


Abstract
Docosahexaenoic acid (DHA; n-3, 22:6) is known to have anticancer activity, but its mechanisms of action remain to be further elucidated. We recently demonstrated that DHA down-regulates superoxide dismutase (SOD) 1 gene expression, thereby weakening cellular antioxidant forces and enhancing cytotoxicity in various human cancer cells. The objective of this study was to investigate the mechanism of the inhibitory effect of DHA on SOD-1 gene expression in human cancer cells. A reporter gene assay indicated that DHA suppresses SOD-1 gene transcription in a time- and concentration-dependent manner in human cancer cells. Pretreatment with vitamin E did not block the inhibitory effect of DHA, indicating that this suppression does not depend on lipid peroxidation. The suppressive effect of DHA on SOD-1 gene transcription could be mimicked by the peroxisome proliferator-activator receptor (PPAR) α ligand clofibrate but not the PPARγ ligand troglitazone, suggesting the involvement of PPARα signaling. Deletion analysis of the key DNA binding elements in the SOD-1 gene promoter identified the distal hypoxia response element (HRE), but not the peroxisome proliferator response element or nuclear factor-κB element, as essential for the suppressive effects of DHA. Coimmunoprecipitation confirmed that PPARα, but not PPARγ, forms a complex with hypoxia-inducible factor (HIF)-2α in cancer cells. Chromatin immunoprecipitation analysis indicated that both DHA and clofibrate reduce HIF-2α binding to the HRE. Thus, we have identified the distal HRE in the SOD-1 gene promoter that mediates the suppression on the transcription of this gene by DHA, and we have demonstrated the involvement of PPARα and HIF-2α signaling in this event.
DHA has been shown to induce anticancer activity in multiple experimental model systems. Many cellular mechanisms have been proposed to play a role in DHA-induced cell death or growth inhibition. Lipid peroxidation is one well established mechanism to explain the action of DHA on cancer cells. Accumulation of the lipid peroxidation products causes peroxidative damage, ultimately leading to the death of cells. The involvement of lipid peroxidation in DHA-induced growth inhibition and cytotoxicity of cancer cells was demonstrated by the measurement of the peroxidative products in DHA-treated cells and by the abilities of antioxidants, such as vitamin E, to block the cytotoxic effects of DHA (Gonzalez, 1995). Moreover, the killing of malignant cells by DHA can be accelerated by increasing cellular oxidative stress (Bégin et al., 1988).
The balance between oxidants and antioxidants is critical for biological systems to maintain normal function. In eukaryotes, antioxidant defenses are provided by small molecules, such as glutathione, vitamin E, or vitamin C, and enzymes, such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (Pacifici and Davies, 1991). SOD and catalase reduce superoxide anions and hydrogen peroxides, respectively. Glutathione peroxidase uses glutathione to reduce hydrogen peroxides. The majority of the total cellular SOD consists of SOD-1, which is mainly located in the cytoplasm and nucleus and requires copper and zinc for its activity (Crapo et al., 1992). SOD-2 is located primarily in the mitochondrial matrix and requires manganese for its activity. Both overexpression and underexpression of SOD-2 have been described in various types of human cancer (Oberley and Oberley, 1997; Kinnula and Crapo, 2004), and it has been suggested that SOD-2 is a tumor suppressor gene. Because all cells actively produce superoxide during routine metabolic processes, the altered expression of SOD-2 in many cancer cells (Fridovich, 1995) suggests that malignant cells may be more dependent for survival upon SOD-1 than normal cells. If so, targeted inactivation of SOD-1 is likely to result in preferential tumor cell killing. Indeed, a well designed study demonstrated that drug therapy, believed to inhibit SOD-1, results in increased levels of superoxide, free radical-mediated damage to mitochondria, and induction of apoptosis in cancer cells (Huang et al., 2000). Furthermore, inhibition of SOD-1 attenuated angiogenesis and selectively induced apoptosis of tumor cells (Juarez et al., 2006).
The rat and human SOD-1 gene promoters contain PPREs (Yoo et al., 1999; Rojo et al., 2004). There is considerable evidence indicating that DHA is a natural ligand for PPARs, which bind to PPREs and regulate expression of several genes (Desvergne and Wahli, 1999). Additional transcription factors that might be implicated in regulation of SOD-1 expression by DHA are HIF and NF-κB, which have DNA binding elements present in the human SOD-1 gene promoter (Rojo et al., 2004). Our recent results indicate that DHA down-regulates expression of the SOD-1 gene, thereby enhancing oxidative stress and more effectively killing cancer cells (Ding et al., 2004). The down-regulation of SOD-1 gene expression by DHA was observed several human cancer lines (Ding and Lind, 2007). This indicates that not only could DHA initiate lipid peroxidation due to its possessing multiple double bonds, which increases its oxidation potential, but also that DHA could suppress the antioxidant enzyme system, thereby enhancing oxidative stress. To understand how DHA regulates gene expression of antioxidant enzymes, the present study investigated the detailed cellular mechanisms of DHA-induced suppression of SOD-1 gene expression in human cancer cells. The A2780 line, a well established ovarian cancer line, was chosen as our model system because it is one of the solid tumor lines showing a down-regulation of SOD-1 expression by DHA (Ding and Lind, 2007). We report here that DHA targets SOD-1 gene transcription through an interaction of HIF-2α and PPARα signaling that acts via the distal HRE in the SOD-1 gene promoter.
Materials and Methods
Materials. Dual-Luciferase Reporter kit was purchased from Promega (Madison, WI). Antibodies were obtained from the following resources: PPARα, PPARγ, and HIF-1α were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was from ProMab Biotechnologies, Inc. (Albany, CA); and HIF-2α was from Novus Biologicals, Inc. (Littleton, CO). The pGL-3-HRE-Lu reporter construct was kindly provided by Dr. Konstantin Salnikow (Radiation Oncology Branch, National Cancer Institute, Frederick, MD) (Salnikow et al., 1999; Surazynski et al., 2008). The QuikChange site-directed mutagenesis kit was from Stratagene (La Jolla, CA). 4-Hydroxynonenal (4-HNE) was from Cayman Chemical (Ann Arbor, MI). DHA, clofibrate, troglitazone, lipopolysaccharide (LPS), and other reagents were analytic grade and were purchased from Sigma-Aldrich (St. Louis, MO).
Cell Culture. The ovarian carcinoma cell line A2780 was provided by Dr. Stephen Howell (University of California, San Diego, CA). Cells were cultivated in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin, at 37°C in humidified air with 5% CO2. The cells were subcultivated twice a week in 75- or 150-mm flasks following a protocol by American Type Culture Collection (Manassas, VA). Other cell lines, including Panc-1 (pancreatic cancer), MCF-7 (breast cancer), Raji (lymphoma), and CEM (leukemia), were from American Type Culture Collection and cultured according to American Type Culture Collection's instructions.
DNA Deletion. The SOD-1 promoter reporter constructs were kindly provided by Dr. Antonio Guadrado (Autonoma University of Madrid, Madrid, Spain) (Rojo et al., 2004). These include p1499-sod1, p750-sod1, p552-sod1, and p355-sod1. The QuikChange site-directed mutagenesis kit (Stratagene) was used to delete DNA binding elements present in the p1499-sod1 promoter, including NF-κB and HRE. The deletion was performed according to the manufacturer's protocol. The primers used for the deletions are as follows: NF-κB-del forward primer, 5′-GAAAATTGCAGGGGAAGGATTGAGGTGTAGCGAC-3′; NF-κB-del reverse primer, 5′-GTCGCTACACCTCAATCCTTCCCCTGCAATTTTC-3′; HRE-del forward primer, 5′-GCCAGACAAAAACGCTCTGTAGGGTTGTGG-3′; and HRE-del reverse primer, CCACAACCCTACAGAGCGTTTTTGTCTGGC-3′. The sequence containing a putative NF-κB (5′-AAAAGGTAAGTCCCGG-3′, -480 to -464 of the human SOD-1 gene promoter; Kim et al., 1994) and a putative HRE (5′-AGGTGATGCCTAGAAGCCAACTAGTTGCCGTTTGGTTA-3′, -626 to -589 of the human SOD-1 gene promoter; Kim et al., 1994) were deleted as confirmed by DNA sequencing.
Transient Transfection and Luciferase Activity Assay. A2780 cells (2 × 106) were plated in 100-mm cell culture dishes with 8 ml of RPMI 1640 medium. The cells proliferated to 60 to 80% confluence after 24 h of culture. Then, 3 μg of DNA of the reporter constructs was transfected into A2780 cells using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. In some cases, an additional reporter construct encoding Renilla reniformis luciferase was cotransfected with the SOD-1 gene promoter constructs to determine the transfection efficiency in each well. Twenty-four hours after the transfection, cells were lifted and randomly plated into 24-well plates at 250,000/well. This ensures a randomly even distribution of transfected cells. At 48 h of transfection, cells were treated with DHA, clofibrate, troglitazone, or other reagents at indicated concentrations and durations. Luciferase activity was assayed using the Dual-Luciferase Reporter kit from Promega. In short, cells were washed with cold phosphate-buffered saline twice and harvested by adding 100 μl of lysis buffer. The lysate was quickly centrifuged at 13,000g for 1 min, and the insoluble material was removed. The firefly luciferase reporter was measured first by adding Luciferase Assay Reagent II (50 μl) to the cell extracts from each sample (50 μl). The R. reniformis luciferase reaction was initiated sequentially by adding Stop and Glo Reagent to the same sample. The firefly luciferase activity was normalized by the R. reniformis luciferase activity or by the amount of protein used for each determination. The data are expressed as percentages of luciferase activity detected in untreated cells.
Western Blot Analysis. The protein samples (40 μg each) were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto polyvinylidene difluoride membranes. After overnight blocking in 5% milk at 4°C, blots were incubated with primary antibodies (PPARα, PPARγ, HIF-1α, HIF-2α, and GAPDH) at appropriate dilution ratios for 2 h at 22°C. After three 10-min washes with Tris-buffered saline with 0.1% Tween 20, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at 22°C. Chemiluminescent substrate (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) was used to detect binding of the primary antibodies to their cognate antigens, and images were captured using an X-ray developer (SRX-101; Konica Minolta, Tokyo, Japan).

DHA down-regulates human SOD-1 promoter activity. The p1499-sod1 reporter construct was transfected into A2780 cells, and the cells were treated with 100 μM DHA for 1, 4, and 20 h (A), with various concentrations of DHA for 4 h (B), or with 100 μM linolenic acid (LA) (n-6; 18:2) for 1 and 4 h (C). Panc-1 cells were also transfected and treated with 150 μM DHA for 4 h (D). The cell lysates were prepared, and luciferase activity was assayed. Data are expressed as percentages of untreated cells (bars, S.E.M.; n = 3). ⋆, P < 0.01, compared with untreated cells, using one-way ANOVA followed by Dunnett's analysis.

Vitamin E blocks the cytotoxicity of DHA but not inhibition of SOD-1 promoter activity. A2780 cells were pretreated with 100 μM vitamin E (Vit E) for 15 min before addition of 100 μM DHA for 4 h (A) or various concentrations of DHA for 72 h (C). Cells were also treated with 100 μM 4-HNE for 4 h (B). Luciferase activity and cell viability were assayed as described under Materials and Methods. Data are expressed as percentages of untreated control cells (bars, S.E.M.; n = 3).

Clofibrate down-regulates human SOD-1 promoter activity. The p1499-sod1 reporter construct was transfected into A2780 cells, and the cells were treated with 500 μM clofibrate, 20 μM troglitazone, or 1 μg/ml LPS for 16 h. The cell lysates were prepared, and luciferase activity was assayed. Data are expressed as percentages of untreated cells (bars, S.E.M.; n = 3). ⋆, P < 0.01, compared with untreated cells, using one-way ANOVA followed by Dunnett's analysis.

Human SOD-1 gene promoter. Top, a diagram showing different DNA fragments of the human SOD-1 gene promoter that have been cloned into pGL-3 luciferase reporter construct. Major DNA binding elements in the promoter are indicated. Bottom, relative promoter activity detected in A2780 cells. Cells were transfected with different SOD-1 promoter reporter constructs along with the R. reniformis luciferase construct. Luciferase activity was assayed, and the firefly luciferase activity was normalized by the R. reniformis luciferase activity. Data are expressed as percentages of the luciferase activity detected in p1499-sod1-transfected cells (bars, S.E.M.; n = 3).
Coimmunoprecipitation. Cells growing in 100-mm dishes were washed twice with cold phosphate-buffered saline and harvested by adding 150 μl of IP buffer containing 10 mM Tris-HCl, pH 7.4, 50 mM NaCl, 0.5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and 1% Triton X-100. Cells were sonicated for 1 to 3 min on ice and centrifuged at 16,000g for 30 min to remove insoluble material. Supernatants were collected, and protein concentrations were determined. In a typical IP reaction, the supernatant contained approximately 100 μg of protein from one sample. Supernatants were precleared by adding 25 μl of agarose-coupled protein A and gently rotated for 60 min at 4°C. The agarose beads were removed by centrifugation at 2000g for 5 min, and the precleared supernatants were collected. Appropriate primary antibodies or IgG was added to the supernatants (1:100 ratio), and the reaction was incubated at 4°C overnight with gentle rotation. The antibody-protein complexes were captured by adding 50 μl of agarose-coupled protein A and rotating at 4°C for an additional 2 h at 4°C. The supernatants were then removed by centrifuging at 2000g for 5 min. The pellets were washed with IP buffer four times and solubilized with 2× SDS-PAGE sample buffer at room temperature for 60 min. The samples were centrifuged, and the supernatants were collected. The immunoprecipitated proteins were subjected to Western blot analysis.
Chromatin Immunoprecipitation Assay. The ChIP assay was done as described previously (Ding et al., 2006). In brief, A2780 cells were treated with 150 μM DHA, 500 μM clofibrate, or 20 μM troglitazone for 16 h. Formaldehyde was then added directly to the medium (final concentration, 1%) to fix the cells at 37°C for 10 min. Cells were washed, removed from the dish, pelleted, and lysed on ice in the SDS buffer. The lysate was sonicated to shear DNA into sizes ranging from 200 to 1000 bp, and the DNA was precleared overnight at 4°C with salmon sperm DNA-saturated protein A agarose. The complex of protein and DNA was precipitated overnight at 4°C using HIF-2α antibody, and the precipitants were incubated at 65°C for 4 h to reverse cross-links. The precipitants were then digested with protein kinase K, and DNA was extracted with phenol/chloroform and precipitated with ethanol. DNA was analyzed by polymerase chain reaction amplification using primers specific to the promoter regions of interest, and the polymerase chain reaction products were separated on a 1% agarose gel containing ethidium bromide and visualized under ultraviolet light. The primers used for DNA amplification of the distal HRE fragment in the SOD-1 gene promoter were as follows: forward, 5′-GCCTTTAGGCCAGAC-3′ (-649 to -635); and reverse, 5′-CTGAGTTTGGCCACAGCGTC-3′ (-333 to -352 (Kim et al., 1994).
Statistical Analysis. One-way ANOVA was performed to assess differences among groups of data using Prism 4 (GraphPad Software Inc., San Diego, CA).
Results
DHA Down-Regulates SOD-1 Gene Transcription. We recently demonstrated that DHA reduces SOD-1 gene expression at both mRNA and protein levels in human cancer cells (Ding et al., 2004; Ding and Lind, 2007). Because DHA can enter the nucleus and regulate gene transcription (Huang et al., 2002), we hypothesized that DHA down-regulates SOD-1 gene expression by targeting SOD-1 gene transcription regulation. To test this hypothesis, an SOD-1 promoter reporter construct (p1499-sod1) was transfected into A2780 cells, and the effects of various DHA doses and treatment times on promoter activity were measured. DHA inhibited SOD-1 gene promoter activity in a time- and concentration-dependent manner (Fig. 1). The inhibitory effects were most pronounced after 4 h of treatment and lasted for at least 20 h. These results, combined with our previous reports (Ding et al., 2004; Ding and Lind, 2007), clearly show that DHA targets SOD-1 gene transcription in human cancer cells. An additional cell line, Panc-1, was tested to determine whether the inhibitory effects of DHA on SOD-1 gene transcription are detectable in cell lines other than A2780. Clearly, DHA also inhibited SOD-1 gene transcription in this cell line (Fig. 1D).

The distal HRE in the human SOD-1 gene promoter is essential for DHA- and clofibrate-induced suppression of SOD-1 gene transcription. A2780 cells were transfected with p750-sod1, p552-sod1, and p355-sod1 (A) or p1499-sod1, HRE-del, and NF-κB del (B and C) reporter constructs. Cells were treated with 500 μM clofibrate or 100 μM DHA for 16 h. The cell lysates were prepared, and luciferase activity was assayed. Data are expressed as percentages of untreated cells (bars, S.E.M.; n = 3). ⋆, P < 0.01, compared with untreated cells, using one-way ANOVA followed by Dunnett's analysis.
To understand whether the inhibitory effect is DHA-specific, cells were treated with 100 μM linolenic acid (n-6; 18:2), a long-chain n-6 PUFA, for 4 h. This long-chain n-6 PUFA was unable to reduce SOD-1 gene transcription (Fig. 1C). Because lipid peroxidation is a well established mechanism explaining the cytotoxicity of DHA (Gonzalez, 1995; Ding et al., 2004), we tested whether it contributes to the inhibitory effect of DHA on SOD-1 gene transcription. The antioxidant vitamin E blocked the cytotoxicity of DHA (Fig. 2C) but did not alter DHA-induced suppression of SOD-1 gene transcription (Fig. 2A), suggesting that the lipid peroxidation products do not contribute to the mechanism by which DHA represses SOD-1 expression. This was further supported by the fact that 4-HNE, a lipid peroxide product, did not suppress SOD-1 gene transcription (Fig. 2B).
PPARα Ligand Mimics the Effects of DHA on SOD-1 Gene Transcription. DHA is an established ligand to PPAR and binding of DHA to PPAR regulates gene transcription in mammalian model systems (Diep et al., 2000). To understand whether PPARα or PPARγ is involved in the inhibitory effect of DHA on SOD-1 gene expression, cells were treated with 20 μM troglitazone, a PPARγ agonist (Adams et al., 1997), and 500 μM clofibrate, a PPARα agonist (Göttlicher et al., 1992), for 20 h. The concentrations of troglitazone and clofibrate were chosen based on previous studies (Canuto et al., 2003; Hashimoto et al., 2004; Yang et al., 2007). Luciferase activity assay indicated that clofibrate suppresses SOD-1 gene transcription, whereas troglitazone does not (Fig. 3), suggesting that PPARα, and not PPARγ, is involved in transcriptional control of SOD-1 gene expression. In contrast, LPS, an activator to the NF-κB signaling pathway, was able to up-regulate SOD-1 gene transcription, a result consistent with a previous report (Marikovsky et al., 2003). There is a putative PPRE in the human SOD-1 gene promoter (Rojo et al., 2004), which probably mediates the suppressive effects of DHA and clofibrate on SOD-1 gene transcription through a PPARα signaling pathway. To test this possibility, A2780 cells were transfected with a series of deletion mutants of the SOD-1 gene promoter reporter constructs, as indicated in Fig. 4. It was surprising that the inhibitory effects of both DHA and clofibrate were evident in cells transfected with the p750-sod1 reporter construct that does not retain the PPRE but that they disappeared in cells transfected with the p552-sod1 that lacks the PPRE and distal HRE or the p355-sod1 reporter that lacks the PPRE, distal HRE, and NF-κB sites (Fig. 5A). This indicates that the PPRE is not required in DHA-induced transcription suppression of the SOD-1 gene. Thus, the critical elements that mediate DHA suppression are located within the 200-bp fragment of the promoter between -755 bp and -552 bp, suggesting that the distal HRE is a likely candidate.

DHA and clofibrate suppress HRE-mediated gene transcription. A2780 cells were transfected with the pGL-3-HRE-Lu reporter construct. Cells were treated with 500 μM clofibrate or 150 μM DHA for 16 h. The cell lysates were prepared, and luciferase activity was assayed. Data are expressed as percentages of untreated cells (bars, S.E.M.; n = 3). ⋆, P < 0.01, compared with untreated cells, using one-way ANOVA followed by Dunnett's analysis.

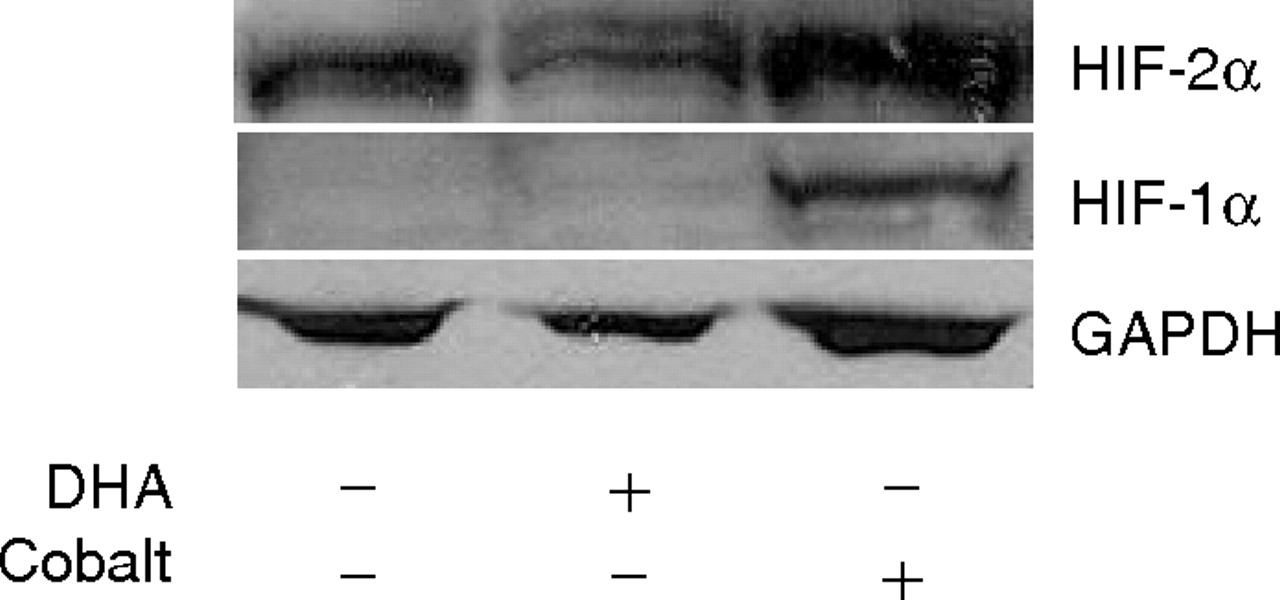
HIF-2α is constitutively expressed in A2780 cells. A2780 cells were treated with 100 μM DHA or 150 μM cobalt for 16 h. The cell lysates were prepared and blotted with antibodies against HIF-1α, HIF-2α, and GAPDH. Shown are representative gels of three separate experiments.

PPARα complexes with HIF-2α in A2780 cells. A2780 cells were treated with 100 μM DHA for 16 h, and cell lysates were prepared. The cell lysates were precipitated with HIF-2α (p-HIF-2α) antibody or IgG (p-IgG) as described under Materials and Methods. The precipitants as well as the total cell lysates were separated onto SDS-PAGE and blotted with antibodies against PPARγ, PPARα, HIF-2α, and GAPDH. Shown are representative gels of three separate experiments.
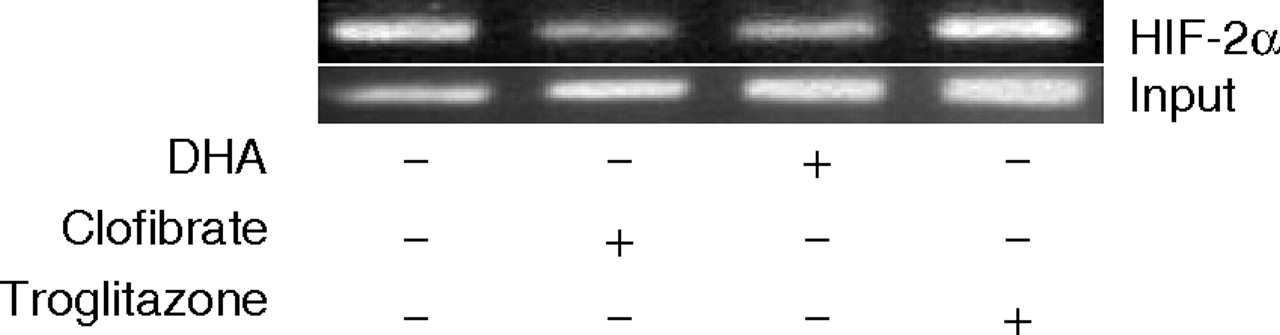
DHA and clofibrate reduce HIF-2α binding to the human SOD-1 gene promoter. A2780 cells were treated with 500 μM clofibrate or 150 μM DHA for 16 h. ChIP assay was performed as described under Materials and Methods. The amplified DNA fragments were separated on 1% agarose gel and visualized under ultraviolet light. Shown are representative gels of two separate experiments.
HRE Mediates the Inhibitory Effects of DHA and Clofibrate on SOD-1 Gene Transcription. To locate the DNA binding elements that mediate the inhibitory effects of DHA and clofibrate on SOD-1 gene transcription, deletions of the NF-κB and distal HRE DNA binding elements in the intact SOD-1 gene promoter were constructed (Fig. 5C). Although deletion of the NF-κB element did not reverse inhibition by DHA and clofibrate, deletion of the distal HRE element completely reversed the inhibitory effects on SOD-1 gene transcription (Fig. 5, A and B), indicating the key role of this HRE in the transcription suppression of the SOD-1 gene by DHA and clofibrate. To further confirm this observation, the pGL-3-HRE-Lu construct (Salnikow et al., 1999) containing only HRE binding elements in the promoter was transfected into A2780 cells, and the effects of DHA and clofibrate on the reporter activity were analyzed. Cobalt chloride was included as a positive control because it enhances HRE-mediated transcription (Bianchi et al., 1999). Consistent with the DHA inhibition of SOD-1 promoter activity in Fig. 5, both DHA and clofibrate suppressed HRE-mediated luciferase activity (Fig. 6).
PPARα Complexes with HIF-2α to Mediate the Effects of DHA on SOD-1 Gene Transcription. Western blot analysis with specific antibodies confirmed that PPARγ and PPARα are expressed in A2780 cells (Tuller et al., 2009). Furthermore, HIF-2α is constitutively expressed in this cell line, whereas HIF-1α is not detectable in the presence and absence of DHA. Treatment of the cells with cobalt induced HIF-1α expression, consistent with a previous report (Bianchi et al., 1999), which serves as a positive control (Fig. 7). To understand whether PPARα might act with HIF-2α to regulate HRE-responsive gene expression, we applied coimmunoprecipitation using cellular proteins extracted from DHA-treated cells. It is interesting that in the HIF-2α precipitants, PPARα, but not PPARγ, was clearly detected (Fig. 8). DHA decreased the levels of HIF2α in ovarian cancer cells, as well as in several other human cancer cell lines, including breast cancer (MCF-7), lymphoma (Raji), and leukemia (CEM) lines (data not shown). Consequently, the interaction of PPARα and HIF-2α was affected by DHA in these cells (Fig. 8). We then performed ChIP analysis to determine whether HIF-2α is capable of binding to the distal HRE of the SOD-1 promoter, thereby regulating SOD-1 gene transcription. A2780 cells were treated with 150 μM DHA and 500 μM clofibrate for 20 h. Specific primers covering the DNA fragment surrounding the distal HRE of the human SOD-1 gene promoter were used to amplify HIF-2α antibody-precipitated DNA. This showed that HIF-2α is able to bind the human SOD-1 promoter, and this binding was indeed significantly attenuated in DHA- and clofibrate-treated cells, indicating that the HIF-2α signaling mediates transcription suppression of the SOD-1 gene (Fig. 9).
Discussion
The present study demonstrates that DHA down-regulates SOD-1 gene transcription through an HRE-mediated mechanism, involving an interaction of PPARα and HIF-2α signaling. This provides a novel mechanism by which DHA weakens the antioxidant status of cancer cells, resulting in enhanced oxidative stress, growth inhibition, or apoptotic cell death (Ding et al., 2004). The concentration of DHA used in the present study has been frequently used in in vitro model systems (Collett et al., 2001; Leonardi et al., 2005; Merendino et al., 2005; Ng et al., 2005). A previous study in human subjects reported that the highest tolerated dose of EPA plus DHA is 188 mg/kg/day orally (Burns et al., 1999), which is considerably higher than the usual (“physiological”) adult intake. There are no data published to indicate what molar concentration of DHA is achieved in plasma and cells when given at these pharmacological doses. However, a recent study reported that daily consumption of 4.4 g of EPA plus DHA over 6 weeks resulted in a plasma DHA concentration at 51 μg/ml (155 μM) in humans, which is three times higher than the basal level (Garg et al., 2006). Based on these previous reports, we believe that the concentrations of DHA used in our study are physiologically relevant.
Targeting SOD-1 has been recognized as a potential strategy for cancer therapy (Huang et al., 2000). We recently demonstrated that DHA suppression of SOD-1 gene expression contributes to its cytotoxicity in human cancer cells (Ding et al., 2004). The cellular mechanisms of the action of DHA that lead to reduced expression of the SOD-1 gene remain unknown. In the present study, we approached this question by focusing on transcriptional regulation of the SOD-1 gene, because DHA is an established ligand to nuclear receptors and is known to regulate gene transcription in mammalian systems (Desvergne and Wahli, 1999). It was found that DHA suppresses SOD-1 gene transcription, whereas linolenic acid (n-6; 18:2) does not affect SOD-1 promoter activity. Furthermore, this suppression of SOD-1 gene transcription could not be reversed by pretreatment of the cells with vitamin E, suggesting that lipid peroxidation products are not involved in this process. These findings support a new model for DHA to exert its cytotoxic effects on tumor cells. On the one hand, DHA is a long-chain n-3 PUFA that has high oxidative potential due to its possession of multiple double bonds (Hawkins et al., 1998). Indeed, lipid peroxidation is a well accepted mechanism of the action of DHA to kill cancer cells (Bégin et al., 1988; Gonzalez, 1995). On the other hand, DHA is also a nuclear receptor ligand that can enter the nucleus by binding to fatty acid-binding proteins (Huang et al., 2002) and modulating gene transcription. It is important that we have shown that DHA reduces expression of antioxidant enzymes, resulting in weakened antioxidant forces and more effective cancer cell killing (Ding et al., 2004). Thus, by initiating lipid peroxidation and inhibiting antioxidant power, DHA is able to more effectively kill cancer cells. Because DHA is a natural product and has potential to be used for cancer prevention or treatment (Bougnoux, 1999; Hardman, 2004), understanding the detailed mechanisms of the action of DHA is important in the development of novel strategies that use DHA as an anticancer agent.
Several DNA binding elements such as PPRE, NF-κB, and HRE are described previously in the human SOD-1 gene promoter, which may contribute to SOD-1 gene transcription regulation (Rojo et al., 2004). Deletion analysis of the human SOD-1 gene promoter enabled us to locate the DNA binding element that is responsible for DHA-induced suppression of SOD-1 gene transcription. Although we initially assumed that the PPRE and the NF-κB DNA binding elements present in the SOD-1 gene promoter are probably involved in the inhibitory effect of DHA on SOD-1 gene expression, the experimental results disagreed with this assumption. In fact, several lines of evidence support the key role of the distal HRE of the SOD-1 gene promoter in mediating the suppressive effect of DHA in our model system. First, although the PPARα ligand clofibrate mimicked the inhibitory effects of DHA on SOD-1 gene transcription, deletion of both the PPRE and the NF-κB elements did not reverse or attenuate the inhibitory effects of DHA and clofibrate. This suggests that these two DNA binding elements are not required for the transcription suppression of the SOD-1 gene by DHA. In addition, deletion of the distal HRE element in the SOD-1 gene promoter completely reversed the inhibitory effects of DHA and clofibrate, indicating the essential role of the HRE in mediating this event. Second, application of the HRE-Lu allowed us to further determine the involvement of this DNA binding element in DHA- and clofibrate-induced transcription suppression. It was found that DHA and clofibrate inhibited HRE-mediated promoter activity to an extent similar to their effects on the SOD-1 gene transcription, providing direct evidence of DHA targeting HRE-mediated transcription. Third, the essential role of HRE in mediating the effects of DHA on the SOD-1 gene was further supported by the findings that HIF-2α binds to the SOD-1 gene promoter as shown via ChIP analysis and that both DHA and clofibrate can reduce this binding. A direct interaction of PPARα, but not PPARγ, with HIF-2α was also evident in this model system. Our experimental results thus suggest that PPARα and HIF-2α signaling regulate the inhibitory effect of DHA on SOD-1 gene transcription via the distal HRE element.
Although the interaction of the PPARα and NF-κB signaling pathways has been well established in several model systems (Staels et al., 1998; Delerive et al., 1999, 2000; Okamoto et al., 2005), the direct interaction of PPARα and HIF proteins to regulate gene transcription has not been described previously. Our results indicate that PPARα and HIF-2α form a complex that might bind to the HREs and regulate gene expression. To the best of our knowledge, this is the first report of physical interaction between the HIF-2α and PPARα transcription factors. This novel observation of cross-talk between the PPAR and HIF signaling pathways provides insight into our understanding of the regulation of the SOD-1 gene. These observations are consistent with a recent study reporting that n-3 PUFAs, including EPA and DHA, down-regulate HIF signaling in colon cancer cells (Calviello et al., 2004), and a previous report indicating that HRE mediates the induction of SOD-1 gene transcription (Yoo et al., 1999).
Footnotes
- Received May 1, 2009.
- Accepted June 15, 2009.
This study was supported by the Oklahoma Center for the Advancement of Science and Technology [Grant HR04-021]; the American Cancer Society [Grant IRG-05-066-01]; and the College of Medicine, University of Oklahoma Health Sciences Center [Grant COM 22134].
ABBREVIATIONS: DHA, docosahexaenoic acid; SOD, superoxide dismutase; PPRE, peroxisome proliferation-responsive element; PPAR, peroxisome proliferator-activated receptor; HIF, hypoxia-inducible factor; NF-κB, nuclear factor-κB; HRE, hypoxia-response element; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; 4-HNE, 4-hydroxy nonenal; LPS, lipopolysaccharide; PAGE, polyacrylamide gel electrophoresis; IP, immunoprecipitation; ChIP, coimmunoprecipitation; bp, base pair(s); ANOVA, analysis of variance; PUFA, polyunsaturated fatty acid; Lu, luciferase; EPA, eicosapentaenoic acid.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}