular signals, some of which seem to be independent of changes in transmembrane sodium transport and are, rather, more analogous to that of receptor tyrosine kinases. Unfortunately, the measurement of CTS is mainly based on “in-house” immunoassays, which makes the comparison of results obtained in different laboratories difficult (Manunta et al., 2009; Nicholls et al., 2009). We propose that the development of reliable commercial methods of measurement of CTS levels is critical for the future advancement of this field.
The α-subunit of the Na+/K+-ATPase represents the specific receptor for CTS, and α isoforms of Na+/K+-ATPase exhibit differential sensitivity to various CTS. Therefore, this family of isoforms essentially can be viewed as multiple receptors for multiple endogenous CTS. Accordingly, changes in the levels of endogenous CTS are associated with levels of expression of Na+/K+-ATPase as well as with the changes in its sensitivity to endogenous ligands. Although radiolabeled ouabain and digoxin are widely used in the studies of CTS, the absence of commercially available labeled bufadienolides remains a limiting factor in the studies of receptor functions of the Na+/K+-ATPase.
Although our understanding of the functions of CTS is still quite incomplete, the importance of this class of hormones seems to be considerable. Endogenous CTS exhibit physiological functions that go far beyond regulation of sodium transport, natriuresis, and blood pressure and include regulation of cell growth and differentiation, apoptosis and proliferation, and glucose metabolism and control of central nervous functions. Dysregulation of these hormones seems to play an important role in a number of disease states ranging from hypertension to cancer. We propose that expanding our understanding of this class of hormones will lead to novel and effective therapeutic strategies of great relevance to optimizing health and curing diseases.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health National Institute on Aging.
Footnotes
-
↵1 Abbreviations: CTS, Cardiotonic steroid(s); TM, transmembrane; MBG, marinobufagenin; PKC, protein kinase C; ANP, atrial natriuretic peptide; GFR, glomerular filtration rate; EGFR; epidermal growth factor receptor; EO, endogenous ouabain; ACTH, adrenocorticotrophin; Dahl-S, Dahl salt-sensitive rats; PST2238, rostafuroxin; BP, blood pressure; RAS, renin-angiotensin system; CHF, congestive heart failure; ATII, Angiotensin II; AT, angiotensin; ERK, extracellular signal-regulated kinase; PI(3)K, phosphoinositide 3-kinase; ROS, reactive oxygen species; NHE3, Na+/H+ exchanger isoform 3; ESRD, end-stage renal disease; LV, left ventricular; α-hANP, α-human atrial natriuretic peptide; OSA, obstructive sleep apnea; PaCO2, arterial tension of carbon dioxide; PKG, protein kinase G; NCX1, Na+/Ca2+ exchanger 1; SEA0400, [2-[4-[(2,5-difluorophenyl)methoxy]phenoxy]-5-ethoxyaniline]; KB-R7942, 2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea methanesulfonate.
-
This article is available online at http://pharmrev.aspetjournals.org.
-
doi:10.1124/pr.108.000711.
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





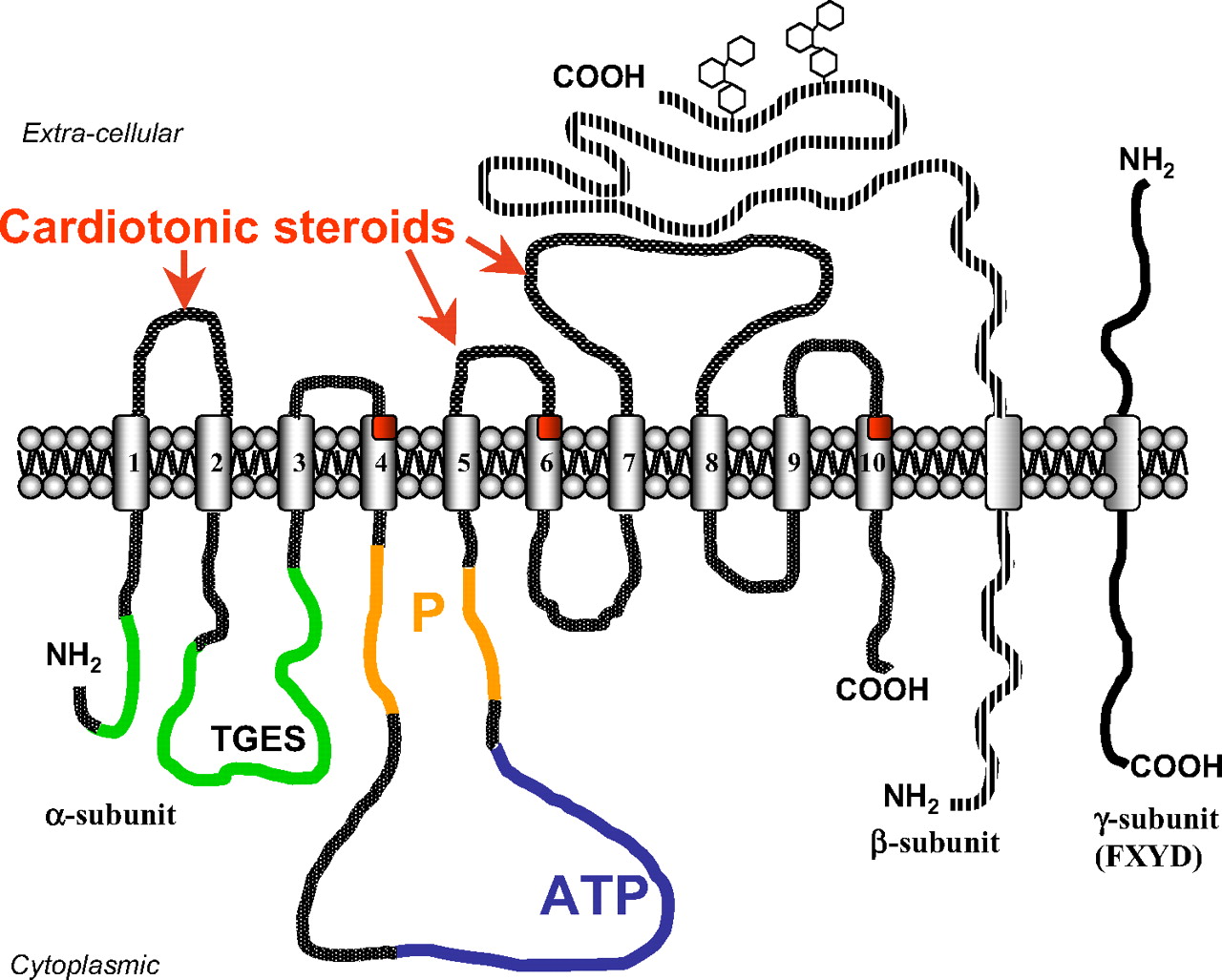{kind=link}
{kind=link}
{kind=link}
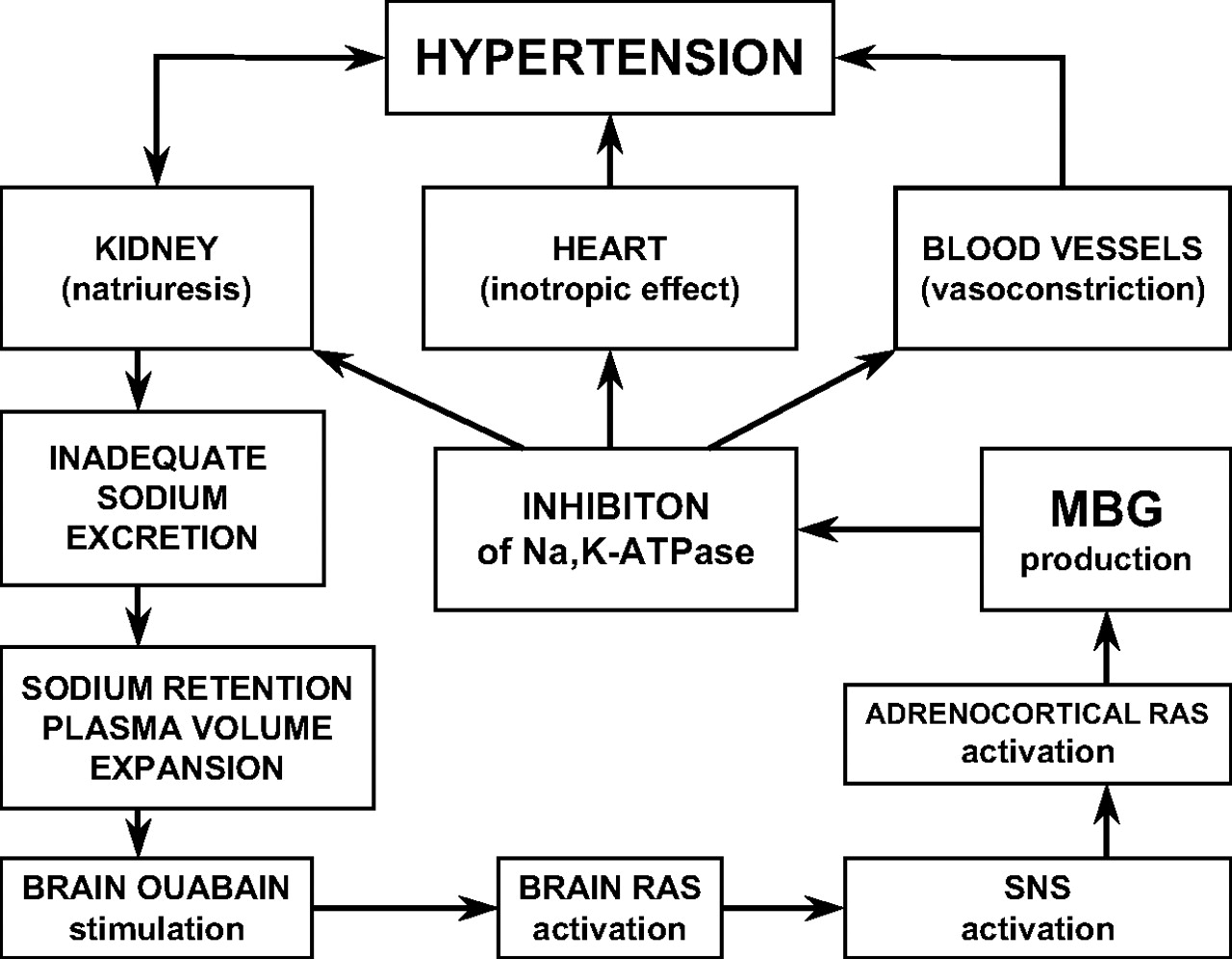{kind=link}
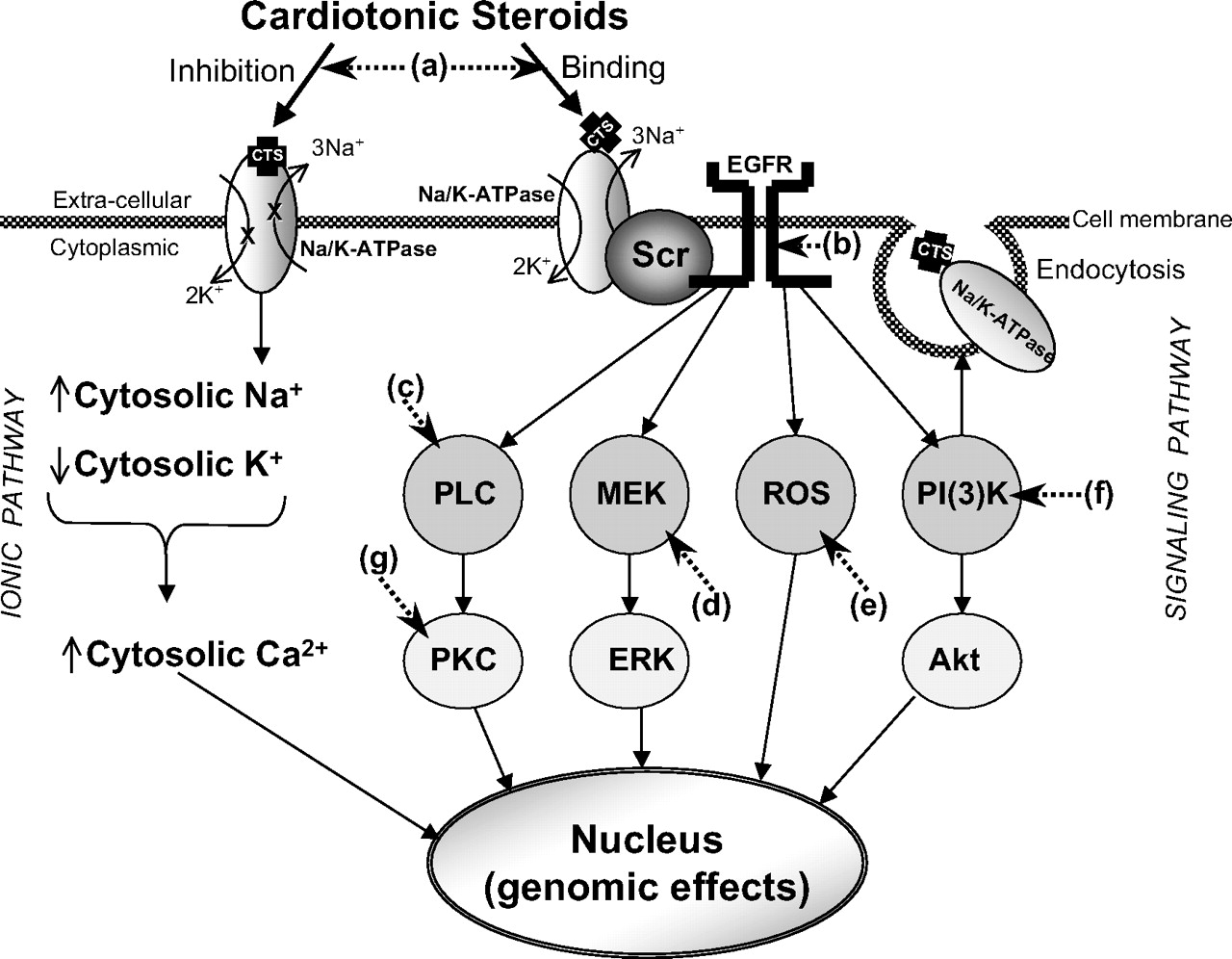{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Na+/K+-ATPase
- III. Endogenous Digitalis
- IV. Salt-Sensitive Hypertension, Concept of Natriuretic Hormone, and Elucidation of Nature and Roles of Endogenous Cardiotonic Steroids
- V. Endogenous Cardiotonic Steroids and Cell Signaling
- VI. Effects of Endogenous Cardiotonic Steroids on Kidney and Sodium Metabolism
- VII. Chronic Renal Failure and Uremic Cardiomyopathy
- VIII. Congestive Heart Failure
- IX. Preeclampsia
- X. Other Conditions
- XI. Possible Strategies for Pharmacological Antagonism of Endogenous Cardiotonic Steroids
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters