


Abstract
Peroxisome proliferator-activated receptor (PPARs) modulate target gene expression in response to unsaturated fatty acid ligands, such as arachidonic acid (AA). Here, we report that the AA metabolite 15-hydroxyeicosatetraenoic acid (15-HETE) activates the ligand-dependent activation domain (AF2) of PPARβ/δ in vivo, competes with synthetic agonists in a PPARβ/δ ligand binding assay in vitro, and triggers the interaction of PPARβ/δ with coactivator peptides. These agonistic effects were also seen with PPARα and PPARγ, but to a significantly weaker extent. We further show that 15-HETE strongly induces the expression of the bona fide PPAR target gene Angptl4 in a PPARβ/δ-dependent manner and, conversely, that inhibition of 15-HETE synthesis reduces PPARβ/δ transcriptional activity. Consistent with its function as an agonistic ligand, 15-HETE triggers profound changes in chromatin-associated PPARβ/δ complexes in vivo, including the recruitment of the coactivator cAMP response element-binding protein binding protein. Both 15R-HETE and 15S-HETE are similarly potent at inducing PPARβ/δ coactivator binding and transcriptional activation, indicating that 15-HETE enantiomers generated by different pathways function as PPARβ/δ agonists.
Peroxisome proliferator-activated receptor-β/δ (PPAR β/δ) is a ligand-regulated transcription factor that modulates target gene expression in response to certain fatty acids and fatty acid derivatives (Forman et al., 1997; Desvergne et al., 2006). PPARβ/δ forms heterodimers with the nuclear receptor RXR that bind to peroxisome proliferator response elements (PPREs) in target genes. A major function of the ligand in this context is to induce a conformational change in PPARβ/δ that results in the displacement of interacting corepressors, such as silencing mediator for retinoid and thyroid (SMRT) receptors and SMRT/histone deacetylase-associated repressor protein (SHARP), by specific coactivators, such as sequential recruitment of steroid receptor coactivator-1 (SRC-1) and p300/CBP, resulting in transcriptional activation (Yu and Reddy, 2007; Zoete et al., 2007). PPARβ/δ can regulate genes also by different mechanisms. Thus, PPARβ/δ ligands repress pro-inflammatory gene expression by releasing the hematopoietic transcriptional repressor Bcl-6 from a complex with PPARβ/δ (Lee et al., 2003). In another study (Matsusue et al., 2006), it was reported that PPARβ/δ can repress genes by sequestering RXR from other RXR-dependent nuclear receptors.
PPARβ/δ plays an important role in the regulation of energy homeostasis, lipid catabolism, and glucose homeostasis (Desvergne et al., 2006) but also has essential functions in developmental processes, differentiation, and wound healing. Mice lacking PPARβ/δ show an aberrant development and malfunction of the placenta (Peters et al., 2000; Barak et al., 2002; Nadra et al., 2006) and exhibit a defect in wound healing (Michalik et al., 2001). PPARβ/δ is critical for the survival, differentiation, and proliferation of keratinocytes (Peters et al., 2000; Di-Poï et al., 2002; Burdick et al., 2006), and promotes the differentiation of Paneth cells in the intestinal crypts (Varnat et al., 2006). However, PPARβ/δ also plays a role in cancer and inflammation: it modulates intestinal tumorigenesis with diverging effects in different mouse models (Peters et al., 2000; Barak et al., 2002; Di-Poï et al., 2002; Gupta et al., 2004; Wang et al., 2004; Burdick et al., 2006), inhibits chemically induced skin carcinogenesis (Kim et al., 2004; Bility et al., 2008), exerts an essential function in the tumor stroma (Abdollahi et al., 2007; Müller-Brüsselbach et al., 2007), and has potent anti-inflammatory activities (Kilgore and Billin, 2008). Therefore, PPARβ/δ represents a highly relevant drug target for the treatment of major human diseases, which has helped lead to the development of several synthetic drug candidates with subtype selectivity and high-affinity binding, such as GW501516 and L165,041 (Peraza et al., 2006).
One of the fatty acids that induces PPARβ/δ activity to a moderate extent is AA. It has been shown that AA is a low-affinity ligand that interacts with the PPARβ/δ LBD (Xu et al., 1999), raising the possibility that the agonistic effect of AA is due directly to its interaction with PPARβ/δ. On the other hand, metabolites of AA may also account for this effect. Prostanoids are major AA metabolites generated by the combined action of cyclooxygenases and prostaglandin or thromboxane synthases. Prostaglandin I2 (PGI2; prostacyclin) has indeed been postulated to act as a PPARβ/δ agonist (Gupta et al., 2000; Hatae et al., 2001), but this issue remains controversial (Yu et al., 1995; Forman et al., 1996; Fauti et al., 2006).
Another major group of eicosanoid metabolites is generated by the lipoxygenases (Pidgeon et al., 2007), but lipoxygenase products of AA acting as bona fide PPARβ/δ ligands have not yet been described. 15-Hydroxyeicosatetraenoic acid (15-HETE) has been reported to activate PPARβ/δ in a reporter assay in keratinocytes, but it remains unclear whether this involves a direct interaction with the PPARβ/δ LBD or indirect mechanisms (Thuillier et al., 2002).
In the present study, we have systematically addressed this question using mouse fibroblasts as a model system. We show that the agonistic effect of AA is due largely to its lipoxygenase-mediated oxidation to 15-HPETE and the subsequent enzymatic conversion to 15-HETE. Consistent with this finding, 15-HETE enabled the interaction of PPARβ/δ with coactivator peptides in vitro, and interacted with PPARβ/δ in a competitive ligand-binding assay. Furthermore, 15-HETE induced the PPARβ/δ target gene Angptl4 (Mandard et al., 2004) in a clearly PPARβ/δ-dependent manner. Both enantiomers of 15-HETE, 15R-HETE and 15S-HETE, showed similar agonistic properties, indicating that different pathways converge on PPARβ/δ. Whereas 15S-HETE is generated by LOX pathways, 15R-HETE is synthesized by cytochrome P450 or acetylated COX-2 (Clària et al., 1996; Clària and Serhan, 1995; Gilroy, 2005; Romano, 2006; Titos et al., 1999). Collectively, our findings demonstrate that 15-HETE enantiomers produced by different signaling pathways function as ligands for PPARβ/δ and induce its transcriptional activity.
Materials and Methods
Chemicals.
GW501516, 9-cis-retinoic acid, and the LOX inhibitors NDGA and EDBCA were purchased from Axxora (Lörrach, Germany) and prostaglandins D2, E2, and F2 from Cayman Europe (Tallinn, Estonia). GW1929 was obtained from Cayman Europe; LXA4 from BIOMOL (Hamburg, Germany); and GW7647 and diclofenac from Sigma-Aldrich (Steinheim, Germany). All other eicosanoids were from Cayman Europe and Axxora (Lörrach, Germany).
Mouse Strains.
Pparb/d-null and wild-type mice have been described previously (Peters et al., 2000). Pparb/dck mice (Barak et al., 2002) harboring a floxed Pparb/d exon 4 were kindly provided by R. Evans.
Cell Culture.
Pparb/d-null, wild-type, and floxed fibroblasts were established from fetal lungs and cultured as described previously (Müller-Brüsselbach et al., 2007). WPMY-1 cells were obtained from the American Type Culture Collection (Manassas, VA). A CHO cell line with a stably integrated pFR-Luc reporter gene (Stratagene/Agilent Technologies, Waldbronn, Germany), which expresses luciferase under the control of five Gal4 DNA binding sites and stably expresses a Gal4-hPparb fusion protein, was generated by successive electroporation of the two vector constructs. After selection in G418, the cells were cloned by limited dilution, and positive clones were identified using a charge-coupled device camera. The cell clone showing the best response to synthetic PPARβ/δ ligands was chosen for this study. All cells were maintained in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin in a humidified incubator at 37°C and 5% CO2.
Plasmids.
pCMX-mPparδ (Forman et al., 1997) and Gal4-mPparδ (Shi et al., 2002) were kindly provided by Dr. R. Evans. 3×FLAG-PPARβ/δ was generated by cloning the coding sequence of mPPARβ/δ N-terminally fused to a triple FLAG tag (Müller-Brüsselbach et al., 2007) into pcDNA3.1(+) zeo (Invitrogen, Karlsruhe, Germany). pCMX-empty has been described previously (Umesono et al., 1991). LexA-PPARβ/δ, 7L-TATA initiator module (TATAi), and 10×Gal4SVGL3 have been described previously (Jérôme and Müller, 1998; Fauti et al., 2006). LexA-PPARα and LexA-PPARγ were constructed in a fashion analogous to the construction of LexA-PPARβ/δ. pSG5-hRxRa containing the full-length RxRa cDNA was kindly provided by Dr. A. Baniahmad. The PPRE-TATAi plasmid was constructed by inserting a PPRE containing fragment of the third intron of the human ANGPTL4 gene (Mandard et al., 2004) into TATAi-pGL3 (Jérôme and Müller, 1998). The pUC18 plasmid was obtained from New England Biolabs (Frankfurt am Main, Germany), and pcDNA3.1 was obtained from Invitrogen (Karlsruhe, Germany).
Transfections and Luciferase Reporter Assays.
Transfections were performed with polyethylenimine (average mol. wt., 25,000; Sigma-Aldrich). Cells were transfected on 12-well plates at 70 to 80% confluence in DMEM plus 2% FCS with 2.5 μg of plasmid DNA and 2.5 μl of polyethylenimine (1:1000 dilution, adjusted to pH 7.0 and preincubated for 15 min in 100 μl of phosphate-buffered saline for complex formation). Four hours after transfection, the medium was changed and cells were incubated in normal growth medium for 24 h. Luciferase assays were performed as described previously (Gehrke et al., 2003). Values from three independent experiments were combined to calculate averages and standard deviations.
Retrovirally Transduced Cells Expressing FLAG-PPARβ.
3×FLAG-PPARβ/δ was cloned into the retroviral vector pLPCX (Clontech, Mountain View, CA). Phoenix cells expressing ecotropic env were transfected with 3×FLAG-mPPARb-pLPCX as described elsewhere (http://www.stanford.edu/group/nolan/retroviral_systems/retsys.html). Culture supernatant was used to infect Pparb-null fetal mouse lung fibroblasts that had previously been established from Pparb-knockout mice by standard procedures. Cells were selected with puromycin (2 μg/ml; Sigma), resulting in a cell population expressing 3×FLAG-mPPARβ/δ at moderate levels.
Quantitative PCR.
cDNA was synthesized from 1 μg of RNA using oligo(dT) primers and the Omniscript kit (QIAGEN, Hilden, Germany). qPCR was performed in a Mx3000P real-time PCR system (Stratagene, La Jolla, CA) for 40 cycles at an annealing temperature of 60°C. PCR reactions were carried out using the Absolute QPCR SYBR Green Mix (ABgene, Hamburg, Germany) and a primer concentration of 0.2 μM according to the manufacturer's instructions. L27 was used as normalizer. Comparative expression analyses were statistically analyzed by Student's t test (two-tailed, equal variance). The following primers were used: Pparb: forward, 5′-CTCCATCGTCAACAAAGACG; reverse, 5′-TCTTCTTTAGCCACTGCATC; Angptl4: forward, 5′-CTC TGG GGT CTC CAC CAT TT; reverse, 5′-TTG GGG ATC TCC GAA GCC AT; L27: forward, 5′-AAA GCC GTC ATC GTG AAG AAC; reverse, 5′-GCT GTC ACT TTC CGG GGA TAG.
siRNA Transfections.
For siRNA transfection, cells were seeded at a density of 5 × 105 cells per 6-cm dish in 4 ml of DMEM with 10% FCS and cultured overnight. Four hundred picomoles of siRNA in 500 μl of OptiMEM (Invitrogen) and 10 μl of Lipofectamine 2000 (Invitrogen) in 500 μl of OptiMEM were separately incubated for 5 min at room temperature, mixed, and incubated for another 20 min. The siRNA-lipid complex was added to the cells cultured in DMEM without FCS (time = 0), and the medium was changed to normal growth medium after 6 h. Cells were passaged and replated 48 h after transfection at a density of 5 × 105 cells per 6-cm dish. Transfection was repeated 72 h after start of the experiment, and cells were passaged after another 24 h. Forty-eight hours after the last transfection, cells were stimulated and harvested after 3 h for RNA isolation. The following siRNAs were used: Pparb siRNA1 (CCGCATGAAGCTCGAGTATGA; QIAGEN); Pparb siRNA2 (CAAGTTCGAGTTTGCTGTCAA; QIAGEN); control siRNA (Alexa Fluor 488-labeled nonsilencing duplex siRNA; QIAGEN).
Chromatin Immunoprecipitation Analysis.
WPMY-1 cells were grown to confluence. After stimulation, the cells were fixed by addition of 37% formaldehyde to a final concentration of 1%. Incubation time was 10 min at room temperature. Glycine was added to a final concentration of 125 mM for 5 min. After two washes with ice-cold phosphate-buffered saline, the cells were pelleted for 5 min at 1200 g. Pellets were resuspended in cold hypotonic lysis buffer [5 mM PIPES, pH 8.0, 85 mM KCl, 0.5% (v/v) NP40, and protease inhibitor cocktail (Sigma)] at a ratio of 1 ml per 107 cells and incubated on ice for 20 min. Nuclei were pelleted by centrifugation as before and resuspended in radioimmunoprecipitation assay buffer [10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% (v/v) NP40, 1% sodium deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA, and protease inhibitors] at a ratio of 1 ml per 107 nuclei. Soluble chromatin was prepared by sonication with a microtip using a sonifier (S-250D; Branson Ultrasonics Corporation, Danbury, CT) set to 1-s pulse, 2-s pause. Eighty pulses were applied. After centrifugation at 16,000g for 15 min in a tabletop centrifuge, the supernatant was collected. An aliquot was incubated overnight with proteinase K and RNase A at 65°C and loaded on a 1% agarose gel to estimate shearing efficiency. The supernatant was precleared by addition of protein A Sepharose beads (Invitrogen), which were previously blocked in radioimmunoprecipitation assay buffer with 1 mg/ml bovine serum albumin, 0.4 mg/ml sonicated salmon sperm DNA (Stratagene), and protease inhibitors, coupled to rabbit IgG (Sigma I5006). After rotation for 1 h at 4°C, the beads were removed by centrifugation. The supernatant was used for immunpreciptiations. Four micrograms of antibody [rabbit IgG pool, Sigma; α-PPARβ/δ, α-CBP, Santa Cruz Biostechnology (Santa Cruz, CA); α-acetylated H4, Millipore (Billerica, MA)] were added to 300 μl of precleared chromatin corresponding to 3 × 106 nuclei and incubated overnight at 4°C with mild rotation. After addition of blocked Sepharose beads, incubation time was 1 h at 4°C with mild rotation. The beads were washed once with 1 ml of chilled mixed micelle buffer [20 mM Tris, pH 8.1, 150 mM NaCl, 2 mM EDTA, 0.1% (w/v) SDS, and 1% (v/v) Triton X-100], once with buffer 500 [20 mM Tris, pH 8.1, 500 mM NaCl, 2 mM EDTA, 0.1% (w/v) SDS, and 1% (v/v) Triton X-100], twice with LiCl detergent buffer (10 mM Tris, pH 8.1, 250 mM LiCl, 1% (v/v) NP40, 1% (w/v) sodium deoxycholate, and 1 mM EDTA), and twice with room-temperature Tris-EDTA. Complexes were eluted twice with 250 μl of elution buffer [1% SDS (w/v) and 100 mM NaHCO3]. Supernatants were pooled; adjusted to 180 mM NaCl, 35 mM Tris, pH 6.5, and 9 mM EDTA; and incubated with 20 μg of proteinase K and 10 μg of RNase A for 65°C overnight. DNA was purified using a PCR purification kit (QIAGEN). Eluates were quantified by qPCR employing the ΔCt method relative to an amount equivalent to 1% of DNA used for immunoprecipitation. Standard deviations were calculated from triplicate measurements considering Gaussian error propagation. The following primers were used: ANGPTL4: PPRE, forward: CCT TAC TGG ATG GGA GGA AAG; reverse, CCC AGA GTG ACC AGG AAG AC.
Time-Resolved Fluorescence Resonance Energy Transfer Assays in Vitro.
TR-FRET (Stafslien et al., 2007) was performed with the LanthaScreen TR-FRET PPARβ/δ competitive binding assay and the LanthaScreen TR-FRET PPARα, PPARβ/δ, and PPARγ coactivator assays according to the instructions of the manufacturer (Invitrogen). Incubation time was 60 min for all assays shown in this study. All assays were validated for their robustness by determining the respective Z′-factors (Zhang et al., 1999). Measurements were performed on a VICTOR3V Multilabel Counter (WALLAC 1420; PerkinElmer Life and Analytical Sciences, Rodgau, Germany) with instrument settings as described in the manufacturer's instructions for LanthaScreen assays. The following peptides were used for the coactivator recruitment assay: PGC1α, EAEEPSLLKKLLLAPANTQ; C33, HVEMHPLLMGLLMESQWGA; CBP, AASKHKQLSELLRGGSGSS; PPARγ-interacting protein, VTLTSPLLVNLLQSDISAG; TRAP220, NTKNHPMLMNLLKDNPAQD.
Quantification of HETEs in Cell Culture Supernatants by Liquid Chromatography/Tandem Mass Spectrometry.
Cell culture supernatants were spiked with ∼1 ng of deuterated internal standards and acidified with 20 μl of saturated NH4Cl solution containing 1.25 M HCl. The analytes were extracted with 1 ml of diisopropyl ether and, after complete drying, resolved in acetonitrile/water [1:1 (v/v)]. For determination, a 10-μl aliquot was injected into the LC/tandem mass spectrometry. The LC/MS analysis was carried out on a mass spectrometer (API3000; Applied Biosystems, Foster City, CA) equipped with two Series 200 LC Micro Pumps (PerkinElmer Life and Analytical Sciences, Waltham, MA), a CTC HTC PAL autosampler (CTC Analytics, Zwingen, Switzerland), a turbo-ion interface and a HSID interface (Ionics Mass Spectrometer Group Inc., Bolton, ON, Canada). The flow rate was set to 200 μl/min, and a C18 column (5 cm × 2 mm, 3-μm particles) was used. A generic LC gradient of 10 min was used for sample separation. Solvents were water/acetonitrile (95:5) (A) and acetonitrile/water (95:5) (B), both containing 0.2% acetic acid. The gradient profile used was 40% solvent A for 0 to 2 min linearly increasing to 100% within 7 min. Mass spectrometer conditions were: scan type, multiple reaction monitoring with negative polarity; ion-spray voltage, −4200 V; temperature, 350°C; collision gas, 4 psi; all potentials (declustering, focusing, entrance and exit potential) were optimized for each ion. Collision energy was 20 eV, and the following transitions were used for quantitation: 319.2/114.9 (5-HETE), 327.2/115.9 (d8-5-HETE), 319.2/154.2 (8-HETE), 319.2/207.8 (12-HETE), 327.2/214.1 (d8-12-HETE), 319.2/218.9 (15-HETE), and 327.2/182.0 (d8-15-HETE). Dwell time was 300 ms each. All chemicals and solvents were obtained from Merck (Darmstadt, Germany).
Results
Effect of Arachidonic Acid Derivatives on the Transcriptional Activity of PPARβ/δ.
We analyzed the effect of AA and its metabolites on the transcriptional activity of PPARβ/δ in NIH3T3 fibroblasts using luciferase reporter constructs harboring multiple Gal4 or LexA binding sites upstream of a TATAi. The transcriptional activator in this system is a fusion construct consisting of the PPARβ/δ LBD and a Gal4 (Fig. 1A) or LexA (Fig. 1B) DNA binding domain. With both reporter systems, we observed an induction of approximately 2-fold by 20 μM AA.

Effects of lipoxygenase inhibitors and specific eicosanoids on the transcriptional activity of the PPARβ/δ ligand binding domain. NIH3T3 cells were transiently transfected with an expression vector encoding a Gal4-PPARβ/δ (A) or LexA-PPARβ/δ fusion protein (B) together with a LexA or Gal4 binding luciferase reporter plasmid. Cells were preincubated with the pan-LOX inhibitor NDGA (A) or the 12/15-LOX inhibitor EDBCA (B) at the indicated concentrations for 24 h and then for another 24 h with 20 μM AA before harvesting. Control cells (−) received solvent only. C, CHO cells stably expressing a Gal4-PPARβ/δ fusion protein and harboring a stably integrated Gal4-responsive luciferase reporter gene were treated with the indicated compounds for 20 h. Values represent the average of triplicates; error bars show the standard deviation. Significant differences between control cells and cells treated with inhibitor are indicated by an asterisk (paired t test; P < 0.001).
The observed induction by AA was not influenced by the COX inhibitor diclofenac, and no transcriptional activation was seen when specific prostaglandins (PGD2, PGE2, PGF2) were applied (data not shown). This confirms previous findings that AA-derived prostanoids have no detectable effect on the activity of PPARβ/δ in human embryonic kidney 293 and NIH3T3 cells (Yu et al., 1995; Forman et al., 1996; Fauti et al., 2006). PGE2 has been described as an activator of PPARβ/δ, but this is an indirect effect involving PGE2-mediated induction of PI3K-AKT signaling and may therefore be dependent on the cell type and experimental system (Wang et al., 2001). Because NIH3T3 cells synthesize 5-, 8-, 12-, and 15-HETE at readily detectable levels (up to ∼60 ng/ml after 6-h culture, corresponding to ∼0.2 μM; Table 1), we tested the effect of LOX inhibitors and found a clear concentration-dependent inhibition on both the basal and AA-induced transcriptional activity of PPARβ/δ by the pan-LOX inhibitor NDGA (Fig. 1A). We also analyzed the 12-/15-LOX inhibitor EDBCA and observed a very similar effect compared with NDGA (Fig. 1B). These effects were specific, because no inhibition of transcription by NDGA or EDBCA was observed in the absence of LexA-PPARβ/δ (data not shown).
HETE synthesis by NIH3T3 fibroblasts
Concentrations shown are those in the culture medium 6 h after addition of arachidonic acid.
These observations raised the possibility that 12-/15-LOX products are activators of PPARβ/δ. We therefore tested the effects of 12-HETE and 15-HETE alongside a number of other eicosanoids on the transcriptional activity of PPARβ/δ. This experiment was performed with CHO cells harboring a stable Gal4-driven luciferase reporter gene and stably expressing a Gal4-PPARb/d fusion protein. Figure 1C clearly shows that 15-HETE was the only compound that was more potent than AA at activating the transcriptional activity of PPARβ/δ in this cellular system.
15-HETE Induces Coactivator Peptide Binding to PPARβ/δ.
The observations described above raise the possibility that the LOX products 12- or 15-HPETE and/or their direct metabolites 12- or 15-HETE are agonists of PPARβ/δ. To test this hypothesis we investigated the effect of different LOX products on PPARβ/δ coactivator interaction in vitro by TR-FRET. In this assay, the interaction of the PPARβ/δ LBD (indirectly labeled by terbium) with the coactivator peptide C33 (labeled with fluorescein) is determined (Stafslien et al., 2007). C33 was previously identified as a peptide strongly interacting with coactivator binding sites of PPARs in response to agonist binding, similar to PGC1α- or steroid receptor coactivator-derived peptides (Chang et al., 1999). This assay measures the intensity of terbium-induced fluorescence emission of the fluorescein moiety of the C33 peptide, expressed as the ratio of fluorescein- and terbium-derived fluorescence.
As shown in Fig. 2A, only AA, 15-HETE, and 15-HPETE were able to induce a significant FRET, whereas 8-HETE, 12-HETE, and 12-HPETE were unable to do so. These data suggest that the activation of PPARβ/δ by different HETEs reported in a previous study (Thuillier et al., 2002), including 8-HETE and 12-HETE, was due to indirect effects, perhaps involving HETE membrane receptors triggering signaling pathways impinging on PPARβ/δ-activated transcription. 15-HETE was clearly the most efficacious compound tested, giving rise to a comparable FRET signal at approximately 25% the concentration of AA or 15-HPETE. Titration analysis of lower concentrations of 15-HETE showed a statistically significant effect already at 0.1 μM and a half-maximal signal at approximately 0.55 μM (Fig. 2B).

Ligand-induced binding of a coactivator-derived peptide to the PPARβ/δ LBD in vitro. Interaction of fluorescein-labeled coactivator peptide and recombinant GST-Pparb bound by a terbium-labeled anti-GST antibody was determined by TR-FRET. A, the indicated metabolites or synthetic ligands were used at the indicated concentrations. B shows a titration analysis of lower concentrations of 15-HETE. Results are expressed as the ratio of fluorescence intensity at 520 nm (fluorescein emission excited by terbium emission) and 495 nm (terbium emission). All data points represent averages of triplicates (± S.D). In B, all FRET ratios obtained for concentrations ≥0.1 μM are statistically significant (Bonferroni-Holm adjusted t test).
15-HETE Competes for PPARβ/δ LBD Binding.
We next analyzed the interaction of AA, 15-HETE, and 15-HPETE with the LBD of PPARβ/δ in a TR-FRET-based competitive ligand binding assay. In this assay, the terbium-labeled PPARβ/δ LBD interacts with Fluormone Pan-PPAR Green (Invitrogen) as PPAR ligand, which produces FRET. Displacement of the fluorescent Fluormone Pan-PPAR Green by unlabeled ligand results in a quantifiable attenuation of FRET. The data obtained with this assay are shown in Fig. 3A. In agreement with the results of the coactivator recruitment assay, all three compounds were able to displace the PPARβ/δ bound ligand.

Competitive in vitro ligand binding assay for PPARβ/δ. Displacement of the Fluormone Pan-PPAR Green PPAR ligand from recombinant GST-Pparb was determined by TR-FRET. AA and the indicated metabolites were used at the concentrations shown in the graph. A, binding of 15-HETE and 15-HPETE compared with that of AA. B, binding of 15-HETE enantiomers and their metabolites. Results are expressed as the ratio of fluorescence intensity at 520 nm (fluorescein emission excited by terbium emission) and 495 nm (terbium emission). All data points represent averages of triplicates (± S.D).
15-HETE Induces PPARβ/δ-Mediated Transcription.
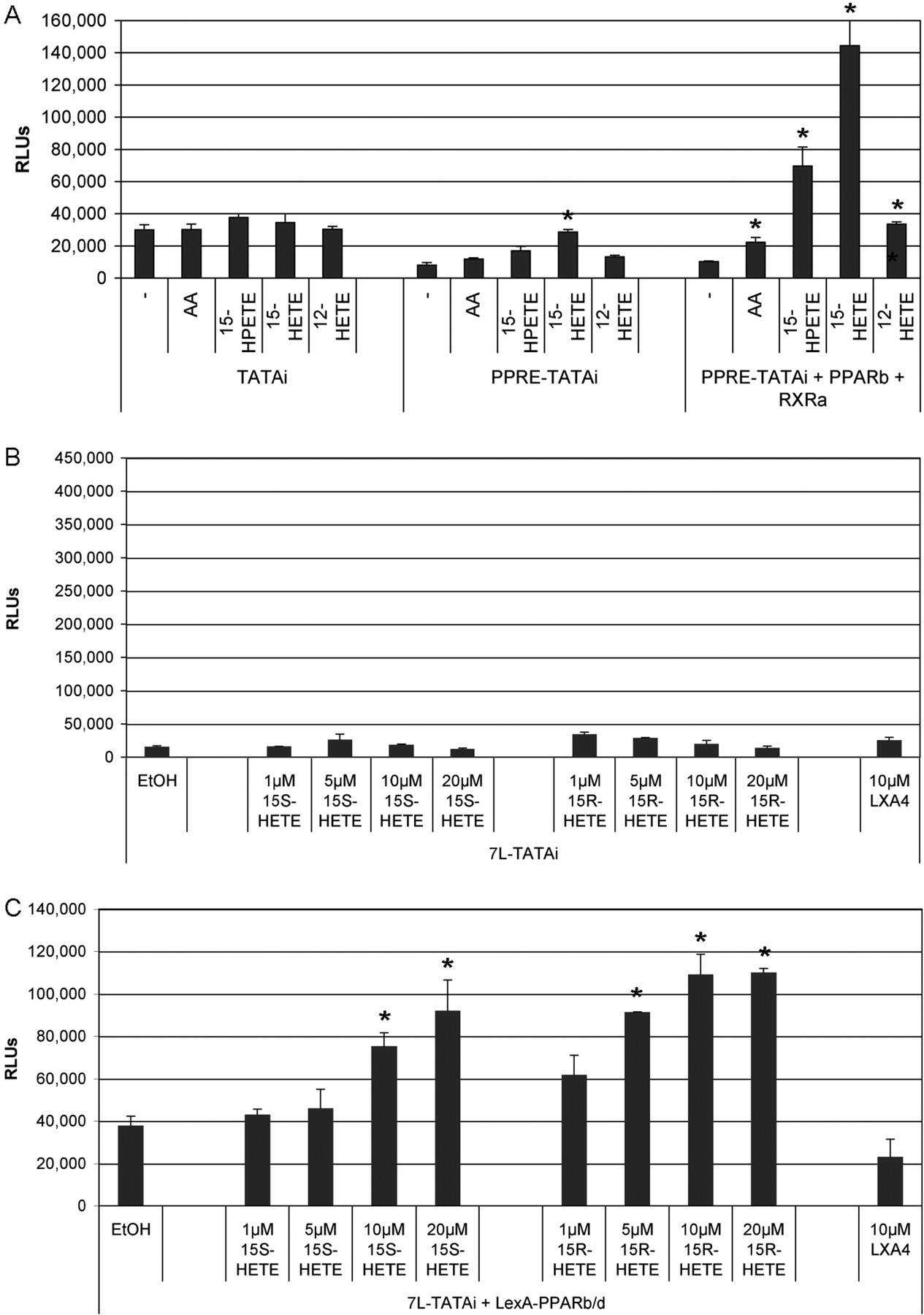
We next analyzed the effect of 12-HETE, 15-HETE, and 15-HPETE on a luciferase reporter construct driven by a PPAR-responsive minimal promoter (i.e., a PPRE-containing fragment from the first intron of the ANGPTL4 gene) (Mandard et al., 2004) linked to the TATAi module described above. For this and most of the subsequent experiments, we used NIH3T3 fibroblasts, because they essentially lack endogenous PPARα and PPARγ (data not shown), which, because of their partially redundant function, could obliterate PPARβ/δ-mediated effects. The data in Fig. 4A show that the basal level activity of the TATAi vector (left) was reduced by the presence of a PPRE (center), most likely as a result of the recruitment of PPARβ/δ repressor complexes (unpublished observations). The addition of AA, 12-HETE, 15-HETE, or 15-HPETE led to insignificant changes in basal activity, but a clear induction by both 15-HETE and 15-HPETE was seen with PPRE-TATAi (4.7- and 3.7-fold, respectively). In contrast, no significant induction was observed with AA or 12-HETE. Cotransfection of PPARβ/δ and RXRα expression vectors (Fig. 4A, right) clearly enhanced the effect. 15-HETE and 15-HPETE strongly induced PPRE-dependent transcription (21- and 11-fold, respectively), whereas the induction by both AA and 12-HETE was comparably weak (2- to 3-fold) albeit significantly higher than the solvent control (1.3-fold). These data confirm the data obtained by the TR-FRET analysis described above (Fig. 2).

Effects of different HPETEs and HETEs on the transcriptional activity of PPARβ/δ. A, NIH3T3 cells were transiently transfected with a PPRE-lacking control plasmid (left bars) or a PPRE-driven luciferase reporter plasmid (middle and right bars) together with either empty vector (left bars and middle) or expression vectors encoding PPARβ/δ and RXRα (right bars). The concentration of 12-HETE, 15-HETE, and 15-HPETE was 10 μM, AA was 20 μM. B and C, analysis of 15-HETE enantiomers and their metabolites in the LexA-based system described in Fig. 1. In B, the empty vector pcDNA3.1 was cotransfected instead of the LexA-Pparb expression plasmid (C). Values represent the average of triplicates; error bars show the standard deviation. Significant differences between solvent (EtOH) and ligand-treated cells are indicated by an asterisk (paired t test; P < 0.001).
Induction of Endogenous PPARβ/δ Target Genes by 15-HETE.
Angptl4 is an established PPAR target gene (Mandard et al., 2004) and was therefore tested for its inducibility by 15-HETE. As shown in Fig. 5B, Angptl4 was clearly induced in all three cell lines used for this analysis (i.e., NIH3T3 cells, WPMY-1 myofibroblasts, and HaCaT keratinocytes). HaCaT cells showed the strongest response with a 16-fold induction at 5 μM, compared with 5.1-fold in WPMY-1 cells and 3.4-fold in NIH3T3 cells. It is noteworthy that a clear induction of Angptl4 expression was seen already at 1 μM 15-HETE (3.1-fold in HaCaT cells; 1.8-fold in NIH3T3 cells). Maximum induction by 15-HETE was comparable with that seen with the synthetic agonist GW501516 at 1 μM (Fig. 5A). Because NIH3T3 cells lack other PPAR subtypes, these observations strongly suggest that the observed induction by 15-HETE is mediated by PPARβ/δ. Neither GW501516 nor 15-HETE altered Pparb/d expression levels (data not shown), indicating that the observed induction of Angptl4 expression by these compounds is due to an increased PPARβ/δ activity.

Inducibility of the PPARβ/δ target gene Angptl4. A, regulation of Angptl4 by 15-HETE and its enantiomers 15S-HETE and 15R-HETE in comparison to GW501516 (0.3 μM) in NIH3T3 cells. B, regulation of Angptl4 by 15-HETE at different concentrations in NIH3T3, HaCaT, and WPMY-1 cells. Cells were treated for 3 h, RNA was isolated and analyzed by qPCR using L27 for normalization. Induction values were calculated relative to solvent (EtOH)-treated cells and represent averages of triplicates (± S.D) normalized. *, values significantly different (P < 0.001) between solvent (EtOH) and ligand treated cells.
In agreement with these observations, chromatin immunoprecipitation analyses of human WPMY-1 cells showed recruitment of the coactivator CBP to the PPARβ/δ-bound ANGPTL4 PPRE upon stimulation with either GW501516 or 15-HETE (Fig. 6A). Concomitant with CBP recruitment to the PPRE, we observed acetylation of histone H4, but not of H3, at both the PPRE (Fig. 6B) and the TSS (data not shown) with either compound. Either GW501516 or 15-HETE also led to an increased binding of PPARβ/δ to the ANGPTL4 PPRE, which is consistent with previous observations made with synthetic ligands (Mandard et al., 2004). In contrast, specific binding of CBP, PPARβ/δ, or acetylated H4 to an irrelevant ANGPTL4 upstream region at −12 kilobase pairs relative to the transcription start site was not detectable (data not shown). These similarities in chromatin alterations triggered by both compounds provide further evidence that 15-HETE is an agonistic PPARβ/δ ligand.

Altered transcription factor binding and histone acetylation on the ANGPTL4 promoter after treatment with 15-HETE of WPMY-1 myofibroblasts. Cells were treated with 15-HETE (10 μM), GW501516 (0.3 μM) or solvent [ethanol for 15-HETE, dimethyl sulfoxide (DMSO) for GW501516] for 1 h (A) or 3 h (B). Chromatin immunoprecipitation was carried out using antibodies against CBP (A) or PPARβ/δ and acetylated histones H3 and H4 (B). An unspecific IgG pool from rabbit was used as a negative control. DNA was amplified with primers encompassing the ANGPTL4 PPRE. Relative amounts of amplified DNA in immunoprecipitates were calculated by comparison with 1% of input DNA. Results are expressed as percentage input and represent averages of triplicates (± S.D).
Induction of the PPARβ/δ Target Gene Angptl4 by 15-HETE Is Dependent on PPARβ/δ.
To obtain unequivocal evidence that Angptl4 induction by 15-HETE is indeed mediated by PPARβ/δ, we performed a series of experiments using genetically modified fibroblasts or siRNA technology. First, we analyzed the effect of 15-HETE on Angptl4 expression in mouse fibroblasts with disrupted Pparb/d alleles and in cells with restored PPARβ/δ expression. For this experiment, fetal lung fibroblasts from Pparb/d-null mice were infected with a retrovirus expressing FLAG-tagged PPARβ/δ or a control retrovirus (pLPCX). The data in Fig. 7 show a clear PPARβ/δ-dependent induction of the Angplt4 gene by 0.3 μM GW501516 (6.9-fold), 10 μM 15-HPETE (5.6-fold), and 10 μM 15-HETE (10.7-fold), respectively, but only a marginal effect (1.5-fold) by 20 μM AA (Fig. 7B). In the Pparb/d-null cells infected with the control vector, expression of Angptl4 was not induced by any of the compounds (Fig. 7A). As expected, the PPARα ligand GW1929 and the PPARγ ligand GW7647 showed no detectable regulation in either cell line.
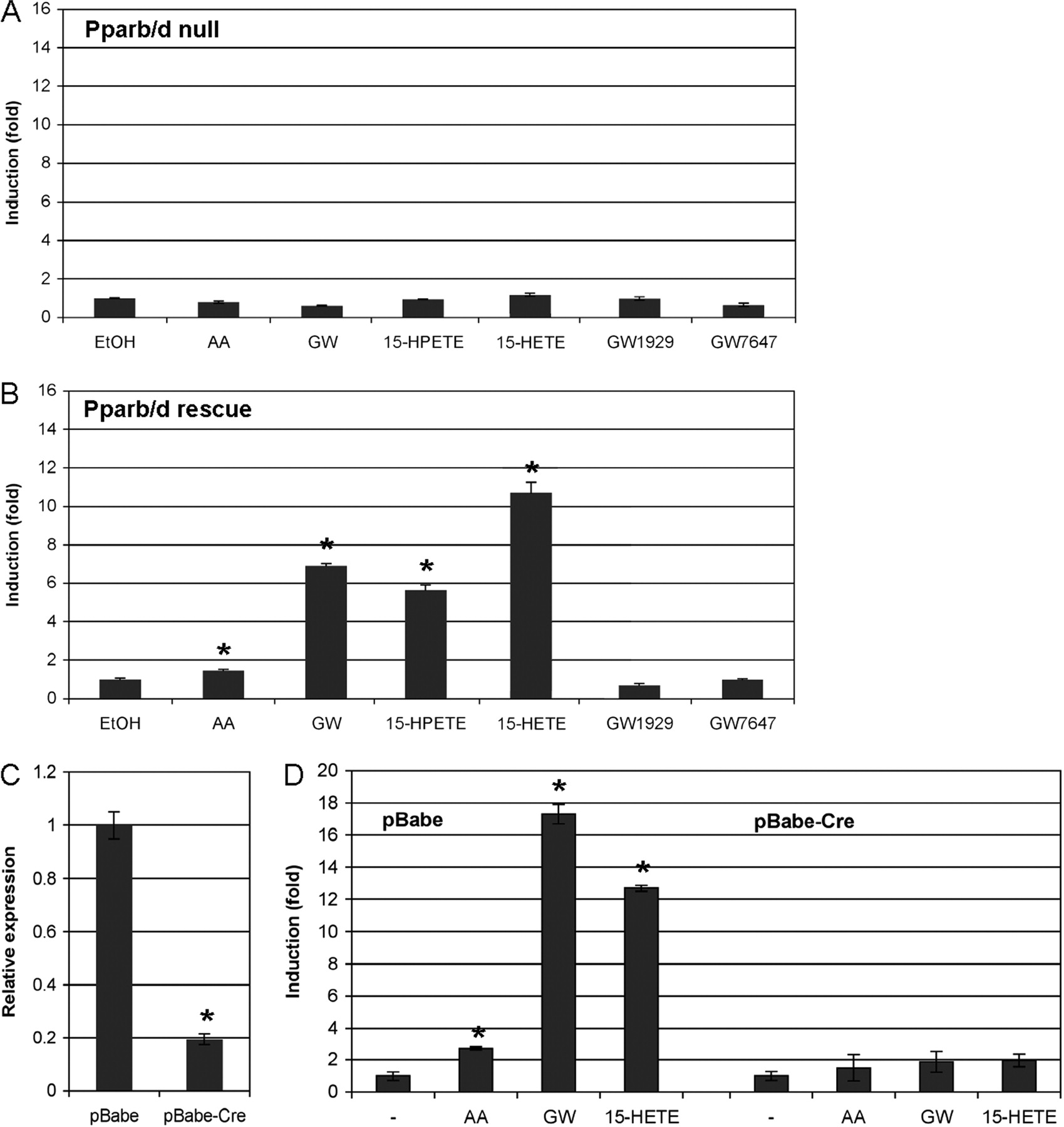
Expression of the PPARβ/δ target gene Angptl4 in fibroblasts lacking PPARβ/δ (A), in the same cells with restored PPARβ/δ expression (B) and after the retrovirus-mediated deletion of floxed Pparb/d alleles (C and D). Fetal lung fibroblasts from Pparb/d-null mice (Müller-Brüsselbach et al., 2007; Peters et al., 2000) were infected with a control retrovirus (pLPCX; A) or a retrovirus expressing FLAG-tagged PPARβ/δ (3xFlag-Pparb; B). C and D, fetal lung fibroblasts established from a mouse strain with a floxed Pparb/d allele (Barak et al., 2002) were infected with either the empty retroviral vector (pBabe; left) or a Cre-expressing retrovirus (pBabe-Cre; right) (Li et al., 1997). C, reduction of Pparb/d expression after deletion of the floxed alleles. D, effect of Pparb/d deletion on Angptl4 expression. Cells were treated with solvent (EtOH), AA (20 μM), 15-HPETE (10 μM), 15-HETE (10 μM), GW501516 (GW; 0.3 μM), the selective PPARγ ligand GW1929 (0.3 μM), or the selective PPARα ligand GW7647 (0.3 μM) for 6 h and analyzed for expression of Angptl4 (A, B, and D) and Pparb/d (C) by qPCR. Induction values were calculated relative to solvent-treated cells and represent averages of triplicates (± S.D) normalized. *, values significantly different (P < 0.001) from vector control (A) or pBabe (C), or between solvent (EtOH) and ligand-treated cells (D).
We also performed the converse experiment by making use of fetal lung fibroblasts we established from a mouse strain with a floxed Pparb/d allele (Barak et al., 2002). The floxed alleles can be deleted in cell culture by a Cre-expressing retrovirus (Li et al., 1997). In these cells, the Angptl4 gene was clearly inducible by AA (2.7-fold), which was reduced to a 1.5-fold induction after Cre-mediated deletion of Pparb/d (Fig. 7D). However, a considerably stronger effect was seen with both 15-HETE and GW501516. These compounds induced Angptl4 expression in the control cells infected with the empty retroviral vector (pBabe) by 13- and 17-fold, respectively. In the Cre virus-infected cells, these values were reduced to 2.0- and 1.9-fold, respectively (Fig. 7D).
Finally, we analyzed the effect of 15-HETE on the Angptl4 gene after siRNA-mediated knockdown of PPARβ/δ expression in NIH3T3 cells. Figure 8A shows a 67.4% reduction of Pparb/d mRNA in Pparb/d siRNA-treated cells compared with cells exposed to control siRNA. PPARβ/δ knockdown led to an increased activity of the Angptl4 gene (Fig. 8A, left), presumably because of reduced PPARβ/δ repressor complex recruitment (see also Fig. 4A). Treatment with GW501516, 15-HPETE, or 15-HETE led to the expected Angptl4 induction in the control siRNA-treated cells (4.0-, 1.9-, and 3.0-fold, respectively; Fig. 8B), which was reduced by approximately 50% upon exposure to Pparb/d siRNA, thus confirming Pparβ/δ as the target of GW501516, 15-HPETE, and 15-HETE in the regulation of the Angptl4 gene.

Effect of siRNA-mediated PPARβ/δ knockdown on Angptl4 inducibility in NIH3T3 cells. A, efficacy of siRNA treatment. Cells exposed to Pparb/d-specific siRNA show a 67% reduction of Pparb/d expression and an increased basal Angptl4 mRNA expression (presumably due to the removal of repressor complexes). B, effect of control and Pparb/d siRNA treatment on Angptl4 induction by GW501516 (0.3 μM), 15-HPETE (10 μM), and 15-HETE (10 μM). Cells were treated for 3 h, and RNA levels were quantified by qPCR. Induction values were calculated relative to solvent treated cells and represent averages of triplicates (± S.D) normalized. *, values significantly different (P < 0.001) between control and Pparb/d siRNA-treated cells.
Both Enantiomers of 15-HETE Activate PPARβ/δ.
There are two enantiomers of 15-HETE that are produced by different synthetic pathways, including 15-LOX, cytochrome P450-dependent mechanisms, and aspirin-triggered acetylated COX-2 (Clària et al., 1996; Clària and Serhan, 1995; Gilroy, 2005; Romano, 2006; Titos et al., 1999). The 15S-HETE and 15R-HETE enantiomers can be further converted to lipoxins (LXA4, LXB4) and 15-epi-lipoxins (15-epi-LXA4, 15-epi-LXB4), respectively. We therefore sought to investigate these compounds for their ability to induce the transcriptional activity of PPARβ/δ.
We first addressed this question using the TR-FRET-based competitive ligand binding assay. The data in Fig. 3B clearly show that both 15-HETE enantiomers were able to compete for binding to PPARβ/δ to an extent very similar to that of the enantiomer mixture [15-HETE; also referred to as (±)15-HETE]. In contrast, no significant effect was observed with any of the lipoxins or epi-lipoxins tested.
We next analyzed the inducibility of the transcriptional activity of PPARβ/δ in the LexA-based luciferase assay. Consistent with the data obtained by TR-FRET, both enantiomers induced PPARβ/δ activity to a similar extent (2.5- to 3-fold), whereas the lipoxin LXA4 showed no effect (Fig. 4, B and C). Finally, we tested the effect of 15S-HETE and 15R-HETE on the induction of the PPARβ/δ target gene Angptl4 in NIH3T3 fibroblasts. Both enantiomers induced Angptl4 expression to an extent similar to that of 15-HETE (Fig. 5A). Taken together, these observations strongly suggest that both 15S-HETE and 15R-HETE are agonistic ligands of PPARβ/δ.
Effect of 15-HETE on Different PPAR Subtypes.
Several previous studies have reported an activation of PPARγ by 15-HETE (Nagy et al., 1998; Huang et al., 1999; Schild et al., 2002; Chen et al., 2003). It was therefore of interest to compare the effects of 15-HETE on PPARγ and PPARβ/δ in some of the assays used in the present study. We first addressed this question by analyzing the effect of 15-HETE on the transcriptional activation by the PPARγ and PPARβ/δ LBDs fused to the LexA DNA binding domain. As shown in Fig. 9, 15-HETE significantly activated all PPAR subtypes, but the induction of PPARβ/δ was stronger (∼11-fold) compared with the activation of PPARγ (∼3-fold) and PPARα (∼2-fold). Consistent with this finding, in vitro recruitment of the thyroid hormone receptor-associated protein coactivator peptide to PPARα and PPARγ was detectable but weaker than the interaction with PPARβ/δ (Fig. 10A). However, a precise quantitative comparison in this assay is difficult, because the exact concentrations of functional recombinant PPARs and their occupation by lipid ligands cannot be determined and may thus differ among the three subtypes.

PPAR subtype-selective transcriptional activation by 15-HETE. NIH3T3 were transiently transfected with an expression vector encoding a LexA-PPARα, LexA-PPARβ/δ, or LexA-PPARγ fusion protein together with a LexA-responsive luciferase reporter plasmid (7L-TATAi). Cells were incubated with 15-HETE at the indicated concentrations or with synthetic ligands (1 μM) for 24 h before harvesting. Control cells (−) received solvent only. Values represent the average of triplicates; error bars show the standard deviation. Significant differences between control cells and 15-HETE treated cells are indicated by an asterisk (Bonferroni-Holm adjusted t test).

PPAR subtype selective coactivator peptide recruitment in vitro by 15-HETE. A, comparison of 15-HETE induced binding of TRAP220 coactivator peptide to the PPARα, PPARβ/δ, and PPARγ LBDs. Data are shown as the ratio of fluorescence intensity at 520/495 nm relative to samples without added ligand. The results thus reflect the factor of increased coactivator peptide binding after 15-HETE addition and allow for a direct comparison of the results obtained with different PPAR subtypes. B and C, ligand-induced binding of four different coactivator derived peptides to the PPARβ/δ and PPARγ LBD. GW501516 and GW1929 were used at a concentration of 0.3 μM and 15-HETE at 31.2 μM. Results are expressed as the ratio of fluorescence intensity at 520/495 nm (as in Fig. 2). All data points represent averages of triplicates (± S.D). Significant differences between solvent and ligand treated cells are indicated by an asterisk (paired t test; P < 0.05).
We also analyzed the 15-HETE-triggered interaction of PPARβ/δ and PPARγ with C33 and three other coactivator peptides (PGC1α, CBP, PPARγ-interacting protein). Again, the 15-HETE induced interaction with PPARγ was much weaker compared with the recruitment of all coactivator peptides to PPARβ/δ (Fig. 10, B and C). As expected, the synthetic ligands GW1929 and GW501516 included as positive controls strongly induced the recruitment of all four peptides to PPARγ and PPARβ/δ, respectively (Fig. 10, B and C).
Discussion
Several synthetic drugs with strong agonistic properties and high subtype selectivity have been described for PPARβ/δ (Peraza et al., 2006), but its regulation by natural ligands remains unclear. The description of PGI2 (prostacyclin) (Gupta et al., 2000; Hatae et al., 2001) and all-trans retinoic acid (Schug et al., 2007) as PPARβ/δ agonists are in conflict with data published by us (Fauti et al., 2006; Rieck et al., 2008) and by others (Yu et al., 1995; Forman et al., 1996; Borland et al., 2008). Unsaturated fatty acids have been reported to interact with the PPARβ/δ LBD (Forman et al., 1996; Xu et al., 1999), but their effect on the transcriptional activity of PPARβ/δ is weak (Rieck et al., 2008). Among the fatty acids with a clearly activating effect on PPARβ/δ is AA, but it is unknown whether this results from a direct interaction with the PPARβ/δ LBD or from the formation of a more potent metabolite. In the present study, we addressed this question and have identified a novel AA-dependent but Cox-independent pathway regulating the transcriptional activity of PPARβ/δ.
Using fusion constructs of the PPARβ/δ LBD and heterologous DNA binding domains (LexA, Gal4), we could show that the agonistic effect of AA is LOX-dependent. The pan-LOX inhibitors NDGA (Fig. 1A) and esculetin (data not shown) and the 12- and 15-LOX inhibitor EDBCA (Fig. 1B) clearly diminished the induction of PPARβ/δ by AA. These data demonstrate that the agonistic effect of AA is not due merely to its direct interaction with the PPARβ/δ LBD.
To identify the active metabolite(s) generated by lipoxygenase pathway(s), we analyzed by TR-FRET various LOX-generated AA metabolites for their potential to trigger the interaction of recombinant PPARβ/δ with coactivators peptides in vitro (Fig. 2). This experiment identified 15-HETE as the only metabolite tested that was clearly superior to nonmetabolized AA. However, in an in vitro ligand binding assay, 15-HETE was only marginally more efficient than AA (Fig. 3A), suggesting that the stronger coactivator binding is not attributable to a higher affinity for the PPARβ/δ LBD but rather to a specific conformational change induced by 15-HETE.
The agonistic effect of 15-HETE was selective for PPARβ/δ, because 1) the interaction of PPARβ/δ with the direct precursor of 15-HETE, the 12/15-LOX product 15-HPETE, was considerably weaker, 2) HETE-derived lipoxins LXA4 and LXB4 showed a weaker binding to recombinant PPARβ/δ in vitro than 15-HETE itself (Fig. 3), and 3) the 15-HETE-triggered activation of a LexA-PPARβ/δ and the 15-HETE-induced coactivator peptide recruitment to PPARβ/δ were significantly stronger relative to PPARα and PPARγ (Figs. 9 and 10). This subtype selectivity is in apparent contrast to published data suggesting that 15-HETE activates the transcriptional activity of PPARγ (Nagy et al., 1998; Huang et al., 1999; Schild et al., 2002; Chen et al., 2003), which was also seen in our study, albeit to a lesser extent compared with PPARβ/δ (Fig. 9). In contrast to our work, the studies quoted above did not show a direct interaction of 15-HETE and PPARγ. It can therefore not be excluded that the described effects of 15-HETE on PPARγ are, at least in part, indirect. In addition, the effect of 15-HETE on PPARβ/δ was not assessed in the published studies so that a direct comparison with our data are not possible.
We next performed a series of experiments confirming 15-HETE as an efficient inducer of endogenous PPARβ/δ target genes using the Angptl4 gene as a verified model (Mandard et al., 2004). At a concentration of 3 μM, 15-HETE induced the Angptl4 gene to an extent similar to that of the synthetic agonist GW501516 at 0.3 μM, a concentration leading to maximal induction by this compound (Fig. 5). Consistent with a function as a PPAR agonist 15-HETE triggered the recruitment of PPARβ/δ and the coactivator CBP to PPRE in the ANGPTL4 gene in the human myofibroblast cell line WPMY-1 (Fig. 6A), and triggered the acetylation of histone H4 at the transcription start site of the gene (Fig. 6B). These alterations were basically indistinguishable from those induced by the established PPARβ/δ ligand GW501516. Furthermore, we were able to demonstrate the PPARβ/δ dependence of the 15-HETE-mediated induction of the Angptl4 gene by comparing PPARβ/δ-expressing cells, Pparb/d-null cells, and siRNA-treated cells (Figs. 7 and 8). Taken together, these data provide compelling evidence that 15-HETE functions as an activating ligand for PPARβ/δ.
Another important question concerns the physiological relevance of the proposed 15-HETE/PPARβ/δ signaling pathway. The highest concentration of 15-HETE measured in the culture supernatant of NIH3T3 cells was approximately 200 nM (Table 1), which is close to the concentration that elicits an effect on ANGPTL4 in HaCAT cells (Fig. 5B). However, such comparisons must be considered with great caution, because the concentration reached in the culture medium clearly depends on the time of culture, the cell number, and the volume of the medium. In addition, cell types other than NIH3T3 may synthesize higher amounts of 15-HETE. Moreover, we do not yet know in which physiological scenario the 15-HETE/PPARβ/δ signaling pathway plays a role: Is signaling paracrine? What would be the interacting cell types? Or does this pathway not involve the release of 15-HETE and instead functions intracellularly? What is the role of fatty acid binding proteins, which may increase local concentrations of 15-HETE in a particular subcellular microenvironment? For these reasons, the issue of physiological relevance cannot be completely clarified at present, but this applies to most other studies addressing the role of endogenous PPAR ligands. However, the sensitivity of the response of HaCaT cells to 15-HETE referred to above provides a strong argument to believe that the regulation of PPARβ/δ by 15-HETE is indeed physiologically relevant.
The 15-HETE used for all experiments discussed so far is a mixture of two enantiomers, 15S-HETE and 15R-HETE, that are produced by different pathways. 15S-HETE is formed by a physiological two-step process, involving the 15-LOX-catalyzed oxydation of AA to 15S-HPETE, which is then enzymatically converted to 15S-HETE. 15R-HETE, on the other hand, is produced from AA by a cytochrome P450-dependent mechanism or by aspirin-triggered acetylated COX-2 (Clària et al., 1996; Clària and Serhan, 1995; Gilroy, 2005; Romano, 2006; Titos et al., 1999). In view of these differences in the synthetic pathways, it was important to show that both enantiomers are able to interact with PPARβ/δ (Fig. 3B) and to induce its transcriptional activity (Figs. 4, B and C, and 5A). These results clearly indicate that 15-HETE enantiomers generated by different pathways function as agonistic ligands for PPARβ/δ, which provides further evidence for a central role of PPARβ/δ in eicosanoid-regulated signaling. In this context, it is tempting to speculate that PPARβ/δ may also play a role in mediating the anti-inflammatory effects of aspirin, which would be in agreement with the reported anti-inflammatory function of 15-HETE (van Dijk et al., 1993). This hypothetical connection between aspirin and PPARβ/δ through 15R-HETE will be addressed in future studies.
Even though their role in tumorigenesis is controversial, PPARβ/δ and synthetic PPARβ/δ agonists have been reported to inhibit cancer cell proliferation in multiple studies (for review, see Müller et al., 2008a,b; Peters and Gonzalez, 2009). It is noteworthy that 15-HETE has also been linked to cell proliferation, both in mitogenic and antimitogenic compounds (Moreno, 2009), but its effect on tumor cell proliferation is invariably inhibitory, including prostate cancer (Shappell et al., 2001), lung cancer (Clària et al., 1996), and myeloid leukemia cells (Mahipal et al., 2007), which may point to another link between 15-HETE and PPARβ/δ. In this context, it is also noteworthy that the agonistic effect of 15-HETE on PPARβ/δ was particular high is keratinocytes (HaCaT cells; Fig. 5B), where a growth-inhibitory role for PPARβ/δ is well established (Michalik et al., 2001; Kim et al., 2006; Burdick et al., 2007; Borland et al., 2008; Girroir et al., 2008; Chong et al., 2009).
Acknowledgments
We are grateful to Dr. Jeff M. Peters (Pennsylvania State University, University Park, PA) for Pparb/d-null mice, Dr. Ronald M. Evans (Salk Institute, La Jolla, CA) for Pparb/dck mice and the pCMX-mPparb/d and Gal4-mPparb/d plasmids, Dr. T. Blankenstein (Max-Delbrück-Centrum für molekulare Medizin, Berlin, Germany) for providing the retroviral Cre vector, and Dr. Aria Baniahmad (Friedrich-Schiller-University, Jena, Germany) for expression vectors.
Footnotes
-
This work was supported by the Deutsche Forschungsgemeinschaft [Grants SFB-TR17/A3, Se263/17-1] and the Landes-Offensive zur Entwicklung wissenschaftlich-ökonomischer Exzellenz-Schwerpunkt “Tumor and Inflammation” of the state of Hesse.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.060541.
-
ABBREVIATIONS:
- PPAR
- peroxisome proliferator-activated receptor
- RXR
- retinoid X receptor
- PPRE
- peroxisome proliferator responsive element
- CBP
- cAMP response element-binding protein binding protein
- GW501516
- 2-[2-methyl-4-([4-methyl-2-[4-(trifluoromethyl)phenyl)-1,3-thiazol-5-yl]methylsulfanyl]phenoxy]acetic acid
- L165,041
- [4-[3-(4-acetyl-3-hydroxy-2-propylphenoxy)propoxy]phenoxy]acetic acid
- AA
- arachidonic acid
- LBD
- ligand binding domain
- PG
- prostaglandin
- 15R-HETE
- 15(R)-hydroxy-5Z,8Z,11Z,13E-eicosatetraenoic acid
- 15S-HETE
- 15(S)-hydroxy-5Z,8Z,11Z,13E-eicosatetraenoic acid
- HPETE
- hydroperoxyeicosatetraenoic acid
- COX
- cyclo-oxygenase
- LOX
- lipoxygenase
- NDGA
- nordihydroguaiaretic acid
- EDBCA
- ethyl-3,4-dihydroxy-benzylidene-cyanoacetate
- GW1929
- N-(2-benzoylphenyl)-O-[2-(methyl-2-pyridinylamino)ethyl]-l-tyrosine hydrochloride
- LX
- lipoxin
- GW7647
- 2-[[4-[2-[[(cyclohexylamino)carbonyl](4-cyclohexylbutyl)amino]ethyl]phenyl]thio]-2-methylpropanoic acid
- CHO
- Chinese hamster ovary
- DMEM
- Dulbecco's modified Eagle's medium
- FCS
- fetal calf serum
- PCR
- polymerase chain reaction
- qPCR
- quantitative polymerase chain reaction
- siRNA
- small interfering RNA
- PIPES
- piperazine-N,N′-bis(2-ethanesulfonic acid)
- NP40
- nonidet P40
- TR-FRET
- time-resolved fluorescence resonance energy transfer
- LC
- liquid chromatography
- TATAi
- TATA initiator module
- PGC1α
- peroxisome proliferator-activated receptor γ coactivator 1α
- Angptl4
- angiopoietin-like 4
- Cre
- cyclization recombination.
- Received August 27, 2009.
- Accepted November 10, 2009.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}