


Abstract
Multinuclear platinum compounds have been designed to circumvent the cellular resistance to conventional platinum-based drugs. In an attempt to examine the cellular basis of the preclinical antitumor efficacy of a novel multinuclear platinum compound (BBR 3464) in the treatment of cisplatin-resistant tumors, we have performed a comparative study of cisplatin and BBR 3464 in a human osteosarcoma cell line (U2-OS) and in an in vitro selected cisplatin-resistant subline (U2-OS/Pt). A marked increase of cytotoxic potency of BBR 3464 in comparison with cisplatin in U2-OS cells and a complete lack of cross-resistance in U2-OS/Pt cells were found. A detailed analysis of the cisplatin-resistant phenotype indicated that it was associated with reduced cisplatin accumulation, reduced interstrand cross-link (ICL) formation and DNA platination, microsatellite instability, and reduced expression of the DNA mismatch repair protein PMS2. Despite BBR 3464 charge and molecular size, in U2-OS and U2-OS/Pt cells, BBR 3464 accumulation and DNA-bound platinum were much higher than those observed for cisplatin. In contrast, the frequency of ICLs after exposure to BBR 3464 was very low. The time course of ICL formation after drug removal revealed a low persistence of these types of DNA lesions induced by BBR 3464, in contrast to an increase of DNA lesions induced by cisplatin, suggesting that components of the DNA repair pathway handle the two types of DNA lesions differently. The cellular response of HCT116 mismatch repair-deficient cells was consistent with a lack of influence of mismatch repair status on BBR 3464 cytotoxicity. Because BBR 3464 produces high levels of lesions different from ICLs, likely including intra-strand cross-links and monoadducts, the ability of the triplatinum complex to overcome cisplatin resistance appears to be related to a different mechanism of DNA interaction (formation of different types of drug-induced DNA lesions) as compared with conventional mononuclear complexes rather than the ability to overcome specific cellular alterations.
Cisplatin is one of the most effective antitumor agents with a wide spectrum of activity against human solid tumors (Ozols and Young, 1991; Dancey and Le Chevalier, 1997). However, some common tumors are intrinsically resistant to platinum compounds and the frequent development of resistance in responsive tumors is a major clinical problem and cause for failure in the curative therapy.
Modification of platinum-based compounds is a promising approach for the development of noncross-resistant analogs of cisplatin and a large number of mononuclear platinum compounds have been developed as potential candidates for clinical use (Kelland et al., 1992a; Farrell, 1996a). However, the cellular and molecular mechanisms of these analogs are likely similar to that of cisplatin. An alternative option in the design of platinum-based analogs has been the development of multinuclear platinum compounds. A systematic evaluation of these bifunctional DNA-binding agents identified a novel triplatinum complex (BBR 3464) as the most promising complex of this series (Fig.1). A preclinical efficacy study of this drug indicated a complete lack of cross-resistance between BBR 3464 and cisplatin (Pratesi et al., 1997). This observation has been confirmed in a panel of in vitro-selected cisplatin-resistant cell lines (Giuliani et al., 1997).

Structure of the multinuclear platinum compound BBR 3464.
The cellular basis of BBR 3464 efficacy and the relevance of factors implicated in cisplatin resistance as determinants of response to BBR 3464 are still uncertain. Factors contributing to cisplatin resistance include: decreased drug accumulation (Loh et al., 1992; Mistry et al., 1992); increased content of intracellular thiols such as glutathione (GSH) (Fram et al., 1990; Mistry et al., 1991) and metallothioneins (Kasahara et al., 1991); and increased DNA repair and tolerance to DNA damage (Eastman and Schulte, 1988; Kelland et al., 1992b; Johnson et al., 1997). In particular, the loss of mismatch repair has been related to the acquisition of cisplatin resistance (Aebi et al., 1996; Anthoney et al., 1996). Recently, decreased susceptibility to cisplatin-induced apoptosis has been associated with the cisplatin-resistant phenotype of ovarian carcinoma cell systems (Perego et al., 1996). Multiple mechanisms of resistance to cisplatin can be operative at the cellular level and their relative expression is variable among the different tumor cell lines.
This study was undertaken to examine the mechanisms responsible for the ability of the novel complex to overcome cisplatin resistance in a human osteosarcoma cell system. We previously showed that resistance to cisplatin in U2-OS/Pt cells is associated with a reduced susceptibility to drug-induced apoptosis (Perego et al., 1997). A detailed characterization of this cell system indicated that the development of cisplatin resistance was associated with multiple alterations, including reduced drug accumulation and DNA damage and defects in mismatch repair. Our results show a complete lack of cross-resistance of the osteosarcoma subline to BBR 3464 and support the finding that the efficacy of BBR 3464 against resistant tumors is related to a different mechanism of DNA interaction.
Materials and Methods
Drugs.
Cisplatin and carboplatin were obtained from Boehringer Mannheim (Milan, Italy). BBR 3464 was prepared as NO−3 salt (Farrell et al., 1997), and dissolved in saline before use.
Cell Lines and Growth Conditions.
The U2–0S cell line (HTB 96; American Type Culture Collection, Rockville, MD) was derived from a moderately differentiated sarcoma of the tibia. The cisplatin-resistant cells, designated as U2–0S/Pt, were generated in approximately 1 year by continuous exposure to increasing concentrations of cisplatin up to 1 μg/ml. Both cell lines were grown and maintained as monolayer culture in McCoy’s 5A medium supplemented with 10% fetal calf serum. The hMLH1-deficient human colorectal adenocarcinoma cell line HCT116 and sublines complemented with chromosome 3 (HCT116/chr3) and chromosome 2 (HCT116/chr2) were kindly provided by Dr. R. Boland (San Diego, CA). Cell lines were maintained in Iscove’s modified Dulbecco’s medium (BioWhittaker, Walkersville, MD) supplemented with 10% fetal calf serum. Chromosome-transferred cell lines were grown in the presence of 400 μg/ml geneticin.
Cytotoxicity Assays.
Cell survival after a 96-h exposure to cisplatin or carboplatin was assessed by the tetrazolium dye assay according to the method of Alley et al. (1988), as previously described (Soranzo et al., 1990). Alternatively, the antiproliferative effect of the drugs was assessed by the growth inhibition assay after a 1-h exposure. Cells in the logarithmic phase of growth were seeded in duplicate into 6-well plates. Twenty-four hours after seeding, the drug was added to the medium and cells were incubated for 1 h. Cells were harvested 72 h after the beginning of exposure and counted with a cell counter. IC50 is defined as the inhibitory drug concentration causing a 50% reduction of absorbance at 550 nm or of cell number over that of untreated control.
GSH Content and γ-Glutamyl Transpeptidase (γ-GT) Activity.
Exponentially growing cells were harvested and immediately homogenized in ice-cold 5% trichloroacetic acid. After centrifugation at 5000g for 10 min to remove protein, GSH content was determined according to the method of Ellman (1959). For determination of γ-GT activity, the harvested cells were suspended in 0.01 M Tris-HCl (pH 8.5) and sonicated. The γ-GT new monotest (Boehringer Mannheim GmBH, Mannheim, Germany), which is based on the reaction between glycylglycine and L-γ-glutamyl-3-carboxy-4-nitroanilyde as the substrate, was used. Total proteins were determined with the bicinchonic acid protein assay (Pierce, Rockford, IL).
Northern Blot Hybridization Analysis.
About 20 μg of total RNA prepared by the LiCl guanidine monothiocianate method (Maniatis et al., 1982) was electrophoresed on a formaldehyde-containing 1% agarose gel and was tranferred onto a nylon membrane. DNA probes were32P-labeled with a random primer kit (Amersham, Little Chalfont, U.K.). Hybridization was carried out as previously described (Perego et al., 1994). The expression level was evaluated by densitometric analysis of autoradiograms and compared with the expression level of the control β-actin gene. A ratio between the expression level of resistant and sensitive cells was calculated to define relative expression of the two cell lines. Each experiment was performed at least three times. The probes used were as follows: topoisomerases I and II cDNA fragments were kindly provided by Dr. Leroy Liu (Baltimore, MD); and GSH-S-transferase π, metallothioneins IIa, and DNA polymerase β were obtained from Dr. Jeffrey A. Moscow (Bethesda, MD), Dr. John S. Lazo (Pittsburg, KS), and Dr. Samuel H. Wilson (Bethesda, MD), respectively. Heat shock protein 60 (hsp60) probe was a gift from Dr. Sue Fox (Evanston, IL).
Western Blot Analysis.
Cell lysates from exponentially growing cells were prepared as described previously (Perego et al., 1996). In brief, samples (80 μg/lane) were fractionated by SDS-polyacrylamide gel electrophoresis and blotted on nitrocellulose sheets. Blots were preblocked for 4 h at room temperature in PBS containing 5% (w/v) dried nonfat milk. Filters were incubated overnight at 4°C with monoclonal antibody to MLH1, MSH2, or PMS2 (Oncogene Research Products, Cambridge, MA); a rabbit anti-actin antibody (Sigma Chemical Co., St. Louis, MO) was used as a control for loading. Antibodies binding to the nitrocellulose blots were detected by chemiluminescence procedures (Amersham Pharmacia Biotech Italia, Cologno Monzese, Milan, Italy).
Microsatellite Instability.
Microsatellite loci were amplified by polymerase chain reaction as previously described (Aebi et al., 1996), with minor modifications. Fifty nanograms of DNA was amplified with 0.2 U of KlenTaq polymerase (Bio Nova, Bologna, Italy) in the presence of 2 μCi of [32P]dCTP (Amersham). The polymerase chain reaction products were separated on a 6% polyacrilamide/7 M urea gel and were visualized by autoradiography.
Accumulation Studies.
For drug accumulation studies, 2.5 × 105 cells were seeded into 100-mm tissue culture dishes in triplicate and 48 h later they were exposed to cisplatin or BBR 3464. Cell monolayers were then washed twice with ice-cold PBS and harvested for analyzing for platinum content by atomic absorption spectrometry (model 3300; Perkin Elmer, Norwalk, CT). Cellular platinum levels were expressed as nanograms of platinum per cell number, with cell number determined by counting parallel cultures.
DNA Platination.
Cells grown to near confluence were exposed to cisplatin or BBR 3464 for 1 h. DNA was extracted according to standard procedures involving lysis in the presence of 1 mg/ml proteinase K overnight at 37°C (Maniatis et al., 1982). DNA content was determined spectrophotometrically and platinum content was measured by inductively coupled plasma mass spectroscopy (Bonetti et al., 1996). DNA-bound platinum levels were expressed as picograms of platinum per micrograms of DNA, the amount of DNA being determined spectrophotometrically.
Alkaline Elution.
Cisplatin-induced interstrand cross-link (ICL) in U2-OS and U2-OS/Pt was determined by the alkaline elution method developed by Kohn (1979). Cellular DNA was labeled with 0.08 μCi/ml [2-14C]thymidine (Amersham) for 24 h and the labeled nucleoside precursor was removed 24 h before exposure to drug. Cells were exposed to cisplatin or to BBR 3464 for 1 h and then processed immediately or incubated in fresh medium up to 5 h. To produce a known frequency of γ-ray-induced DNA single-strand breaks before analysis, approximately 106 cells were irradiated with a137Cs source (300 rad). Cells were deposited on a 2.0-μm pore polycarbonate filter (Corning Costar, Acton, MA) and lysed with 2% SDS, 0.02 M disodium EDTA (pH 10), and 0.5 mg/ml proteinase K. For evaluation of DNA-protein cross-links 7 × 105 cells were loaded on 2.0-μm pore PVC filters (Millipore, Bedford, MA) and lysed with 0.2% sarcosyl, 2 M NaCl, and 0.04 M disodium EDTA (pH 10). The DNA was then eluted at a flow rate of 0.035 ml/min with a 0.02 M EDTA solution adjusted to pH 12.15 with tetrapropylammonium hydroxide. In ICL measurments, 0.1% SDS was added to the eluting solution. During a 15-h elution, the lysate was collected in fractions and counted by liquid scintillation. DNA ICL frequency was calculated using the following formula (Kohn et al., 1981, Ewig et al., 1978):
Results
Biological Features of the Cell System.
The human osteosarcoma subline U2-OS/Pt was selected in vitro after continuous exposure to cisplatin. The development of cisplatin resistance was not associated with a significant change in the proliferation rate (doubling time: 24 ± 0.7 versus 25 ± 1.2 h for parental and resistant cells, respectively) and resistance was stable in the absence of the selecting agent for at least 3 months. A marginal increase in GSH content was found in resistant cells (2.4 mmol/g) as compared with parental cells (1.94 mmol/g); this change was correlated with an appreciable increase in γ-GT activity (0.9 versus 1.65 mU/mg total protein).
Response to Cisplatin, Carboplatin, and BBR 3464.
Cellular response to cisplatin and carboplatin was evaluated after a 96-h drug exposure. The degree of resistance to cisplatin was approximately 6-fold, with IC50 values of 0.64 ± 0.5 μg/ml for U2-OS cells and 4.19 ± 0.3 μg/ml for U2-OS/Pt cells. U2-OS/Pt cells were cross-resistant to carboplatin (IC50 of U2-OS cells = 12.5 ± 0.8 μg/ml; IC50 of U2-OS/Pt cells = 90.0 ± 4.0 μg/ml). Cytotoxicity of BBR 3464 and cisplatin on U2-OS and U2-OS/Pt cells (Fig. 2) was assessed after a 1-h drug exposure to allow comparison with other cellular effects (uptake and DNA damage). The degree of resistance to cisplatin (approximately 6-fold) was not dependent on the time of exposure (IC50 of U2-OS cells = 4.1 ± 0.8 μg/ml; IC50 of U2-OS/Pt cells = 21.3 ± 1.0 μg/ml). A marked increase of potency of BBR 3464 in comparison with cisplatin was observed in U2-OS cells (IC501.1 ± 0.7 μg/ml and 4.1 ± 0.8 μg/ml for BBR 3464 and cisplatin, respectively). A complete lack of cross-resistance was found in U2-OS/Pt cells, which presented an IC50(1.2 ± 1.0 μg/ml) comparable to cisplatin-sensitive cells.

Sensitivity of U2-OS and U2-OS/Pt cells to cisplatin or BBR 3464. Sensitivity was assessed by growth inhibition assay with a 1-h drug exposure. Cells were counted 72 h after drug exposure. Values are the mean (± S.D.) of six independent experiments. U2-OS cells were exposed to cisplatin (○) or to BBR 3464 (■); U2-OS/Pt cells were exposed to cisplatin (•) or to BBR 3464 (▪).
Molecular Characteristics of the Cellular Model.
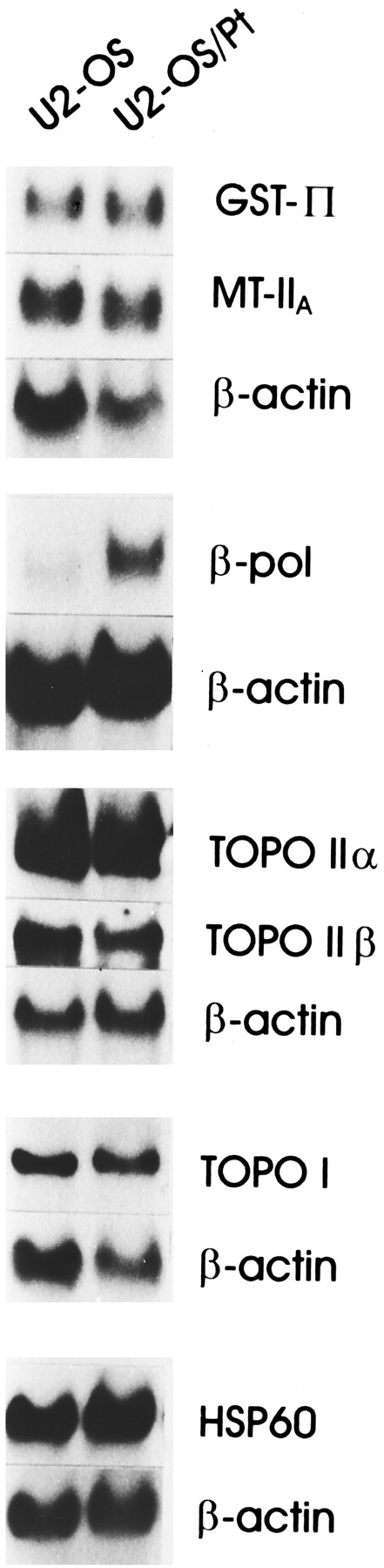
To characterize the molecular determinants of the lack of cross-resistance between cisplatin and BBR 3464, we performed a Northern blot analysis of selected genes thought to be relevant for resistance to platinum compounds, such as DNA polymerase β and DNA topoisomerases (I, IIα, and IIβ). hsp60 was examined because it is induced by several types of environmental stress, including heavy transition metals (Lindquist, 1989) and has been shown to be overexpressed in cisplatin-resistant cells (Kimura et al., 1993). In this cell system, no relevant difference in hsp60 expression was found between U2-OS and U2-OS/Pt cells (ratio = 1.3 ± 0.1). A marked difference (ratio = 3.6 ± 1.4) in RNA levels was detected for DNA polymerase β, an enzyme involved in repair synthesis of damaged DNA (Wang, 1991) (Fig.3). This finding supports an increased ability of the resistant subline to repair cisplatin-induced damage. Because defense factors, including cellular thiol levels, have been shown to contribute to cellular resistance to cisplatin (Kasahara et al., 1991; Mistry et al., 1991), we also examined the expression of metallothioneins IIa and glutathione S-transferase π. However, the levels of these two genes were only slightly different in sensitive and resistant cells (ratio = 1.2 ± 0.2 and 1.4 ± 0.5, respectively) Similarly, no marked difference between sensitive and resistant cells was found for the expression of topoisomerase I, IIα, and IIβ genes (ratio = 1.4 ± 0.2, 0.9 ± 0.1, 0.9 ± 0.2, respectively; Fig. 3).

Northern blot analysis of selected genes in U2-OS and U2-OS/Pt cells. Total RNA was used and filters were hybridized with the indicated probes: metallothionein IIa (MT IIa); GSHS-transferase π (GST π); HSP60, topoisomerase I (topo I); topoisomerase II α (topo II α) and β (topo II β); or DNA polymerase β (β-pol). Control loading is shown by actin. The reported blots are representative of at least three independent experiments.
Expression and Function of Proteins Involved in DNA Mismatch Repair.
To understand other cellular aspects of the observed capability of BBR 3464 to overcome cisplatin resistance in U2-OS/Pt cells, this cell system was characterized with respect to additional mechanisms of resistance to cisplatin. Because the loss of mismatch repair has been associated with cisplatin resistance in cellular models of different tumor type (Aebi et al., 1996; Anthoney et al., 1996), we examined the expression of MSH2, MLH1, and PMS2. Although PMS2 expression was reduced, MSH2 and MLH1 levels were unchanged. A decrease in PMS2 expression was associated with microsatellite instability at the D17S250 locus, thereby reflecting an alteration of the DNA mismatch repair functionality (Fig.4). To better understand the role of mismatch repair status on sensitivity to BBR 3464, an evaluation of the drug cytotoxicity of BBR 3464 on HCT116 cells deficient in MLH1 (HCT116/chr2) or repleted (HCT116/chr3) was undertaken. In HCT116/chr3 cells, a functional copy of MLH1 had been introduced by chromosome 3 transfer, whereas in HCT116/chr2 cells chromosome 2 had been inserted as a control. Differing from what was observed for cisplatin (Aebi et al., 1996) MLH1-deficient HCT116/chr2 cells were not resistant to BBR 3464 (IC50 = 0.06 ± 0.02 μg/ml, 1-h exposure) as compared with the proficient HCT116/chr3 subline (IC50 = 0.057 ± 0.01 μg/ml). Thus, the loss of DNA mismatch repair due to deficiency of MLH1 does not confer resistance to BBR 3464.

Western blot analysis of MLH1, MSH2, and PMS2 (A) and microsatellite instability (B) in U2-OS and U2-OS/Pt cells. The microsatellite locus shown was amplified with D17S250primers.
Cellular Platinum Accumulation.
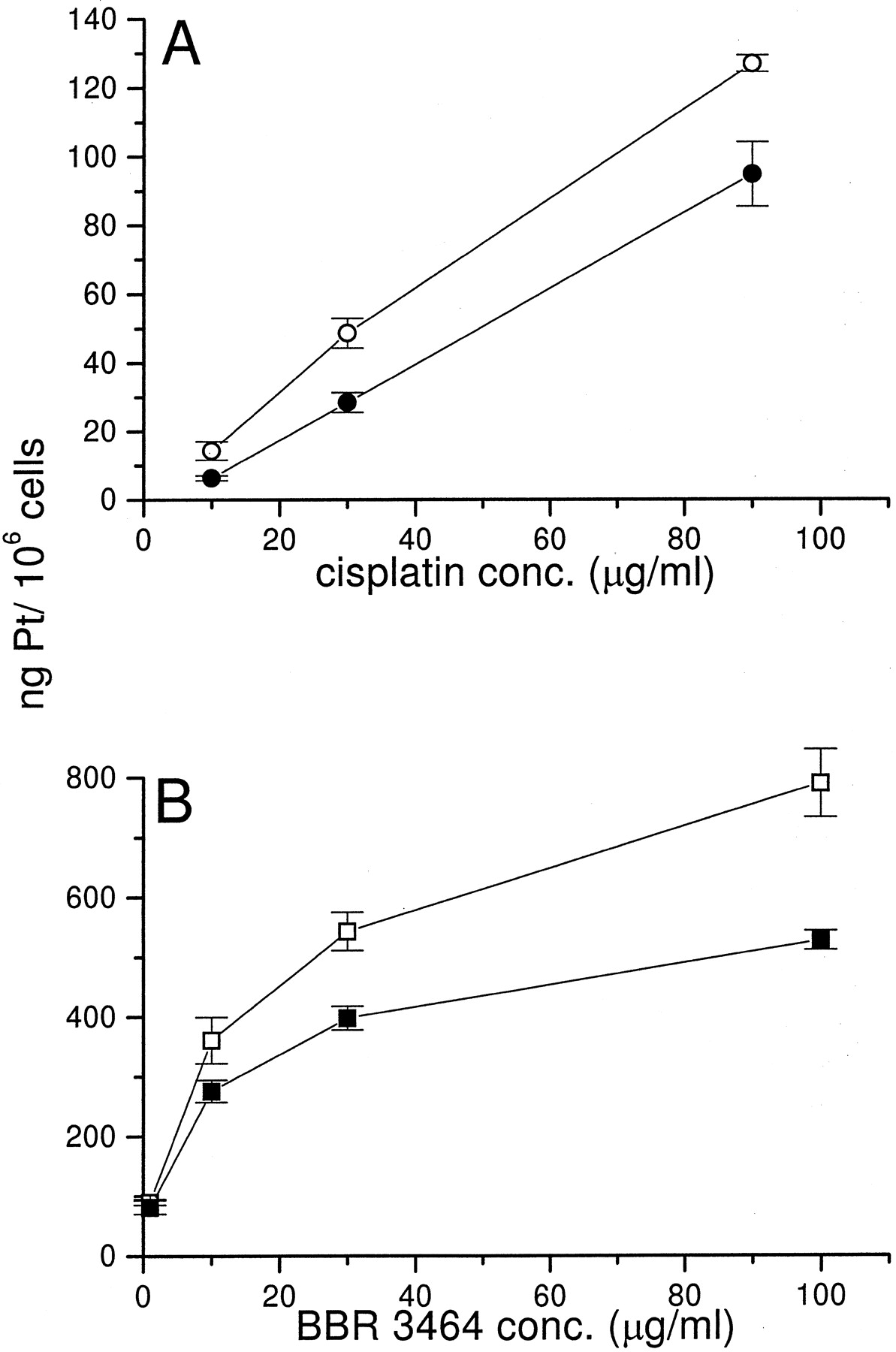
Among the mechanisms of cisplatin resistance, drug transport has been shown to play an important role (Gately and Howell, 1993). A comparison of cellular drug accumulation was performed after the same exposure time (1 h) of cytotoxicity experiments. The exposure of cells to drug concentrations ranging from 10 to 90 μg/ml indicated that cisplatin accumulation was linear in cisplatin-sensitive and -resistant cells. A decrease in platinum content was found in U2-OS/Pt cells as compared with U2-OS cells for all of the used concentrations (Fig.5A). The measurement of platinum level after 1 h of exposure to BBR 3464 revealed that drug accumulation was much higher than that observed for cisplatin both in sensitive and resistant cells (Fig. 5B). In fact, the order of magnitude of platinum accumulation was comparable when the two cell lines were exposed to equitoxic concentrations of BBR 3464 and cisplatin (1 μg/ml in the case of BBR 3464 for both U2-OS and U2-OS/Pt cells and 30 μg/ml for cisplatin in U2-OS/Pt cells). Although BBR 3464 accumulation was reduced in U2-OS/Pt as compared with U2-OS cells, the extent of BBR 3464 accumulation in cisplatin-resistant cells was substantially higher than that achieved by cisplatin. Thus, the observed difference between U2-OS and U2-OS/Pt cells may have a marginal relevance to the final cytotoxic effect.

Cellular accumulation of platinum after a 1-h exposure to cisplatin (Fig. 5A) or BBR 3464 (Fig. 5B) as assessed by atomic absorption spectrometry. U2-OS cells were exposed to cisplatin (○) or BBR 3464 (■); U2-OS/Pt cells were exposed to cisplatin (•) or BBR 3464 (▪). Mean values (± S.D.) of triplicated determinations are shown.
DNA-Bound Platinum and Formation of ICL.
The increased cellular accumulation of platinum in cells exposed to BBR 3464, despite the charged nature of the triplatinum complex, does not rule out the possibility of external drug binding. Therefore, because DNA is recognized as the primary cellular target of platinum drugs, we examined DNA-bound platinum in U2-OS and U2-OS/Pt cells after exposure to cisplatin or to the novel multinuclear compound. Measurement of DNA-bound platinum after 1 h of drug exposure revealed that the amount of platination was higher in U2-OS and U2-OS/Pt cells exposed to BBR 3464 than in the same cell lines exposed to cisplatin (Fig.6).

DNA-bound platinum in U2-OS and U2-OS/Pt cells after a 1-h exposure to BBR 3464 or cisplatin. Platination was measured by inductively coupled plasma mass spectroscopy. U2-OS cells were exposed to cisplatin (○) or BBR 3464 (■); U2-OS/Pt cells were exposed to cisplatin (•) or BBR 3464 (▪). Mean values (± S. D.) of three independent experiments are shown.
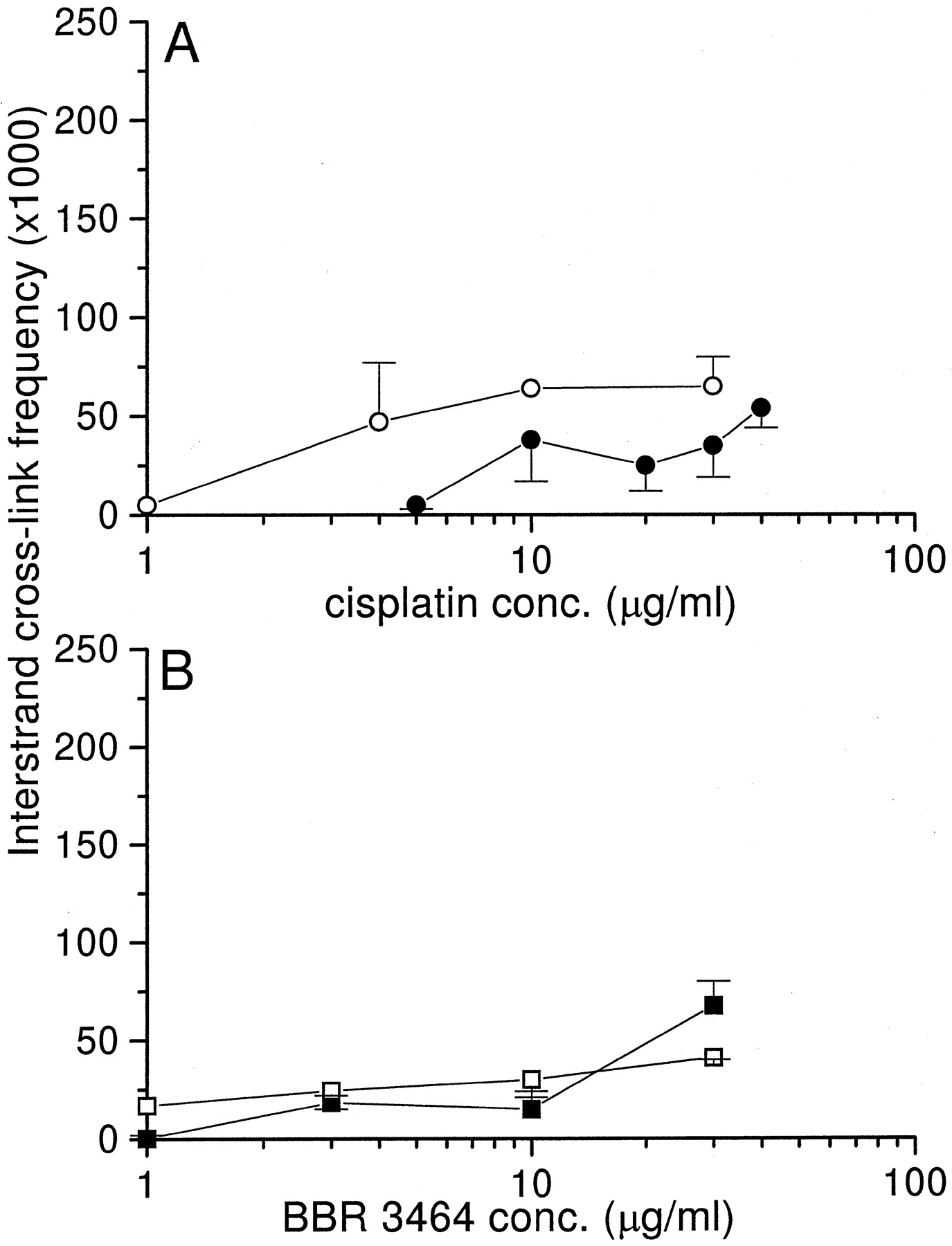
Because the overall platination does not necessarily account for cytotoxic effects (Plooy et al., 1985), an analysis of ICL formation after drug exposure was undertaken in U2-OS and U2-OS/Pt cells. ICL formation was initially studied in the presence of proteinase K to detect ICLs between the drug and DNA. ICL resulting from a 1-h exposure of cells to cisplatin or BBR 3464 is presented in Fig.7. After exposure to cisplatin, a slightly lower level of ICLs than in U2-OS was detected in U2-OS/Pt cells exposed to drug concentrations ranging from 1 to 30 μg/ml. In contrast, ICL frequency was similar in cisplatin-sensitive and -resistant cells exposed to BBR 3464. On the basis of the hypothesis that processing of the ICL could have an impact on drug cytotoxicity, we also performed an analysis of the kinetics of cross-link after drug removal (30 μg/ml). Although a time-dependent increase in the cisplatin-induced ICL was observed in U2-OS and U2-OS/Pt cells, as expected after initial formation of a monoadduct (Fig.8A), BBR 3464-induced ICL remained very low up to 5 h after drug withdrawal (Fig. 8B), suggesting a lack of a substantial conversion of monoadducts to ICLs.

ICL frequency in U2-OS and U2-OS/Pt cells after a 1-h exposure to cisplatin or BBR 3464. ICL frequency was measured by alcaline elution. U2-OS cells were exposed to cisplatin (○) or BBR 3464 (■); U2-OS/Pt cells were exposed to cisplatin (•) or BBR 3464 (▪). Mean values (± S. D.) of six independent experiments are shown.

Kinetics of ICL formation in U2-OS and U2-OS/Pt cells at various times of drug removal. Cells were exposed to 30 μg/ml cisplatin (A) or BBR 3464 (B) for 1 h. ICL frequency was measured by alkaline elution. U2-OS cells were exposed to cisplatin (○) or BBR 3464 (■); U2-OS/Pt cells were exposed to cisplatin (•) or BBR 3464 (▪). Mean values (± S.D.) of six independent experiments are shown.
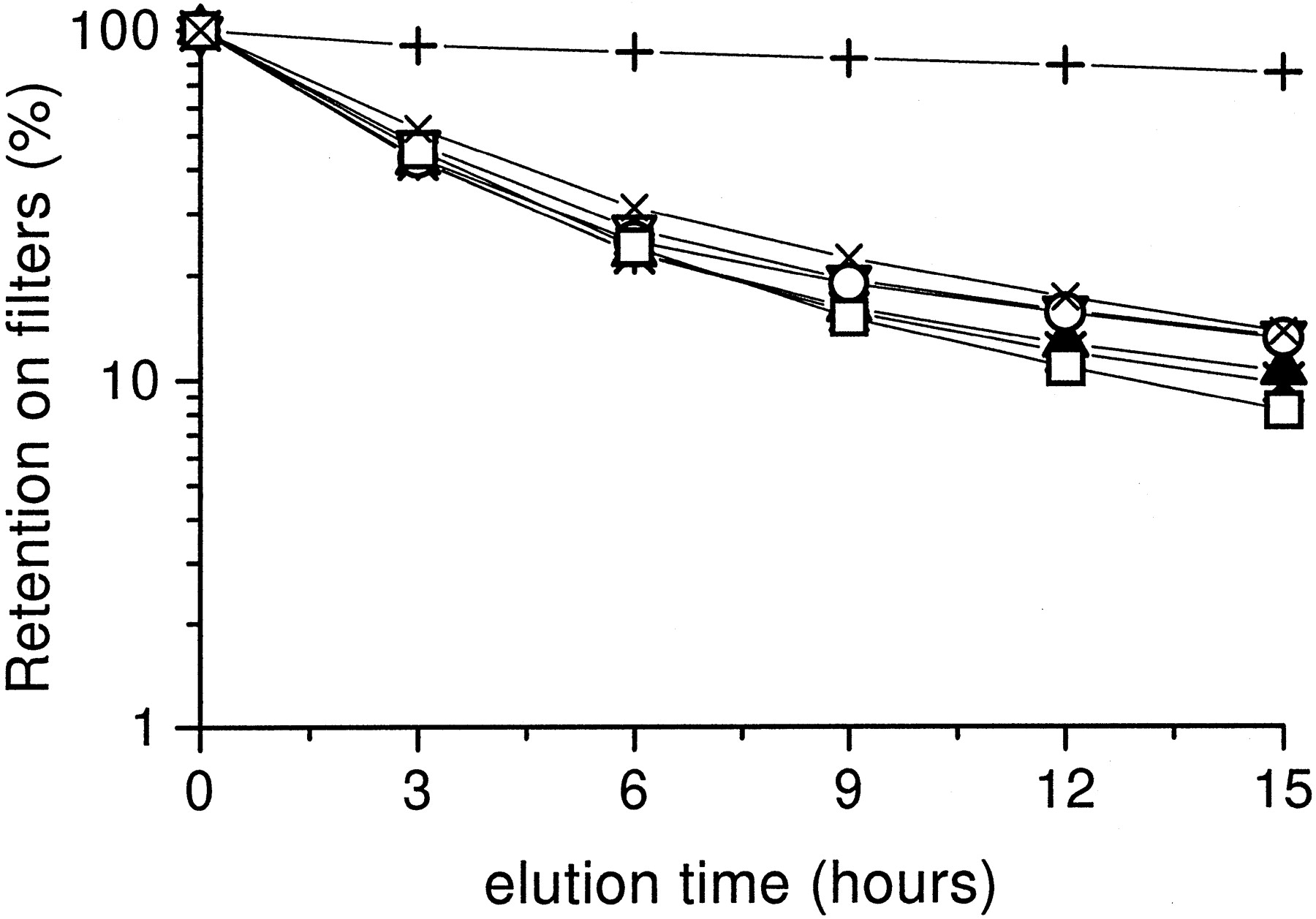
Due to the surprising discrepancy between the extent of DNA platination and ICL formation induced by BBR 3464 compared with cisplatin, cross-link formation was also studied in the absence of proteinase K to explore the relevance of drug-induced DNA protein cross-links. Using drug concentrations that in the procedure with proteinase K do not produce a detectable amount of ICL (not shown), a similar DNA retention was observed after exposure of U2-OS cells to BBR 3464 or cisplatin (Fig. 9), thus suggesting no relevant differences in drug induced DNA-protein cross-links.

DNA-protein cross-link after a 1-h exposure to cisplatin or BBR 3464 in U2-OS cells. Cross-link frequency was measured by alcaline elution in the absence of proteinase K. +, control cells. For irradiated cells (∗) retention overlaps that of cisplatin treated cells. Cells were exposed to 1 (▿) or 3 μg/ml (▴) cisplatin or 0.3 (○), 1 (■), or 3 μg/ml (X) BBR 3464.
Discussion
Although some mononuclear platinum compounds have been reported to overcome specific mechanisms of cisplatin resistance (e.g., accumulation defects; Mellish et al., 1995), these complexes are expected to interact with DNA in a manner similar to conventional cisplatin analogs.
The results presented in this study document unusual features of the cellular pharmacology of the triplatinum complex BBR 3464 that may be relevant for its pharmacological profile, namely, a potent cytotoxicity and the ability to overcome cisplatin resistance (G. Pratesi, P. Perego, D. Polizzi, S. C. Righetti, R. Supino, C. Caserini, C. Manzotti, F. C. Giuliani, G. Pezzoni, S. Tognella, S. Spinelli, N. Farrell, and F. Zunino, submitted). The increased cytotoxic potency of BBR 3464 in comparison to cisplatin was correlated with its cellular accumulation and the extent of DNA binding. This behavior is somewhat surprising considering the large molecular size and the charged nature of this cationic complex. Such observations suggest that the cellular mechanisms underlying the transport of the multinuclear platinum complexes may substantially differ from those of cisplatin.
To better understand the cellular basis of the efficacy of the novel compound against cisplatin-resistant tumors, we have investigated the cellular effects of BBR 3464 in a human osteosarcoma cisplatin-resistant subline. Multiple alterations were found in the resistant phenotype, including reduced drug accumulation, reduced formation of DNA lesions, defects in the DNA mismatch repair and up-regulation of DNA polymerase β. An appreciable reduction of cellular accumulation of BBR 3464 was also observed in U2-OS/Pt cells as compared with U2-OS cells, but overall, the total amount of cellular platinum measured after exposure of both sensitive and resistant cells to the multinuclear platinum complex was markedly higher than that observed in cells exposed to cisplatin. Consistent with accumulation studies, a reduced drug platination and ICL formation was measured in U2-OS/Pt cells after exposure to cisplatin. A reduction of DNA-bound platinum was still found in the U2-OS/Pt cell line exposed to BBR 3464. However, in both cell lines, the extent of DNA-bound platinum was very high as compared with cisplatin-treated cells. This finding is consistent with an increased affinity of BBR 3464 for DNA as expected on the basis of the presence of two reactive platinum centers and on the positive charge (+4) of the complex that should interact with DNA primarily through electrostatic and hydrogen binding. On the basis of the increased DNA binding affinity of BBR 3464 and the predicted drug ability to produce long-distance cross-links, the low frequency of ICL was surprising. The discrepancy between DNA-bound platinum and ICL in cells treated with BBR 3464 could not be explained by an increased DNA-protein cross-link formation. Thus, a plausible explanation of the large amount of DNA-bound BBR 3464 is a preminent formation of intrastrand cross-links or monoadducts. This possibility is in agreement with in vitro observations on drug-oligonucleotide interaction showing no preference of ICL over intra-strand cross-links formation (V. Brabec, J. Kaspàrkovà, O. Vràna, O. Novàkovà, J. Cox, Y. Qu, and N. Farrell, submitted).
The DNA mismatch repair pathway has been shown to be altered in cisplatin-resistant cell lines (Aebi et al., 1996; Anthoney et al., 1996). The described alterations usually regards MLH1 and MSH2. To our knowledge, the U2-OS/Pt cell line is the first cisplatin-resistant subline with a deficiency in PMS2. A deficiency in some components of the mismatch repair system has been implicated in resistance to cisplatin because this system is capable of recognizing cisplatin-induced DNA lesions, likely functioning as a sensor for triggering apoptosis. The lack of cross-resistance of U2OS/Pt and of MLH1-deficient HCT116 cells to BBR 3464 suggests that the integrity of mismatch repair systems is not a determinant of cellular sensitivity to the multinuclear platinum complex.
In conclusion, the ability of BBR 3464 to overcome cisplatin resistance suggests that the cellular basis of lack of cross-resistance is related to a different mechanism of DNA interaction rather than to the ability to overcome specfic alterations associated with cisplatin resistance (e.g., defects in accumulation or decreased adduct formation). The observation of a complete lack of cross-resistance between cisplatin and BBR 3464 in the cisplatin-resistant osteosarcoma cell line emphasizes the pharmacological interest of this new multinuclear platinum compound, which has been selected for clinical development. Our cell system appeared to be well suited to understand specific mechanisms involved in cisplatin resistance as well as to elucidate the cellular determinants of response to BBR 3464. The results are consistent with the view that multinuclear platinum complexes cannot be regarded as cisplatin analogs, but rather as a new class of DNA-binding agents (Farrell, 1996b). Understanding the relevant chemical and biochemical features of these agents may provide a rational basis for improving therapy in resistant tumors.
Acknowledgments
We thank Laura Zanesi for her skillful assistance in typing the manuscript and Dr. S. Aebi for helpful discussion. N.F. acknowledges the support of the American Cancer Society.
Footnotes
-
Send reprint requests to: Dr. Paola Perego, Istituto Nazionale Tumori, Via Venezian 1, 20133 Milan, Italy. E-mail:perego{at}istitutotumori.mi.it
-
This work was partially supported by the Consiglio Nazionale delle Ricerche (Progetto Finalizzato Applicazioni Cliniche della Ricerca Oncologica), by the Associazione Italiana Ricerca sul Cancro and by the Ministero della Sanita’.
- Abbreviations:
- GSH
- Glutathione
- γ-GT
- γ-glutamyl transpeptidase
- ICL
- interstrand cross-link
- HSP60
- heat-shock protein 60
- Received July 27, 1998.
- Accepted November 13, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}