


Abstract
The peroxisome proliferator-activated receptor γ (PPARγ), a member of the nuclear hormone receptor family, represents a possible new target in glioma therapy. Because PPARγ plays a crucial role in regulation of insulin sensitivity, synthetic agonists are already in clinical use for type II diabetes treatment. Beyond these metabolic effects, PPARγ agonists exhibit antineoplastic effects. In this study, we investigated the antineoplastic effects of the PPARγ agonist pioglitazone in glioma cells. Pioglitazone reduced cellular viability of rat, human, and PPARγ-overexpressing glioma cells in vitro in a time- and concentration-dependent manner. No antineoplastic effects were induced by pioglitazone in glioma cells overexpressing a PPARγ mutant. Furthermore, proliferation was reduced by pioglitazone, as measured by Ki-67 immunoreactivity, in vitro. Continuous intracerebral infusion of pioglitazone into gliomas induced by intrastriatal injection of C6 cells reduced tumor volumes by 83%. Oral administration of pioglitazone reduced tumor volumes by 76.9%. Subsequent brain tissue analysis revealed induction of apoptotic cell death. Ki-67 expression and BrdU incorporation revealed a reduction of proliferation in vivo. Reduced invasion of C6 cells and lower matrix metalloproteinase 9 levels in vivo indicate pioglitazone-mediated reduction of invasion. Together, these data indicate that pioglitazone may be of potential use in treatment of malignant gliomas.
Malignant astrocytic gliomas are the most common primary brain tumors. Glioma cells show a high proliferation rate and diffusely infiltrate adjacent brain tissue (Kleihues et al., 2002). These tumors initially respond to radiation and, to a lesser degree, to chemotherapy; however, they invariably recur. Despite substantial efforts, no curative therapy exists, and the median overall survival for patients with the most malignant variant, “glioblastoma”, is poor (Nieder et al., 2005; Stupp et al., 2005; Reardon et al., 2006).
A new antineoplastic approach may lie in targeting the peroxisome proliferator-activated receptor γ (PPARγ) with specific agonists. PPARs are a subclass of nuclear hormone receptors that enable the cell to respond to extracellular stimuli by transcriptionally regulating gene expression. Three isoforms of PPARs have been identified and designated as -α,-β/δ, and -γ and are encoded by different genes. PPARs form heterodimers with the retinoic acid receptor and exhibit ligand-induced transcriptional regulatory activity through sequence-specific PPAR-responsive elements in their target genes (Willson et al., 2000). For more than a decade, work on PPARs was driven by their important role in the regulation of cellular metabolism, especially in tissues known for high rates of β-oxidation, such as liver, heart, muscle, and kidney. Because activation of the PPARγ subtype results in reduced serum glucose (Lemberger et al., 1996) recently developed synthetic PPARγ agonists are already in clinical use as antidiabetic drugs [e.g., pioglitazone (Actos) and rosiglitazone (Avandia)].
Apart from well defined metabolic actions, PPARγ agonists exhibit several antineoplastic effects (Grommes et al., 2004) and induce apoptotic cell death in various malignant cell lineages, including liposarcoma (Tontonoz et al., 1997), breast adenocarcinoma (Elstner et al., 1998; Mueller et al., 1998), prostate carcinoma (Kubota et al., 1998), colorectal carcinoma (Brockman et al., 1998; Sarraf et al., 1998), non-small-cell lung carcinoma (Chang and Szabo, 2000), pancreatic carcinoma (Motomura et al., 2000), bladder cancer (Guan et al., 1999), and gastric carcinoma (Sato et al., 2000). We recently reported that several PPARγ agonists induce apoptosis in rat and human glioma cell lines, and a PPARγ antagonist and BAX antisense oligonucleotides blocked the apoptotic cell death induced by PPARγ ligands (Zander et al., 2002; Grommes et al., 2005). In addition, a recent study has shown that PPARγ agonist-mediated reduction of glioma cell survival is caused by an increased production of reactive oxygen species (Perez-Ortiz et al., 2004). Furthermore, PPARγ agonists moderately inhibited growth of BT4Cn rat glioma cells, an effect that was abolished by the PPARγ antagonist GW9662 (Berge et al., 2001). A significant proportion of glioma tissues from 20 patients expressed PPARγ mRNA (Kato et al., 2002).
The present study demonstrated that the PPARγ agonist pioglitazone (pio) reduced cellular viability of C6 rat glioma and human glioma cell lines (A172, U87) and inhibited C6 glioma cell proliferation, as measured by Ki-67 expression, in vitro. Furthermore, pioglitazone treatment in PPARγ-cDNA overexpressing glioma cells reduced cellular viability in glioma cells, whereas treatment of glioma cells overexpressing the PPARγ mutant E499Q-cDNA, which lacks the transcriptional activity, showed no antineoplastic effects. These findings were confirmed in vivo using a C6 rat glioma model. Here, tumor volumes were reduced by 83% after intracerebral pio administration and by 76.9% with oral pio treatment. In parallel, pio-treated animals exhibited improved clinical outcome, a lower proliferation index (Ki-67), and decreased BrdU-incorporation within the tumor tissue. In addition, in drug-treated animals, tumors exhibited an up-regulation of the proapoptotic proteins Bax and cleaved caspase-3 associated with increased TUNEL-labeling indicative of apoptotic cell death. Furthermore, reduced invasion, as measured in vitro with Boyden chamber experiments and in vivo through matrix metalloproteinase 9 (MMP9) levels, was observed. Pio also induced up-regulation of the astrocytic redifferentiation marker CS-56 in tumor cells in vitro and in vivo, a sign of induced redifferentiation.
Materials and Methods
Materials. Pioglitazone (Takeda Chemical Industries, Osaka, Japan) was dissolved in dimethyl sulfoxide (DMSO) obtained from Sigma (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM), RPMI-1640 medium, penicillin, streptomycin, fetal calf serum, phosphate-buffered saline (PBS), trypsin-EDTA, and Proteinase K were purchased from Invitrogen (Carlsbad, CA). Rabbit Ki-67-antibody was purchased from NeoMarkers (Fremont, CA), rabbit cleaved caspase-3- and rabbit BAX-antibody were purchased from Cell Signaling (Beverly, MA), goat anti-MMP9 was purchased from Santa Cruz Bioechnology (Santa Cruz, CA), and mouse anti-CS-56 was purchased from Sigma. For Western blot analysis, the secondary anti-rabbit-antibody was obtained from GE Healthcare (Little Chalfont, Buckinghamshire, UK). Secondary antibody for immunohistochemistry (Alexa Fluor 488-conjugated goat anti-rabbit IgG) was purchased from Invitrogen.
Cell Culture. Rat C6 glioma and murine cells were grown in DMEM and human glioma cells (U87, A172) were grown in RPMI, supplemented with 10% (v/v) fetal calf serum, 100 U/ml penicillin, and 100 U/ml streptomycin in a 5% CO2 atmosphere. Primary astrocyte cultures were prepared as described previously (McDonald et al., 1998) and grown in DMEM, supplemented with 2.5% (v/v) fetal calf serum, 100 U/ml penicillin, and 100 U/ml streptomycin in a 5% CO2 atmosphere.
Viability Assay. Cellular viability was assessed by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT; Sigma) assay. In brief, C6, U87, and A172 cells (5 × 103/well) or primary astrocytes (5 × 103/well) were seeded in a 96-well plate and exposed to different concentrations of pio (1, 10, and 30 μM; n = 10). DMSO served as vehicle control (0.1% of final concentration). At 1, 3, 5, and 7 days, 10 μl of MTT (5 mg/ml PBS) was added to each well, and plates were incubated at 37°C for 2 h. Medium was then removed and cells were resuspended in 100 μl of DMSO. Cell viability was assessed by colorimetric change using the SpectraMax 340 PC plate reader (Molecular Devices, Sunnyvale, CA) at λ = 550 nm. To ensure stability of pioglitazone at 37°C, the compound was incubated over 21 day at 37°C, and cellular viability of C6 cells was assessed after 5 days. Experiments were performed in triplicate.
Transfection. Transfections were carried out on human A172 glioma cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. PPARγ cDNA constructs were kindly provided as follows: mouse PPARγ1 cDNA, Dr. Ron Evans (The Salk Institute for Biological Studies, San Diego, CA); mouse PPARγ2, Dr. Evan Rosen (Harvard University, Cambridge, MA); and mouse PPARγ2 cDNA mutated at position E499Q31 (a point mutation from Glu499 to Gln499 in the AF-2 activation domain, lacking its transcriptional activity), Dr. Bruce Spiegelman (Harvard University). Forty-eight hours after transfection, cells were treated with 30 μM pioglitazone. Cell viability was assessed after 5 days using the MTT assay described above. Experiments were performed in triplicate and repeated three times.
Animals. Sprague-Dawley rats (Charles River Laboratories, Sulzfeld, Germany) weighting 200 to 250 g and C57BL/6 mice (Charles River Breeding, Cambridge, MA) aged 6 to 8 weeks were used. Animals were housed in groups of two under standard conditions at a temperature of 22 ± 1°C and a 12-h light/dark cycle with free access to food and water. Experiments were carried out in accordance with the declaration of Helsinki and were approved by the local ethical committee for animal experiments.
Brain Tumor Xenograft. Before implantation, 85 to 90% confluent C6 cells were trypsinized, rinsed with DMEM + 10% fetal calf serum, and centrifuged at 1000 rpm for 4 min. The cell pellet was resuspended in DMEM and placed on ice. Concentration of viable cells was adjusted to 1 × 105 cells/1 μl of DMEM. Each rat was anesthetized and placed in a stereotactic frame (David Kopf Instruments, Tujunga, CA), a hole was drilled at anteroposterior 0.0, right -3.0 relative to bregma according to the stereotaxic atlas of König and Klippel (1963). Tumor cells were injected at a rate of 0.5 μl/s, using a 2-μl syringe (Hamilton Co., Reno, NV) with a 26s-gauge needle mounted on a stereotactic holder at a depth of 5 mm. The needle was left in place for a further 5 min to prevent reflux along the needle tract. For intracerebral administration of pio/vehicle (DMSO), the tip of a brain infusion kit (Alzet, Cupertino, CA) was placed at a depth of 5 mm, connected to an osmotic pump (2ML4; Alzet). Drug administration began 4 h after surgery. The osmotic pumps had a 2-ml volume and 2.5 μl/h flow rate. The pumps were filled with 20 μM pio dissolved in PBS or vehicle (DMSO, 0.1% final concentration) dissolved in PBS. Animals were randomly distributed in two groups and treated with pio-filled pumps (n = 26) or vehicle-filled pumps (n = 26). Thereafter, the skull was cleaned, and the incision was sutured. For oral treatment, the drug was pulverized and mixed with Purina chow to give concentrations of 120 ppm pioglitazone. Control animals received Purina chow without additions. Rats were allowed free access to the chow. Tumors were allowed to grow and animals were weighed daily. The intracerebrally treated animals were sacrificed after 3 days (n = 5/group), 6 days (n = 5/group), 9 days (n = 5/group), 14 days (n = 5/group), and 21 days (n = 6/group), and the orally treated animals were sacrificed after 21 days (n = 8/group). C57BL/6 mice (13 animals/group) received a single right frontal intracerebral injection of 5.0 × 104 cells of the murine glioma cell line GL261 and were treated with oral pioglitazone (100 ppm) mixed with mouse diet. Control animals received a single intracerebral injection of the equivalent number of GL261 cells without treatment.
Clinical Assessment. After striatal injection of C6 cells and intracerebral treatment with pio or vehicle for 21 days, the animals were examined for neurological deficits. Neurological function was quantified by evaluation of hemiparesis, cycling, and immobility (Krajewski et al., 1986; Schabitz et al., 1999). Hemiparesis was assessed by forelimb flexion and immobility by the loss of ability to walk a distance greater than 15 cm. One point was given for each negative finding, and a total score was then computed. The scores of the treated and untreated group were averaged. A blinded observer performed neurological examinations.
Sections. Brains were serially sectioned at 10 μm using a cryostat (Jung CM1800; Leica, Wetzlar, Germany). Sections for hematoxylineosin (HE)-staining were placed onto uncoated slides. Sections intended for use in immunoreactivity assays were placed onto coated slides (Fisher Scientific, Houston, TX). Sections were routinely HE-stained for histomorphological assessment and measurement of tumor volume as well as immunohistochemically was processed for Ki-67, BAX, cleaved caspase-3 expression, MMP9, CS-56, and BrdU incorporation.
Tumor Volume. Images of HE-stained sections containing tumors were captured with a SPOT model 1.3 camera (Diagnostic Instruments, Inc., Sterling Heights, MI) using a 1× objective and images processed using NIH Image 1.62 software (http://rsb.info.nih.gov/nih-image/). The tumor area of each section was manually outlined by a blinded observer using the freehand selection tool to measure tumor area in millimeters squared. The area was then multiplied by the section thickness (10-μm section; 10 sections/HE stain) to achieve a section volume measurement. Volumes of all sections were added to calculate the total volume of each tumor. Tumor volumes for five or six animals of every group were measured.
Western blot. For Western blot analysis, tumor-containing hemispheres were homogenized in Tris-HCl [50 mM Tris-HCl, pH 8, 120 mM NaCl, 5 mM EDTA, 0.5% (v/v) Nonidet P-40, and 160 mM phenylmethylsulfonyl fluoride] and sonicated. Homogenates were collected by centrifugation (15 min, 11,000g, 4°C) and protein concentration of the supernatant was determined using the Bio-Rad protein assay (Bio-Rad Laboratories, Munich, Germany). Lysates (20-40 μg) were separated on a 7% (w/v) SDS-polyacrylamide gel under reducing conditions and transferred to a PVDF membrane (Millipore, Bedford, MA). Nonspecific binding was blocked by incubation with 5% (w/v) skimmed milk in TBS for 2 h. After incubation with the primary antibody overnight at 4°C [rabbit anti-BAX, 1:1000; rabbit anti-cleaved caspase-3, 1:1000; mouse anti-CS-65, 1:1000, goat anti-MMP-9, 1:1000; in TBS containing 0.1% (v/v) Tween 20], membranes were washed three times in TBS/Tween for 5 min and subsequently incubated for 120 min in TBS/Tween containing secondary peroxidase-conjugated antibody at room temperature (anti-rabbit, 1:1000; anti-mouse, 1:1000; anti-goat, 1:1000; respectively). Signals were visualized by chemoluminescence (Pierce, Rockford, IL) and band intensities were quantified with NIH Image 1.62 software.
Immunohistochemistry. Frozen brain sections were first blocked with 5% normal goat serum or normal horse serum in PBS and subsequently incubated with rabbit anti-Ki-67 antibody (1:200 dilution), mouse anti-CS-56 (1:100 dilution), goat anti-MMP-9 (1:100 dilution), or rabbit anti-cleaved caspase-3 antibody (1:200 dilution) at 4°C overnight. Sections were washed extensively with PBS before incubation with secondary antibodies. Incubation was carried at room temperature for 1 h. After washing with PBS three times, stained slides were mounted with PBS/glycerol (1:1) and viewed under a fluorescent microscope (Leica).
C6 glioma cell cultures were treated with pio or vehicle for 2 days and 5 days and fixed in 4% paraformaldehyde, rinsed in TBS, and incubated with rabbit anti-Ki-67 (1:200 dilution) at 4°C overnight and processed as described above. Counterstaining was carried out with propidium iodide (1:40; Sigma) or DAPI-staining (1:500 in PBS).
For determination of the proliferation-index, Ki-67 positive cells in pio- and vehicle-treated cells and animals were counted, the percentage of Ki-67 positive cells in 1 × 103 tumor cells [propidium iodide staining used in the in vitro experiments (data not shown); DAPI stain used in the in vivo experiments] calculated and statistically compared. The percentage of cleaved caspase-3-positive cells was assessed by counting the immunopositive cells in 1 × 103 tumor (DAPI stain) cells and statistically compared. Negative control cells showed no immunoreactivity.
TUNEL Assay for Apoptosis. Frozen brain tissue sections were examined for apoptosis using the terminal deoxynucleotidyl transferase-mediated dUTP transferase nick-end labeling (TUNEL assay). Apoptosis was evaluated using the DeadEnd Fluorometric TUNEL System (Promega, Madison, WI), according to the manufacturer's instructions. The percentage of TUNEL-positive cells was assessed by counting the immunopositive cells in 1 × 103 tumor (DAPI stain) cells and statistically compared.
BrdU Incorporation. At 13 days of treatment, animals were i.p. injected with 300 mg/kg of 5-bromodeoxyuridine (BrdU; Sigma), a thymidine analog that incorporates into the DNA of dividing cells during S phase and can be detected immunohistochemically. Twenty-four hours later, these animals were anesthetized and perfused with phosphate-buffered saline (PBS). Brains were dissected, frozen, cut, and immunostained in the following manner. Sections were fixed in 4% paraformaldehyde, washed in PBS, and incubated in 2 N HCl for 10 min at room temperature. Sections were again washed in PBS and incubated in BrdU primary antibody (rat monoclonal 1:100; Abcam, Cambridge, UK) in PBS containing 5% normal goat serum overnight at 4°C. After washing with PBS, sections were incubated in rhodamine-labeled anti-rat secondary antibody (1:100; Jackson ImmunoResearch Laboratories Inc., West Grove, PA) in PBS containing 5% normal goat serum for 1 h at room temperature. The number of dividing cells present in the tumor of these BrdU-stained sections was then counted and compared with total amount of tumor cells stained with DAPI.
Boyden-Chamber Assay. C6 cell migration in the presence or absence of pio was assessed using a Boyden chamber (AP48; Neuro-Probe, Gaithersburg, MD) with an 8-μm polycarbonate polyvinyl pyrrolidone-free filter (GE Osmonics, Inc., Minnetonka, MN). Cells were suspended in DMEM containing 2.5% fetal calf serum and pio at 30 μM or vehicle (DMSO). C6 glioma cells were pretreated for 24 h in the presence or absence of 30 μM pio. Cells (5 × 103 in 50 μl pio/medium or vehicle/medium) were then plated in the wells of the upper compartment of the chamber (six wells/condition), and the wells of the lower compartment were filled with DMEM. Incubation was performed at 37°C in 5% CO2 for 4 h. After incubation, cells on the upper surface of the filter, which had not migrated, were gently scraped off, and the filters were then fixed in methanol and subsequently stained with DAPI (1:500 in PBS; Sigma). Cells that had migrated to the lower surface of the filters were counted using public domain software NIH Image 1.62. The total numbers of migrating cells under pio-treated and vehicle-treated conditions were compared statistically. Experiments were performed in duplicate.

Pioglitazone reduced cellular viability of rat and human glioma cells and proliferation in vitro. Viability of rat C6 glioma cells (A), human U87 and A172 glioma cells (B), or primary astrocytes (C) at 1, 3, 5, and 7 days incubated with the PPARγ agonist pioglitazone (pio) (1, 10, 30 μM for C6 and primary astrocytes; 30 μM for U87 and A172) was assessed using the MTT assay. Asterisks indicate significant level in Student's t test, ***, p < 0.0001 (n = 10). Cellular viability of human glioma cells A172 after transfection with vector control, two different PPARγ-cDNAs and PPARγ mutant-cDNA (E499Q) after 5 days of pio treatment (D) was assessed by MTT assay. Data are expressed as percentage of viable cells relative to untreated control cultures. Experiments were performed in triplicate and repeated three times. Asterisks indicate significant level in Student's t test: *, p < 0.05 (n = 10); **, p < 0.005 (n = 10).
Statiatical Evaluation. Cellular viability data, tumor volumes, immunopositive cells, clinical scores, proliferation index, and densitometric results of Bax and cleaved caspase-3 Western blots were analyzed by Student's t test using Prism version 3.00 (GraphPad Software, San Diego, CA).
Results
Reduced Cellular Viability of Rat C6 Glioma and Human Glioma Cells. Viability of rat C6 glioma cells and human glioma cells (U87, A172) was assessed after incubation with increasing concentrations (1, 10, 30 μM) of the PPARγ agonist pio at days 1, 3, 5, and 7 (Fig. 1, A and B). Ten and 30 μM pio significantly reduced the cellular viability of C6 rat glioma cells in a concentration- and time-dependent manner (Fig. 1A). A significant reduction of cellular viability in C6 cells was observed at 5 and 7 days after treatment with 30 μM pio and at 7 days after treatment with 10 μM pio (Fig. 1A). At 7 days, 30 μM pio reduced viability to 31 ± 3.7%, and 10 μM pio reduced viability to 52.1 ± 6.4% (Fig. 1A). In the human glioma cell lines U87 and A172, 10 and 30 μM pio significantly reduced cellular viability at 3, 5, and 7 days (Fig. 1B, Supplemental Fig. 4).
At all time points and concentrations evaluated, viability of primary astrocytes was not affected by pio treatment (Fig. 1C), indicating that PPARγ ligand-mediated cell death is restricted to neoplastic astrocytic cells.
To assess the role of PPARγ in the antineoplastic effects of pio, we overexpressed PPARγ in the human glioma cell line A172. Pio treatment and overexpression of two different PPARγ cDNA, PPAR1 and -2, reduced cellular viability of A172 cells measured by MTT compared with medium and vector control cells. After transfection of A172 cells with a PPARγ mutant cDNA lacking transcriptional activity, the decrease in effects on cellular viability were no longer observed at 5 days after transfection (Fig. 1D).
Ki-67 Immunoreactivity and Proliferation Index Is Reduced after Pioglitazone Treatment in Vitro. To further characterize the pio-induced effects, Ki-67 expression, a marker for tumor proliferation and malignancy, was evaluated. C6 cells were treated with 30 μM pio or vehicle and Ki-67 immunoreactivity was assessed at 2 days (Supplemental Fig. 1, A and B) and 5 days (Supplemental Fig. 1, C and D). Pio reduced the number of Ki-67 immunopositive cells compared with vehicle treatment at both time points. Thus, the fraction of Ki-67-positive proliferating cells, expressed as proliferation index, was significantly reduced after 5 days of pio treatment (Supplemental Fig. 1E).
Significantly Reduced Tumor Volume and Growth in the Rat Glioma Model. To confirm the antineoplastic effects of pio in vivo, C6 glioma cells were injected in rat striata and treated with pio 20 μM or vehicle via an osmotic pump for 3 days (n = 5/group), 6 days (n = 5/group), 9 days (n = 5/group), 14 days (n = 5/group), or 21 days (n = 6/group) starting 4 h after initial tumor cell injection. In vitro studies demonstrated that the biological activity of pioglitazone remains stable when incubated at 37°C over 21 days (reduction of cellular viability to 31 ± 3.7% before incubation and 31.7 ± 7.4% after incubation; supplemental Fig. 6).
The pio-treated animals showed a dramatic reduction of tumor volume at 21 days of treatment (Fig. 2, B, D, and E) compared with vehicle-treated animals (Fig. 2, A, C, and E). Vehicle-treated animals exhibited a median tumor volume of 0.321 ± 0.11 cm3, whereas pio-treated animals revealed tumor volumes of 0.055 ± 0.03 cm3, reflecting an 83% reduction (Fig. 2G). At 3 and 6 days no significant differences in tumor volumes could be observed (Fig. 2F). However, at 9, 14, and 21 days, tumor volumes decreased significantly in pio-treated animals (Fig. 2F). These effects were also observed in the orally treated animals, where pio treatment reduced tumor volumes by 76.9% after 21 days (Fig. 2H).

Intracerebral and oral administration of pioglitazone significantly reduced tumor growth in the rat glioma model. C6 glioma cells were injected in rat striata and treated with pio (20 μM) or vehicle (DMSO) via an osmotic pump over 3 days (n = 5/group), 6 days (n = 5/group), 9 days (n = 5/group), 14 days (n = 5/group), and 21 days (n = 6/group). Pio-treated animals showed significantly smaller tumor volumes (B, D, E, and F) compared with untreated animals (A, C, E, and F). A and B, coronal section of a representative part of the tumor. C and D, higher magnification of the striatum at the tumor/brain tissue border. A and C, striatum of a vehicle-treated animal at 21 days, with HE staining. Bars, 1 mm (A); 100 μm (C). B and D, striatum of a pio-treated animal at 21 days, with HE staining. Bars, 1 mm (B); 100 μm (D). E, representation of tumor area in treated (pio) and untreated (vehicle) animals at 21 days. The schematic shows coronary brain slices with black color representing tumor tissue and white color representing normal brain tissue. The graph shows tumor area per slide level. F, tumor volumes in square centimenters of pio-treated animals compared with vehicle-treated animals. Data are presented as mean ± S.E. of five animals per group (3, 6, 9, or 14 days) or six animals per group (21 days). Asterisks indicate significant level in Student's t test: ***, p < 0.001 (n = 5); **, p < 0.005 (n = 5 or 6). G, after 21 days of intracerebral pio treatment, tumor volumes of the C6 glioma model are reduced by 83% compared with vehicle-treated animals. H, in the group treated intracerebrally, the tumor volumes are reduced by 76.9%. Data are presented as mean ± S.E. of six animals per group. Asterisks indicate significant level in Student's t test: *, p < 0.05; **, p < 0.001 (n = 6).
Similar results were observed in a mouse model as well. C57BL/6 mice (13 animals/group) received a single right frontal intracerebral injection of the murine glioma cell line GL261 and were treated with oral pioglitazone (100 ppm) mixed with mouse diet. Control animals received a single intracerebral injection of the equivalent number of GL261 cells without treatment. At day 30, the number of surviving animals was 3.6-fold higher in the pio-treated group (Supplemental Fig. 5)
Improved Clinical Outcome. To investigate whether the reduction of tumor volumes results in improved neurological outcome, animals were assessed and scored for signs of hemiparesis, cycling, and immobility as described previously (Krajewski et al., 1986; Schabitz et al., 1999). Pio-treated animals had a significantly better clinical outcome overall. They exhibited less hemiparesis, no cycling, and less immobility (Supplemental Fig. 2) than vehicle-treated control animals.
Animals were also closely monitored for weight loss, a nonbehavioral indicator of intracellular tumor growth (Redgate et al., 1991). Daily weighing of animals showed that the vehicle-treated group exhibited a significant weight loss at 17 days, whereas pio-treated animals showed no weight loss (Supplemental Fig. 2E). Together, these clinical evaluations suggest that pio-induced reduction of tumor volume improved clinical outcome.

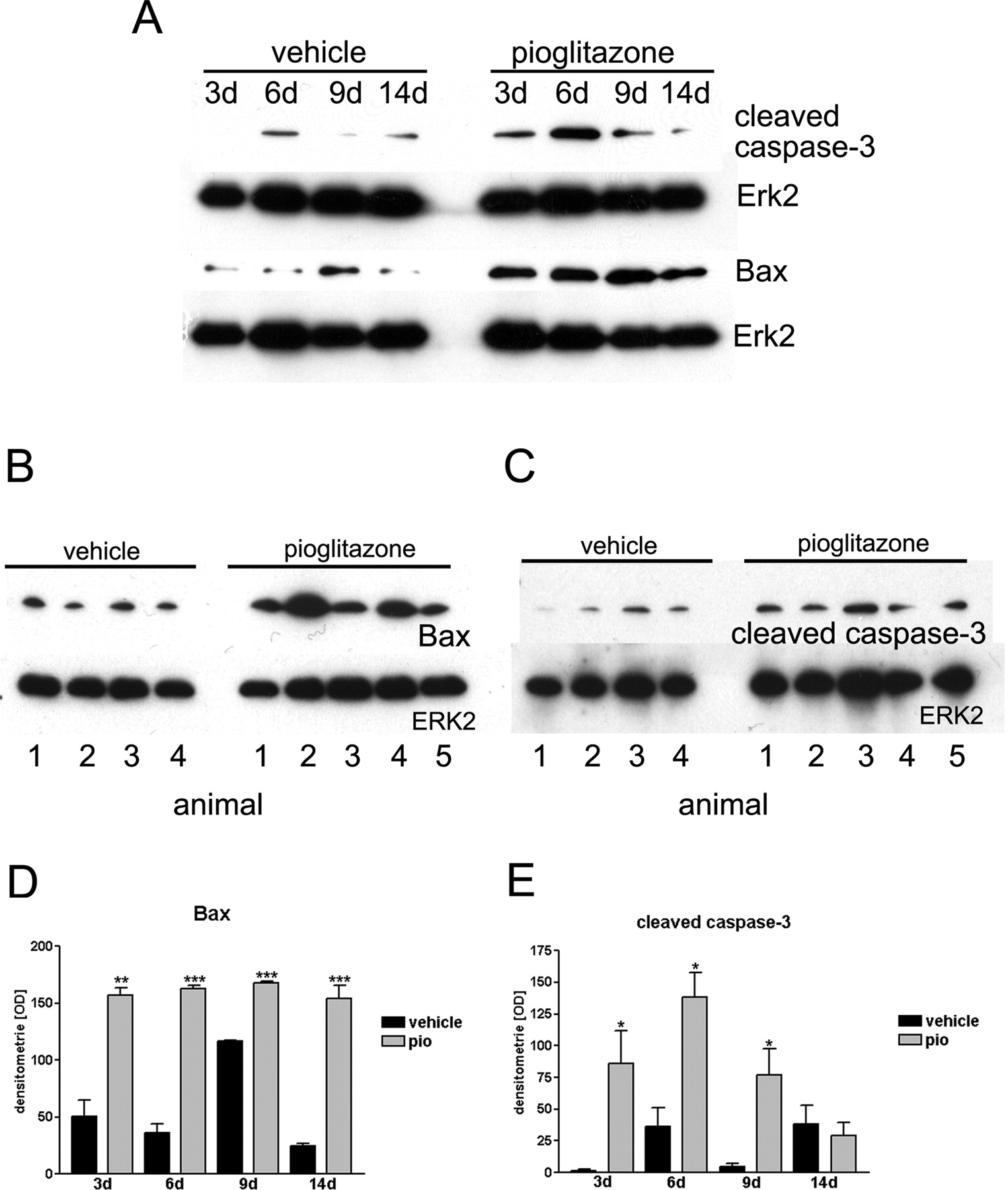
Pioglitazone treatment induced Bax- and cleaved caspase-3-up-regulation in vivo. A, Bax and cleaved caspase-3 expression in whole-hemisphere lysates of C6 rat glioma animals and control at 3, 6, 9, and 14 days of treatment. Vehicle, vehicle-treated animals; Pio, pio-treated animals. Erk2 served as a loading control. B and C, Bax and cleaved caspase-3 expression at 6 days in four (vehicle) and five (pio) different animals. D and E, densitometric analysis of Bax or cleaved caspase-3 levels at 3, 6, 9, or 14 days. Data are presented as mean ± S.E. of three different Western blots. Black bars, vehicle treatment; gray bars, pio treatment. Asterisks indicate significant level in Student's t test: *, p ≤ 0.03 (n = 3); **, p ≤ 0.002 (n = 3); ***, p ≤ 0.001.
Induction of Apoptotic Cell Death in Vivo. To assess whether pio induces apoptotic cell death in vivo, Bax- and cleaved caspase-3-expression were evaluated and TUNEL staining was performed. The expression of the proapoptotic proteins Bax and cleaved caspase-3 was detected in whole-hemisphere lysates of C6 glioma animals and control animals. Analysis at different time points revealed that both proteins were expressed in the pio-treated animals and in control animals (Fig. 3A). Protein levels of either Bax or cleaved caspase-3 were up-regulated in the early phase of pio treatment. Densitometric analysis showed a significant difference in Bax-protein levels at 3, 6, 9, and 14 days (Fig. 3D) and in cleaved caspase-3 protein levels at 3, 6, and 9 days (Fig. 3E) in response to pio treatment. At 6 days of pio treatment, Bax and cleaved caspase-3 were up-regulated (Fig. 3B) with a 4.49-fold induction of Bax protein (Fig. 3D) and a 3.83-fold induction of cleaved caspase-3 (Fig. 3E).
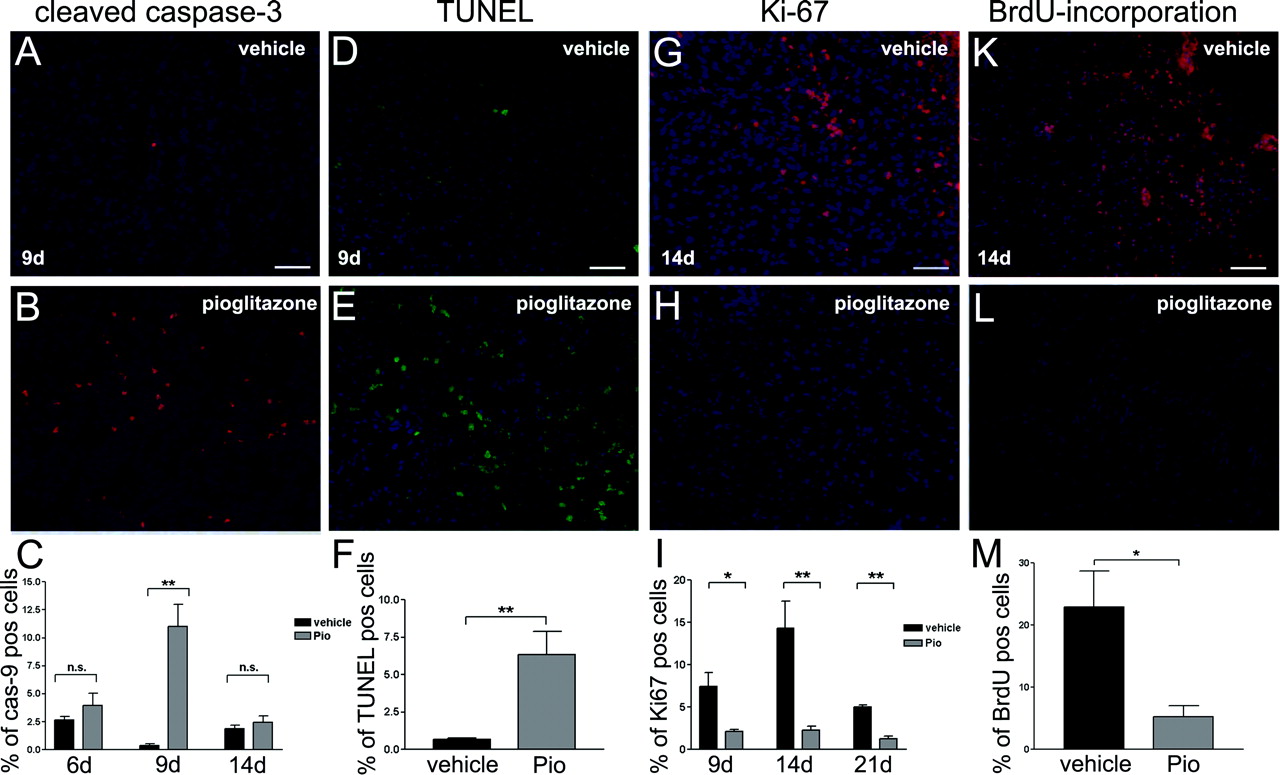
Immunohistochemistry revealed elevated cleaved caspase-3 expression at 6 to 14 days of pio treatment with a significant maximum at 9 days (Fig. 4B) compared with vehicle-treated animals (Fig. 4A). DAPI staining was used to tag single tumor cells and to determine the percentage of cleaved caspase-3 positive cells (Fig. 4C). Increased TUNEL staining was detected at 9 days of pio treatment (Fig. 4E). Very few cells were positively TUNEL-stained in vehicle control cells (Fig. 4D).
Ki-67 Immunoreactivity and Proliferation Index Is Reduced in Vivo. Ki-67 expression was used to evaluate degree of tumor proliferation in vivo. C6 glioma animals showed significant reduction of Ki-67 expression at 9, 14, and 21 days of pio treatment (Fig. 4H) compared with vehicle-treated animals (Fig. 4G). The proliferation index, calculated from the percentage of Ki-67-positive cells in 1 × 103 tumor cells (DAPI), was reduced by pio treatment at 9, 14, and 21 days (Fig. 4I) confirming the results obtained in vitro.
Reduction of BrdU Incorporation. To further confirm that pio treatment reduces proliferation, we assessed BrdU-incorporation in vivo. BrdU-incorporation was reduced in response to pio treatment at 14 days (Fig. 4L) compared with the vehicle-treated groups (Fig. 4K). The percentage of BrdU-positive cells is significantly lower in pio-treated animals (Fig. 4M).
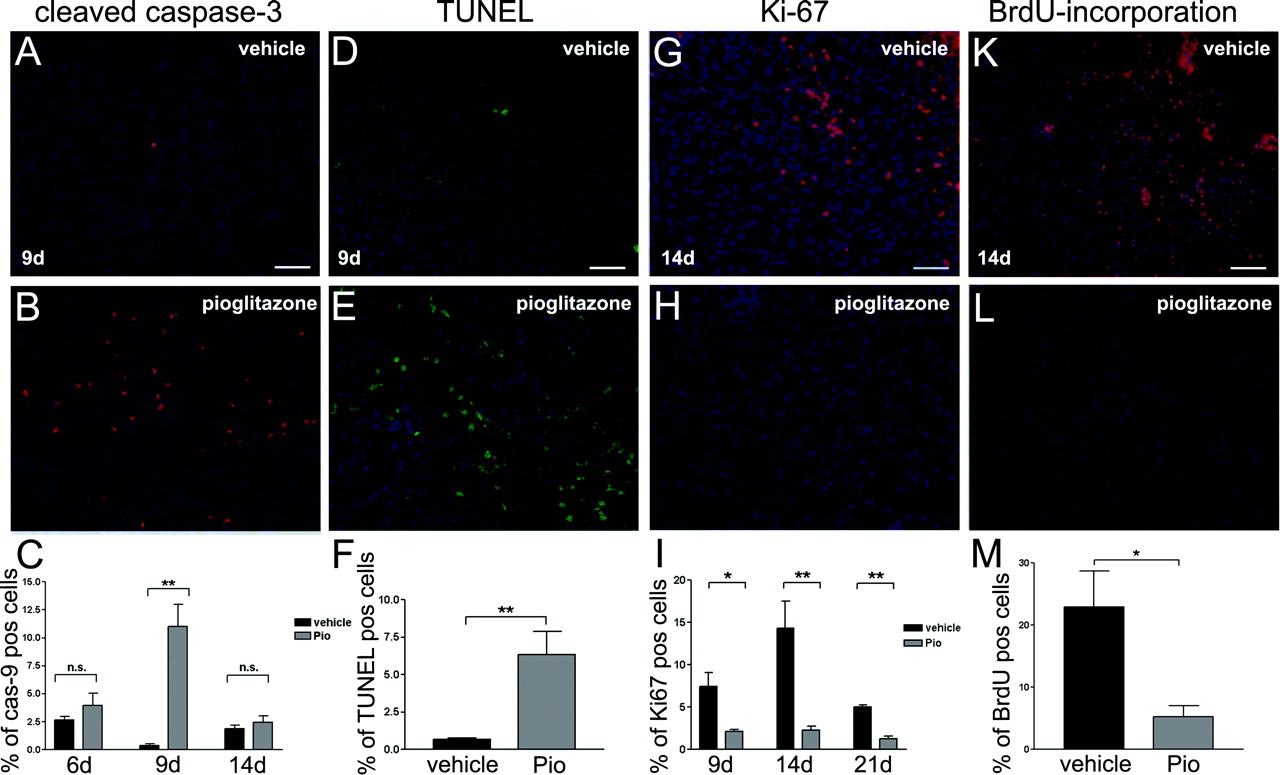
Pioglitazone induced apoptosis and reduced proliferation. Cleaved caspase-3 expression and TUNEL expression in the tumors of C6 rat glioma model animals with and without treatment at day 9 (n = 5). DAPI counter staining to determine number of tumor cells. Bar, 100 μm. Percentage of cleaved caspase-3 positive tumor cells at 6, 9, and 14 days and TUNEL positive tumor cells at 9 days. Data are presented as mean ± S.E. of the 5 animals per group. Black bars, vehicle treatment; gray bars, pio treatment. Asterisks indicate significant level in Student's t test: **, p < 0.008 (n = 5). Ki-67 expression and BrdU incorporation in the tumors of C6 rat glioma model animals with and without treatment at day 14 (n = 5). DAPI counter staining to determine number of tumor cells. Bar, 100 μm. Percentage of Ki-67 positive tumor cells (proliferation-index) at 9, 14, and 21 days and percentage of BrdU positive tumor cells at 14 days. Data are presented as mean ± S.E. of the five animals per group. Black bars, vehicle treatment; gray bars, pio treatment. Asterisks indicate significant level in Student's t test: *, p = 0.0114 (n = 5); **, p ≤ 0.006 (n = 5or n = 5).
Invasion of C6 Rat Glioma Cells after Pioglitazone Treatment in Vitro. In addition to proliferation, the ability of tumor cells to invade healthy nearby tissue is characteristic of the malignancy state of gliomas. Histological evaluation of pio-treated in vivo gliomas revealed more defined tumor margins and decreased invasiveness (Fig. 2, C and D). This led us to examine invasiveness of C6 glioma cells using a Boyden chamber assay. Pio-pretreated (30 μM) (Supplemental Fig. 3, D, E, and F) and untreated (Supplemental Fig. 3, A, B, and C) C6 glioma cells were suspended in the upper chamber containing pio at 30 μM (Supplemental Fig. 3, B and E) or vehicle (Supplemental Fig. 3, A and D). At 4 h, cells that migrated to the other side of an 8 μm filter were stained with DAPI and counted. Pio treatment during the Boyden chamber incubation reduced the invasiveness of C6 cells significantly (Supplemental Fig. 3, A, B, and C). Pretreatment of C6 cells for 24 h with pio further increased inhibition (Supplemental Fig. 3, D, E, and F).
MMP9 and CS-56 Expression in Vivo. To characterize the invasion of pio-treated tumors in vivo, we investigated levels of MMP-9, a protein that plays a major role in glioma invasion (Rao et al., 1993). MMP-9 expression was detected in whole-hemisphere lysates of C6 glioma animals using a goat-anti-MMP-9-antibody. MMP-9 expression was almost completely suppressed in pio-treated animals (Fig. 5A). Erk2 expression served as a loading control (Fig. 5A). Densitometric analysis showed an 81% reduction of MMP-9 expression in the pio-treated group (Fig. 5B). Immunoreactivity of MMP-9 revealed that the epitope is expressed only in the tumor and adjacent brain regions (Fig. 5C). Pio treatment reduced MMP-9 positive staining at tumor margins and even more so in the surrounding healthy brain tissue compared with vehicle treatment (Fig. 5C).
The expression of a marker of astrocytic differentiation, CS-56 (Vitellaro-Zuccarello et al., 2001), was evaluated in the tumors (Fig. 5C). An up-regulation of CS-56 was most prominently observed at the tumor margin toward healthy brain tissue compared with vehicle-treated animals (Fig. 5C). Parallel analysis of tissue lysates by Western blot (Fig. 5D) revealed up-regulation of CS-56 expression in pio-treated animals, which was corroborated by the densitometric measurement, showing an increase of around 56% in CS-56 protein levels (Fig. 5E). Erk2 expression served as a loading control (Fig. 5D).
Discussion
Antineoplastic effects of PPARγ agonists on human tumor cells (Grommes et al., 2005) and rat glioma cell lines (Zander et al., 2002; Grommes et al., 2005) led us to question whether the synthetic PPARγ agonist pio could exert similar effects in rat and human glioma cells as well as in animal glioma models. Pio, a member of the thiazolidinedione class of antidiabetic drugs, was chosen because it is already in clinical use for treatment of type II diabetes mellitus (Actos) and would be readily available for glioma therapy in clinical studies. In the present study pio, demonstrated antineoplastic potency in vitro by decreasing cellular viability of human and rat glioma cells. Similar effects were found in PPARγ-overexpressing glioma cells, whereas pio treatment of glioma cells overexpressing a PPARγ mutant, lacking the transcriptional activation, induced no antineoplastic changes. We demonstrated that the reduction of cellular viability by pio is restricted to neoplastic cell types, as primary astrocytes were not affected by pio treatment. In our hand and in prior studies (Cullingford et al., 1998), normal rat astrocytes express PPARγ at low levels. We have previously shown that primary rat astrocytes are not susceptible to PPARγ agonist-induced cell death (Zander et al., 2002). This is contrary to the apoptotic cell death observed in human fetal astrocytes after treatment with PPARγ agonists and may be due to species differences (Chattopadhyay et al., 2000).
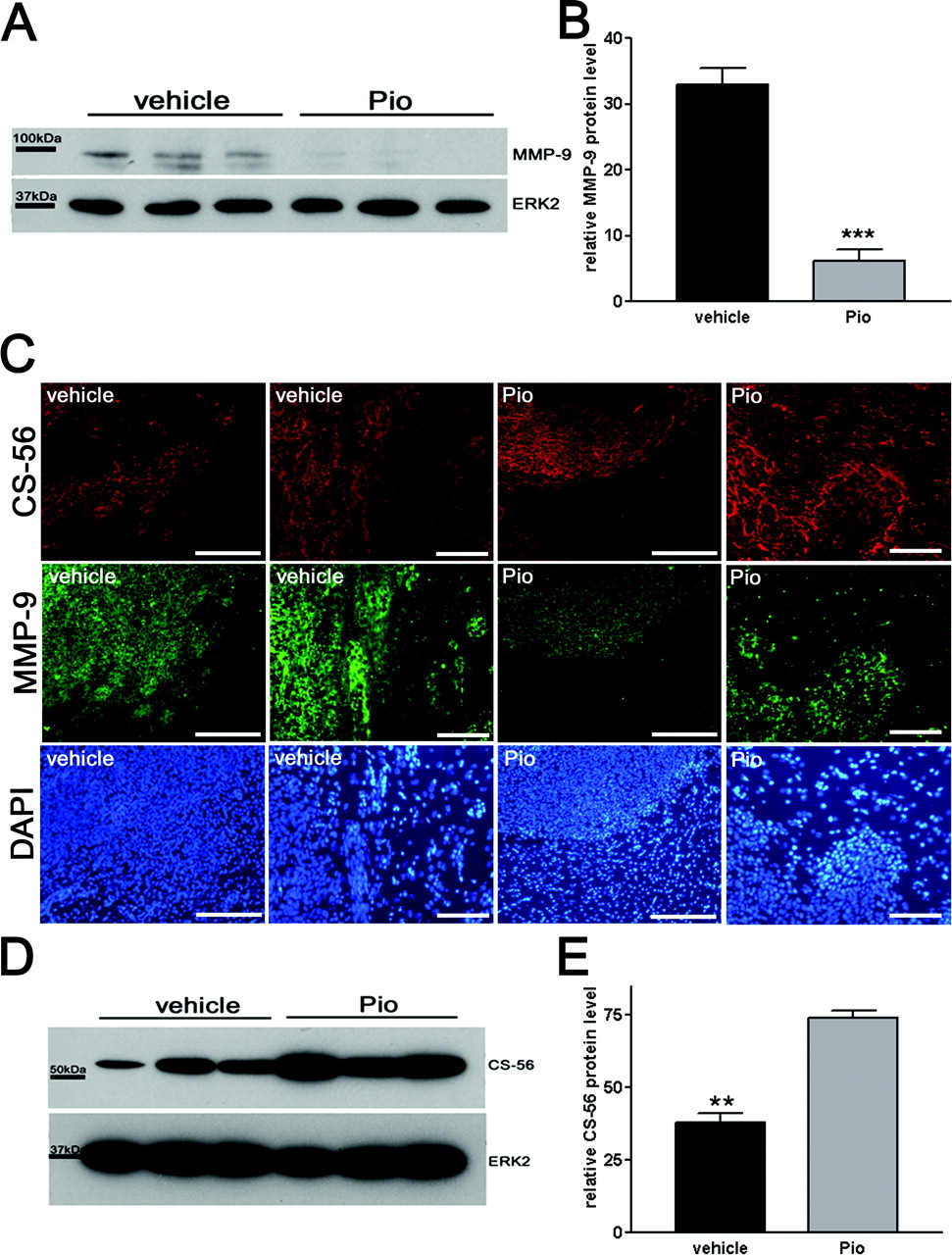
Pioglitazone reduces MMP9 protein levels and induces the astrocytic redifferentiation marker CS-56 in vivo. MMP-9 and CS-56 expression. A, MMP-9 expression in whole-hemisphere lysates of C6 rat glioma model animals using a goat-anti-MMP-9-antibody. Vehicle, vehicle-treated animals (n = 3); Pio, pio-treated animals (n = 3). To ensure equal loading of proteins, each membrane was stripped and reprobed with a rabbit anti-Erk2-antibody. B, densitometry. Data are presented as mean ± S.E. of the seven animals per group. Black bars, vehicle treatment; gray bars, pio treatment. Asterisks indicate significant level in student-t test: ***, p = 0.0009 (n = 3). C, CS-56 (red)/MMP-9 (green) expression at the tumor margin in brain tissue of vehicle-treated (two left rows) and pio-treated (two right rows) animals. Bars, 100 μm. DAPI-staining served as counterstaining. D, CS-56 expression in whole-hemisphere lysates of C6 rat glioma model animals using a mouse-anti-CS-56-antibody. Vehicle, vehicle-treated animals (n = 3); Pio, pio-treated animals. To ensure equal loading of proteins, each membrane was stripped and reprobed with a rabbit anti-Erk2-antibody. E, densitometry. Data are presented as mean ± S.E. of the seven animals per group. Black bars, vehicle treatment; gray bars, pio treatment. Asterisks indicate significant level in Student's t test: **, p = 0.0012 (n = 3).
Of the three glioma cell lines we studied, PPARγ protein levels are highest in U87 compared with A176 and C6 (Zander et al., 2002). Pio reduced cellular viability in human glioma cell line U87 more robustly than in either the A172 or C6 glioma cell lines, which may be related to the relative PPARγ protein levels expressed in these glioma cell lines.
Ki-67 is a nuclear protein expressed in proliferating cells and serves as an important neuropathological marker of human gliomas (Torp, 2002). In astrocytomas, Ki-67 expression is up-regulated and correlates well with tumor grade and clinical prognosis (Parkins et al., 1991; Brown and Gatter, 2002). Pio treatment reduced Ki-67 expression in vitro. Therefore, pio reduces both cellular viability and proliferation, leading to a reduced tumor cell number in vitro.
To verify these results in vivo, C6 cells were injected into rat striata (Stander et al., 1998) and either continuously treated with pio orally or through an intracerebrally placed osmotic pump for 3 weeks. Intracerebral pio treatment reduced tumor volumes by 83% and oral pio treatment by 76.9%. Furthermore, preservation of neurological function and body weight was observed in the pio-treated animals. Tumor volumes showed no significant difference during the early treatment period (3 and 6 days), indicating that pio treatment did not affect initial tumor inoculation. At 9 days, tumor volumes increased in the vehicle group and decreased in the pio-treated animals, indicating that antineoplastic effects of pio appear between days 6 and 9 in vivo, a time frame identical to our in vitro results. Similar antineoplastic effects of pio were seen in a murine animal model.
We investigated Bax protein expression because initial in vitro data indicate that PPARγ agonists mediate their anti-neoplastic effects in rat and human glioma cell lines through a Bax up-regulation leading to apoptosis, which could be abolished by Bax antisense-oligonucleotides (Zander et al., 2002). Primary data revealed that tumor growth reduction by pio was not associated with an up-regulation of proapoptotic proteins after 21 days of treatment (data not shown); therefore, earlier time points were evaluated. Bax as well as its downstream target cleaved caspase-3 peaked in the early phase of the treatment. At 6 days of pio therapy, a significant induction of these proapoptotic proteins was observed, correlating with an increase in cleaved caspase-3 immunoreactivity and TUNEL labeling at day 9. In prior studies, we were able to demonstrate that the PPARγ agonist-induced apoptotic death is BAX-dependent (Zander et al., 2002). Together, these findings indicate that pio-induced apoptosis of rat C6 gliomas occurred from days 6 to 9 and contributed at least in part to the observed reduction in tumor volume.
Reduced tumor growth might also be due to decreased proliferation, as observed in vitro. To assess whether pio also reduced proliferation in vivo, Ki67 expression and BrdU incorporation was determined, because earlier studies showed the ability of PPARγ agonists to reduce BrdU incorportion in various cell lines (Fajas et al., 2003). Both proliferation markers were reduced by pio, indicating that pio not only induced apoptosis but also reduced proliferation.
Histological differences, better defined tumor margins, and fewer invasive cells after pio treatment led us to investigate pio-induced changes of invasion in vitro using the Boyden chamber assay, a reliable test for assessment of C6 cell migration and invasion (Sottocornola et al., 1998). Pio treatment resulted in dramatic reduction of cell migration. Furthermore, MMPs play a critical role in tumor invasiveness and in the malignancy of gliomas (Rao et al., 1993) by mediating basal membrane breakdown. MMP-9, the most abundant MMP in gliomas (Forsyth et al., 1999), is elevated during tumor progression (Rao et al., 1993) because of its secretion by glioma cells (Choe et al., 2002). Tumor growth and formation is blocked by MMP-9 antisense oligonucleotides in vivo (Lakka et al., 2002). In the present study, MMP-9 expression was dramatically reduced by 81% in pio-treated animals, particularly at the tumor margin. MMP-9 down-regulation at the tumor margin may account for the reduced capability of pio-treated tumors to invade brain tissue. Whole-brain slides were used for protein level evaluation through Western blot. Therefore, the ratio of MMP9 protein levels in the tumor to brain tissue is smaller in the treated animals and could account for the lower MMP-9 signal detected by Western blot compared with the immunohistochemical stain.
Treatment of C6 cells with the PPARγ agonist ciglitazone led to increased redifferentiation in prior studies (Zander et al., 2002). Therefore, we investigated protein levels and immunohistochemical localization of CS-56, an astrocytic redifferentiation marker. CS-56 expression reflects levels of chondroitin sulfate proteoglycans, the most abundant component of the extracellular matrix of mammalian brain (Herndon and Lander, 1990), which is involved in development and redifferentiation (Vitellaro-Zuccarello et al., 2001). CS-56 is located predominantly at the tumor border, where it is highly up-regulated by pio treatment both in vitro (data not shown) and in vivo. Pio may, therefore, not only induce cell death and inhibition of proliferation in neoplastic cells but also elicit redifferentiation in the malignant cells. The CS-56 up-regulation at the tumor margin could also reflect glia formation separating tumor tissue from healthy brain tissue because CS-56 is strongly up-regulated in glial scars (Davies et al., 1997).
The concentration of pio required to affect cellular viability is higher than expected according to its known in vitro receptor binding affinity (Sakamoto et al., 2000). PPARγ-independent mechanisms have also been reported for thiazolidinediones (Chawla et al., 2001) and need to be considered. Troglitazone inhibits cholesterol biosynthesis (Wang et al., 1999) or acyl-CoA synthetase in a PPARγ-independent way (Kim et al., 2001). Pio treatment of human glioma cells overexpressing the PPARγ mutant E499Q, lacking the transcriptional activation, did not lead to a reduction of cellular viability, indicating that the described pio effects are indeed displayed in a PPARγ-dependent way.
Although the molecular basis of antineoplastic mechanisms of PPARγ agonists are yet not fully understood, the thiazolidinediones may offer a new therapeutic approach in human glioma therapy because of their negative effects on proliferation and invasion as well as their positive effects on apoptosis and redifferentiation. However, further studies will be required to ascertain optimal mode and timing for application of pio.
Footnotes
-
C.G. was supported by a grant from the German research council (GR 2018/1-1); U.S. and M.T.H. were supported by a grant from the German Cancer research council (10-1795). The work was also supported by the Blanchette Hoker Rockefeller foundation.
-
Conflict of interest statment: G.E.L. has received financial support for research projects on anti-inflammatory actions of PPARγ agonists by Glaxo-SmithKline. Case Western Reserve University holds a US patent on the use of PPARγ agonists in inflammatory indications in the nervous system. The intellectual property and research support do not relate to the antineoplastic actions of the drugs.
-
ABBREVIATIONS: PPAR, peroxisome proliferator-activated receptor; GW9662, 2-chloro-5-nitrobenzanilide; pio, pioglitazone; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling; MMP9, matrix metalloproteinase 9; DMSO, dimethyl sulfoxide; DMEM, Dulbecco's modified Eagle's medium; PBS, phosphate-buffered saline; MTT, [4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; HE, hematoxylin-eosin; TBS, Tris-buffered saline; DAPI, 4,6-diamidino-2-phenylindole; BrdU, 5-bromodeoxyuridine; PBS, phosphate-buffered saline.
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material. - Received January 3, 2006.
- Accepted July 12, 2006.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}