


Abstract
Background/Aim: Oral cancer has been reported to be one of the major cancer-related diseases in human populations and the treatment of oral cancer is still unsatisfied. Fisetin, is a flavonoid from plants and has several biological activities such as antioxidant, anti-inflammatory and anticancer function, but its cytotoxicity in human oral cancer cells is unknown. In the present study, we investigated fisetin-induced cytotoxic effects on HSC3 human oral cancer cells in vitro. Materials and Methods/Results: We used flow cytometric assay to show fisetin induced apoptotic cell death through increased reactive oxygen species and Ca2+, but reduced the mitochondrial membrane potential and increased caspase-8, -9 and -3 activities in HSC3 cells. Furthermore, we also used 4’ 6-diamidino-2-phenylindole staining to show that fisetin induced chromatin condensation (apoptotic cell death), and Comet assay to show that fisetin induced DNA damage in HSC3 cells. Western blotting was used to examine the levels of apoptotic-associated protein and results indicated that fisetin increased expression of pro-apoptotic proteins such as B-cell lymphoma 2 (BCL2) antagonist/killer (BAK) and BCL2-associated X (BAX) but reduced that of anti-apoptotic protein such as BCL2 and BCL-x, and increased the cleaved forms of caspase-3, -8 and -9, and cytochrome c, apoptosis-inducing factor (AIF) and endonuclease G (ENDO G) in HSC3 cells. Confocal microscopy showed that fisetin increased the release of cytochrome c, AIF and ENDO G from mitochondria into the cytoplasm. Conclusion: Based on these observations, we suggest that fisetin induces apoptotic cell death through endoplasmic reticulum stress- and mitochondria-dependent pathways.
Oral cancer is a subtype of head and neck squamous cell carcinoma. Oral squamous cell carcinoma (OSCC) comprises of a large proportion of head and neck SCC (1). In men, oral cancer constitutes approximately 3% of all malignant neoplasms, but in females about 2%. The incidence in the past decade has increased up to almost five-fold in those younger than 40 years old (2). Oral cancer is the fifth common cancer in males in Taiwan and around 13 individuals per 100,000 die annually from oral cancer based on a report in 2017 from the Department of Health, R.O.C. (Taiwan) (3). Currently, primary surgery, definitive radiation therapy and chemotherapy or combination of chemo- and radiotherapy are options for patients with oral cancer. However, these therapies can have a profound effect on the quality of life of survivors (4, 5). Thus, dose-limiting toxicities in patients with cancer restrict the clinical utility of such therapies. Many studies have focused on compounds from natural products for treating patients with oral cancer. Cell cycle progression occurs through multiple cascades regulated by cyclins and cyclin-dependent kinases (CDKs) (6); and the formation of complex of cyclin with CDKs can lead to phosphorylation of substrates involved in cell-cycle progression. Therefore, many anticancer drugs affect cyclins or CDKs, leading to cell-cycle arrest. Apoptosis is a type of programmed cell death with characteristics such as cytoplasmic shrinkage, nuclear chromatin condensation, and DNA fragmentation and apoptotic body in cells after exposure anticancer chemotherapeutic drugs (7-9). Thus, a good strategy for anticancer agent activity is to induce cancer cell apoptosis.
Herbal medicines have been used to treat a variety of human cancer types (10) and some of these herbal medicines and natural plants contain flavonoids (11, 12). These flavonoids are recognized to have anticancer and chemopreventive functions via antioxidant activity, inhibition of cell proliferation and angiogenesis, and induction of cell-cycle arrest and apoptosis (13). Fisetin (3,7,3’,4’-tetrahydroxyflavone), a naturally-occurring flavonoid, is widely distributed among edible vegetables and fruits such as apple, cucumber, grapes, kiwis, onion, persimmon and strawberry, the highest level of fisetin (160 μg/g wet food) being found in strawberries (14). Numerous studies have shown that fisetin has pharmacological activities such as antioxidant (15), anti-inflammatory (16), anti-invasive (17, 18), anti-angiogenesis (19, 20), anti-proliferation (21, 22) and anticancer effects in several cancer types, such as prostate cancer (22, 23), lung adenocarcinoma (20, 24), pancreatic cancer (25), colon cancer (26), cervical cancer (27), melanoma (28) and acute monocytic leukemia cells (29). Recently, it was reported that combination treatment of fisetin and sorafenib effectively inhibited B-Raf proto-oncogene serine/threonine-protein kinase (BRAF)-mutated melanoma cell growth, induced apoptosis, and down-regulated mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) signaling pathways in vitro and in vivo.
Although numerous studies have shown that fisetin induces cell apoptosis in many human cancer cell lines, there is no information available on the effects of fisetin on human oral cancer cell lines. The present study was designed to analyze the anticancer potential of fisetin against human oral cancer HSC3 cells in vitro.
Materials and Methods
Chemicals and reagents. Fisetin of 99% purity, 4’ 6-diamidino-2-phenylindole (DAPI), dimethyl sulfoxide (DMSO), propidium iodide (PI) and trypsin-EDTA were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Minimum essential medium (MEM), fetal bovine serum (FBS), L-glutamine and penicillin-streptomycin were purchased from GIBCO®/Invitrogen Life Technologies (Carlsbad, CA, USA). Fisetin was dissolved in DMSO.
Cell culture. HSC3 human oral cancer cell line was purchased from the Food Industry Research and Development Institute (Hsinchu, Taiwan, ROC). These cells were placed in MEM supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine and were growth at 37°C in a humidified atmosphere consisting of 5% CO2 (30).
Cell viability assays. HSC3 cells (5×104 cells/ml) were seeded and cultured in 12-well plates with MEM for 24 h and then fisetin was added to each well at final concentrations of 0, 20, 40, 60, 80 and 100 μM. After incubation for 24 and 48 hours, cells were harvested, counted and stained with PI (5 μg/ml) followed by immediately analysis by flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA, USA) for total percentage of viable cells as previously described (30).
DAPI staining. Cells (5×104 cells/ml) were plated in a 12-well plate and treated with fisetin (40 μM) for 0, 12, 24 and 48 hours and then fixed in 3% methanol in PBS at room temperature for 20 min. Cells were stained with DAPI solution (2 μg/ml) and photographed under fluorescence microscopy as described previously (31). Nuclear condensation of cells relative to total cells was evaluated, with a minimum of 150 cells/field and at least three fields in each well being counted.
DNA damage measurement. HSC3 cells (5×104 cells/ml) were cultured in 12-well plates for 24 hours and were then treated with fisetin (40 μM) for 0, 12, 24 and 48 hours. Cells were then collected for comet assay as described previously (32).
Measurement of reactive oxygen species (ROS), intracellular Ca2+ and mitochondrial membrane potential (Ψm). The measurement of ROS, Ca2+ and ΔΨm in HSC3 cells after exposure to fisetin were performed by flow cytometric assay. In brief, HSC3 cells (5×104 cells/ml) in 12-well plate were treated with fisetin (40 μM) for 0, 6, 24 and 48 hours. For ROS (H2O2) measurement, cells were isolated and re-suspended with 500 μl of 2’,7’-dichlorfluorescein-diacetate (DCFH-DA) (Sigma Chemical Co.) (10 μM) and kept in the dark for 30 minutes. For intracellular Ca2+ measurement, cells were isolated and re-suspended with 500 μl of fluo-3-acetomethoxyester (Fluo-3/AM) (Invitrogen) (2.5 μg/ml) maintained in the dark for 30 min. For ΔΨm measurement, cells were isolated and re-suspended with 500 μl of 3,3’-dihexyloxacarbocyanine iodide (DiOC6) (Sigma Chemical Co.) (4 μmol/l) and maintained in the dark for 30 minutes. After incubation, all samples were analyzed by flow cytometry as described previously (33-35).

Fisetin reduced HSC3 cell viability. HSC3 cells were treated with 0, 20, 40, 60, 80 and 100 μM of fisetin for 24 (A) and 48 (B) h and then cells were collected to determine the total percentage of viable cells by flow cytometry as described in the Materials and Methods section. ***Significant differences at p<0.001 between fisetin-treated groups and the control as analyzed by Dunnett test.
Measurements of caspase activities. Flow cytometry was used for measuring caspase-3, -8 and -9 activities. In brief, HSC3 cells (5×104 cells/ml) were plated in a 12-well plate and treated with 40 μM of fisetin for 0, 6, 24 and 48 h then cells were harvested and re-suspended in 25 μl of 10 μM substrate solution containing PhiPhiLux-G1D1 for caspase-3, or CaspaLux8-L1D2 for caspase-8 or CaspaLux9-M1D2 (both from OncoImmunin, Gaithersburg, MD, USA) for caspase-9 determination before being incubated at 37°C for 60 min. After incubation, all samples were washed with PBS and caspase activities were analyzed by flow cytometry as described previously (31, 32, 36).
Western blotting analysis. HSC3 cells (1.5×106 cells/dish) were placed in a 10 cm dish for 24 hours and then were incubated with 40 μM fisetin for 0, 12, 24 and 48 hours. Cells were collected and lysed and total protein was determined by Bio-Rad protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Equal amounts of protein extracts were separated by 8-14% (v/v) sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). After blocking, the membranes were hybridized with primary antibodies against myeloid cell leukemia-1 (MCL1), B-cell lymphoma 2 (BCL2), apoptosis regulator Bcl-X (BCLX), BH3-interacting domain death agonist (BID), BCL2-associated agonist of cell death (BAD), BCL2 antagonist/killer (BAK), BCL2-associated X (BAX), apoptosis-inducing factor (AIF), endonuclease G (ENDO G), cytochrome c, apoptotic peptidase activating factor 1 (APAF1), caspase-9, X-linked inhibitor of apoptosis (XIAP), caspase-3, caspase-6, poly-(ADP-ribose) polymerase (PARP), Fas cell surface death receptor (FAS), FAS-ligand, caspase-8, activating transcription factor 6-beta (ATF-6β), calpain 1, caspase-4, glucose-regulated protein 78 (GRP78) and β-Actin at 4°C overnight. After washing, secondary antibodies supplied by GeneTex (Irvine, CA, USA) were used at a 1:10,000 dilution for horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (GTX213110), HRP-conjugated donkey anti-goat IgG (GTX26885) and HRP-conjugated rabbit anti-mouse IgG (GTX213112) for 1 h at room temperature (25°C). Subsequently, proteins were visualized via chemiluminescence signals enhanced using electrochemiluminescence detection (Amersham ECL™; GE Healthcare, Chicago, IL, USA). ImageJ version 1.49o software (National Institutes of Health, Bethesda, MD, USA) was used to quantify changes in protein expression by densitometric analysis using β-actin as the loading control (37).
Confocal laser scanning microscopy assay. HSC3 cells (5×104 cells/ml) were kept on 12-well chamber slides and were treated with fisetin (0 and 40 μM) for 24 hours, then fixed with 4% formaldehyde in PBS for 15 min. Cells were incubated 0.3% Triton-×100 in PBS for 1 h to permeable the cells for blocking non-specific binding sites by using 2% bovine serum albumin. Cells were stained by primary antibodies to cytochrome c, AIF and ENDO G (all in green fluorescence) overnight followed by secondary antibody [fluorescein isothyocyanate-conjugated goat anti-mouse (or rabbit or goat) IgG] and PI (red fluorescence) staining for examination of nuclei. Slides were mounted, examined and photo-micrographed under a Leica TCS SP2 Confocal Spectral Microscope (Leica Microsystems, Bannockburn, IL, USA) (38).
Statistical analysis. All values are presented as the mean±standard deviation of three independent experiments. Differences between groups were analyzed by one-way analysis of variance and Dunnett test for multiple comparisons (SigmaPlot for Windows version 12.0; Systat Software, Inc., San Jose, CA). A value of p<0.05 was considered to indicate a statistically significant difference.
Results
Fisetin reduced the total cell viability of HSC3 cells. The cytotoxic effects were examined in HSC3 cells after exposure to different concentrations of fisetin for 24 and 48 hours. The results showed that fisetin reduces HSC3 cell viability dose- and time-dependently (Figure 1).
Fisetin induced chromatin condensation (apoptotic cell death) in HSC3 cells. After HSC3 cells were exposed to fisetin (40 μM) for 0, 12, 24 and 48 h, they were stained with DAPI and photographed by using fluorescence microscopy. The results showed brighter fluorescence of HSC3 cells after 24 and 48 h of treatment with 40 μM fisetin (Figure 2A); the bright fluorescence represents nicked DNA and chromatin condensation. These effects also occurred in a time-dependent manner (Figure 2B).

Fisetin induced chromatin condensation in HSC3 cells. Cells were treated with 40 μM of fisetin for 0, 12, 24 and 48 h then were stained with 4’ 6-diamidino-2-phenylindole (DAPI) (A) and total head intensity (fold of control) was evaluated (B) as described in the Materials and Methods section. *Significant differences at p<0.05 fisetin-treated groups and the control as analyzed by Dunnett test.

Fisetin induced DNA damage in HSC3 cells. Cells were treated with 40 μM of fisetin for 0, 12, 24 and 48 h then the cells were measured by comet assay (A) for comet tail length (B) as described in the Materials and Methods section. Significant differences at *p<0.05, and ***p<0.001 between fisetin-treated groups and the control as analyzed by Dunnett test.
Fisetin induced DNA damage in HSC3 cells. The comet assay measures DNA damage in cells after exposure to chemicals. HSC3 cells were incubated with 40 μM fisetin for 0, 12, 24 and 48 h and were examined by comet assay. The results showed that fisetin induced a typical comet tail (DNA damage) (Figure 3A) and these effects were time-dependent (Figure 3B). These results suggest that fisetin induces DNA damage in HSC3 cells.
Fisetin induced ROS production and Ca2+ release, and reduced the mitochondrial membrane potential (Ψm) in HSC3 cells. In order to further understand whether fisetin induces cell apoptosis in HSC3 cells through the production of ROS and Ca2+ or dysfunction of mitochondria, cells were treated with 40 μM fisetin for different time periods and cells were collected and analyzed by flow cytometric assay. The results show that after 6-48 hours of treatment, fisetin increased ROS production (Figure 4A) and Ca2+ release (Figure 4B) but reduced Ψm (Figure 4C). These results indicate that ROS, Ca2+ and ΔΨm are involved in fisetin-induced cell apoptosis of HSC3 cells in vitro.

Fisetin induced reactive oxygen species (ROS) and Ca2+ production and reduced the mitochondrial membrane potential (MMP) in HSC3 cells. Cells (5×104 cells/ml) were treated with fisetin (40 μM) for different time periods. Cells were isolated and then were re-suspended in 500 μl of 2’,7’-dichlorfluorescein-diacetate (10 μM) for 30 min for determination of ROS (A), re-suspended in 500 μl of fluo-3 acetomethoxyester (2.5 μg/ml) for 30 min for determination of intracellular Ca2+ concentration (B), and re-suspended in 500 μl of 3,3’-dihexyloxacarbocyanine iodide (4 μmol/l) for 30 min for determination of MMP (C) as described in the Materials and Methods section. Significant differences at *p<0.05, **p<0.01, and ***p<0.001 between fisetin-treated groups and the control as analyzed by Dunnett test.
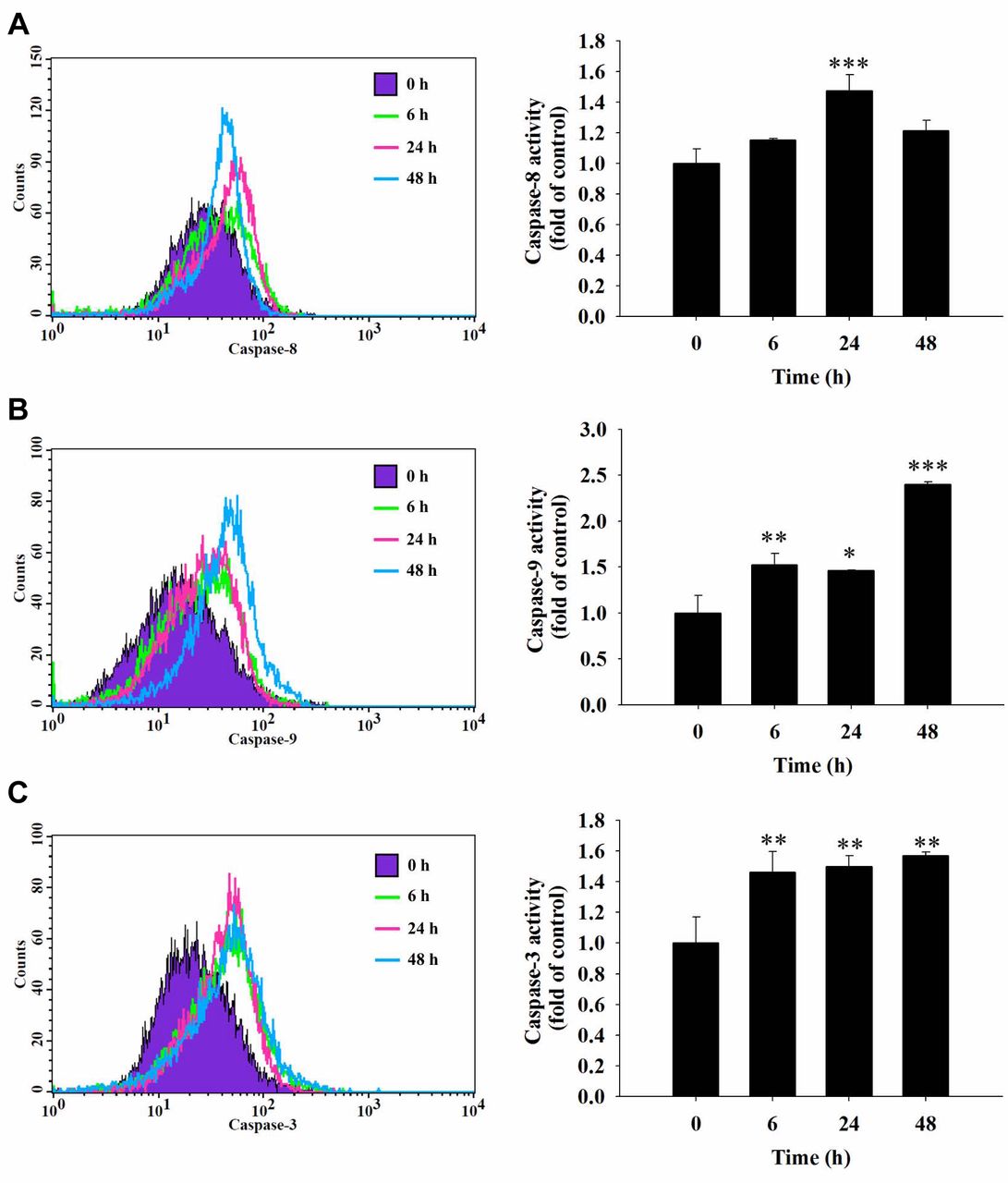
Fisetin increased the activities of caspase-3, -8 and -9 in HSC3 cells. In order to further understand whether fisetin induces apoptotic cell death in HSC3 cells through the activation of caspases, cells were treated with 40 μM fisetin for different time periods and cells were collected and analyzed by flow cytometric assay. The results show that fisetin increased caspase-8 activation at 24 h of treatment (Figure 5A), increased caspase-9 (Figure 5B), and caspase-3 activation (Figure 5C) from 6-48 h of treatment. These results indicate that caspase-3, -8 and -9 are involved in fisetin-induced cell apoptosis of HSC3 cells in vitro.

Fisetin induced the activity of caspase-3, caspase-8 and caspase-9 in HSC3 cells. HSC3 cells (5×104 cells/ml) were plated in a 12-well plate and treated with 40 μM fisetin for 0, 6, 24 and 48 h then cells were harvested and re-suspended in 25 μl of 10 μM substrate solution containing CaspaLux8-L1D2 for caspase-8 (A), CaspaLux9-M1D2 for caspase-9 (B) or PhiPhiLux-G1D1 for caspase-3 activity determinations (C) before being incubated at 37°C for 60 min. After incubation, all samples were washed with PBS and caspase activities were analyzed by flow cytometry as described previously. Significant differences at *p<0.05, **p<0.01, and ***p<0.001 between fisetin-treated groups and the control as analyzed by Dunnett test.
Fisetin altered expression of apoptosis-associated proteins in HSC3 cells. HSC3 cells were treated with 40 μM fisetin for different time periods and then apoptosis-associated proteins were examined and quantified with western blotting. The results show that fisetin significantly increased the expression of BID, BAD, BAK, BAX (Figure 6A), AIF, ENDO G, cytochrome c, APAF1, cleaved form of caspase-9 (Figure 6B), cleaved form of caspase-3, caspase-6 and PARP (Figure 6C), FAS, FAS-ligand and cleaved form of caspase-8 (Figure 6D), cleaved form of ATF-6β, calpain 1, caspase-4 and GRP78 (Figure 6E), but reduced the expression of MCL1, BCL2, BCL-x (Figure 6A), and XIAP (Figure 6C). These results indicate that fisetin induces apoptotic cell death of HSC3 cells through endoplasmic reticulum (ER) stress and mitochondria-dependent pathways.
Fisetin altered the translocation of apoptotic-associated proteins in HSC3 cells. In order to further confirm fisetin effects, we studied the translocation of cytochrome c, AIF, and Endo G in HSC3 cells. Cells were treated with or without 40 μM of fisetin for 24 h, and were then stained by anti-cytochrome c, -AIF and -ENDO G and examined by confocal laser microscopic systems and results. The results showed that fisetin increased cytochrome c (Figure 7A), AIF (Figure 7B) and ENDO G (Figure 7C) release from mitochondria into the cytoplasm when compared to the control group.
Discussion
It is well known that many flavonoids have antitumor or growth-suppressive capacity against various human tumor cell lines and fisetin is a flavonoid. Numerous reports have shown that fisetin induced cytotoxic effects on many human cancer cell lines through cell-cycle arrest and apoptosis (20, 22-29), however, there is no report demonstrating that fisetin induces apoptosis of human oral cancer cells, therefore, we investigated the cytotoxic effects of fisetin on HSC3 human oral cancer cells in vitro. It is well documented that the best strategy for anticancer drugs is to induce cancer cell apoptosis. The induction of apoptosis in cancer cells by fisetin may be one of its critical characteristics as a chemopreventive agent. Herein, we found that fisetin i) reduced the percentage of viable cells (Figure 1); ii) induced chromatin condensation (Figure 2) and DNA damage (apoptotic cell death) (Figure 3); iii) increased ROS and Ca2+ levels and reduced Ψm (Figure 4); iv) increased the activities of caspase-3, -8 and -9 (Figure 5); v) increased expression of pro-apoptotic proteins BAX, BAD and BAK, AIF, ENDO G and cytochrome c, cleaved caspases and reduced anti-apoptotic proteins BCL2 and BCL-x and XIAP (Figure 6); vi) induced release of cytochrome c, AIF and ENDO G from mitochondria into the cytoplasm (Figure 7).
It is well known that apoptotic cell death play a role in maintaining tissue homeostasis and eliminating unnecessary cells. Furthermore, the dysregulation of apoptosis is pivotal to tumorigenesis and cancer development. We used several methods, including DAPI and comet staining, to show that fisetin induced apoptosis of HSC3 cells. DAPI staining in cancer cells has been used as a method to measure apoptotic cell death (39, 40). We found that fisetin increased the productions of ROS and Ca2+ in HSC3 cells (Figure 4A and B). It was reported that chemotherapeutic agents induced necrotic or apoptotic cell death in cancer cells via the generation of ROS (41). But another report showed that fisetin treatment of A375.S2 melanoma cells resulted in a significant decrease in ROS level (28), while another reported that fisetin stimulated generation of ROS in U266 multiple myeloma cells and induced apoptosis through activation of AMP-activated protein kinase pathways in a ROS-dependent manner (42). Thus, based on these observations, fisetin-induced modulation of ROS may be cell type-dependent. It was reported that any stress negatively impacts upon the intracellular calcium level, which can trigger ER stress (43); ER stress sensor protein GRP78 can be useful therapeutic target to enhance tumor cell death (43). Numerous evidence has shown that anticancer drugs induce cell apoptosis through dysfunction of mitochondria or reduction of Ψm (43-45). The results from our flow cytometric assay showed that fisetin reduced Ψm in HSC3 cells (Figure 4C). ROS and mitochondria play an important role in the stimulation of apoptosis, through a mechanism known as the intrinsic signaling pathway (46, 47). It was reported that fisetin plays a key role in the loss of Ψm leading to apoptosis and inhibiting tumor growth (48). The accumulation of excessive ROS leads to nuclear DNA damage, followed by disruption of the Ψm and release of cytochrome c into the cytosol, ultimately leading to apoptosis (47). Thus, we suggest that fisetin induces apoptosis of HSC3 cells via a mitochondria-dependent pathway.
Caspases, cysteinyl aspartate-specific proteases, are involved in anticancer drug-induced cancer cell apoptosis, and can be activated during apoptosis in a self-amplifying cascade (49, 50). Our results revealed that fisetin significantly increased caspase-3, -8 and -9 expression (Figure 5), showing the involve¬ment of caspases in apoptosis of HSC3 cells. In HeLa human cervical ade¬nocarcinoma cells, it was reported that fisetin exposure led to both caspase-8 and caspase-3 activation (27). It is well documented that activations of both caspase-8 and caspase-9 trigger both the extrinsic and intrinsic cell apoptotic pathways (51-53). Our findings were confirmed by western blot analysis, showing the expression of cleaved caspase-3, -8 and -9 (Figure 6B-D) proteins were significantly increased. Activation of caspase-8 is closely related with apoptosis signaling through the extrinsic pathway (54) and activated caspase-8 links to the mitochondrial pathway through cleavage of the BCL2 family members from BID to the truncated p15 BID (tBID) (55). Overall, our findings indicate that activation of the intrinsic apoptotic pathway in HSC3 cells after exposed to fisetin as a result of unresolved ER stress (GRP78 and cleaved form of ATF-6β; Figure 6E) occurs through members of the BCL2 family which control the release of cytochrome c from the mitochondria.
It was reported that BCL2 family proteins are implicated in the regulation of mitochondrial permeability transition pore opening and release of cytochrome c from mitochondria into the cytosol (56) followed by activation of caspase-3 and apoptosis (57-59). Our results indicated that fisetin up-regulated the expression of BAX, BAD and BAK (Figure 6A), which are pro-apoptotic proteins, and caused down-regulation of BCL2 and BCL-xL, which are anti-apoptotic proteins (Figure 6A). Therefore, alterations of BAX/BCL2 lead to dysfunction of mitochondria causing them to release cytochrome c (Figure 6B), and increased the active form of caspase-3 protein that led to apoptosis in HSC3 cells. Moreover, our data show that fisetin up-regulated GRP78 and cleaved form of ATF-6β that induced ER stress in HSC3 cells (Figure 6E).


Fisetin affects expression of apoptosis-associated proteins in HSC3 cells. Cells were treated with 40 μM fisetin for 0, 12, 24 and 48 h and then total proteins were quantitated and apoptosis associated proteins were measured by western blotting as described in the Materials and Methods section. A: Myeloid cell leukemia-1 (MCL1), B-cell lymphoma 2 (BCL2), apoptosis regulator BCL-X (BCLX), BH3-interacting domain death agonist (BID), BCL2-associated agonist of cell death (BAD), BCL2 antagonist/killer (BAK), BCL2-associated X (BAX). B: Apoptosis-inducing factor (AIF), endonuclease G (ENDO G), cytochrome c, apoptotic peptidase-activating factor 1 (APAF1), caspase-9. C: X-linked inhibitor of apoptosis (XIAP), caspase-3, caspase-6 and poly-(ADP-ribose) polymerase (PARP). D: Fas cell surface death receptor (FAS), FAS-ligand and caspase-8. E: Activating transcription factor 6-beta (ATF-6β), calpain 1, caspase-4 and glucose-regulated protein 78 (GRP78).
Our results shown in Figure 6B indicated that fisetin increased the expression of cytochrome c, AIF and ENDO G in HSC3 cells, and by confocal laser microscopy examination we also found that fisetin promoted the release of cytochrome c, AIF and ENDO G in HSC3 cells from mitochondria into the cytoplasm (Figure 7-C). Based on these observations, we suggesting fisetin-induced apoptotic cell death may also occur through mitochondria-dependent pathway in HSC3 cells.
In conclusion, we examined the cytotoxic effects of fisetin on human oral cancer HSC3 cells in vitro and found that fisetin reduced viable cells through the induction of apoptosis. Fisetin triggered apoptotic cell death via the induction of ROS, ER stress and by disrupting the mitochondria membrane potential, which caused cytochrome c, AIF and ENDO G release from mitochondria into the cytosol. Cytochrome c subsequently activated caspase-9 and downstream executioner caspase-3, leading to apoptosis. Apoptosis via the extrinsic pathway, which involves caspases (caspase-8), and the intrinsic pathways leading to AIF and ENDO G release from mitochondria causing apoptosis are summarized in Figure 8.

Fisetin affects the translocation of apoptotic-associated proteins in HSC3 cells. Cells were treated with 40 μM fisetin for 24 h and cells were stained by antitobodies to cytochrome c (A), apoptosis-inducing factor (AIF) (B) and endonuclease G (ENDO G) (C) and then were stained with a secondary antibody and photographed by a Leica TCS SP2 confocal laser microscopic system as described in the Materials and Methods section.

The possible signaling pathways for fisetin-induced apoptosis in HSC3 human oral cancer cells. B-cell lymphoma 2 (BCL2), BH3-interacting domain death agonist (BID), BCL2-associated X (BAX). Apoptosis-inducing factor (AIF), endonuclease G (ENDO G), cytochrome c (Cyto. c), Fas cell surface death receptor (FAS), activating transcription factor 6-beta (ATF-6β), glucose-regulated protein 78 (GRP78), endoplasmic reticulum (ER) stress), calcium (Ca2+), mitochondrial membrane potential (Ψm).
Acknowledgements
This study was supported by the grant 103-48 from Cheng Hsin General Hospital and by the grant SKH-8302-103-NDR-09 from Shin Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan. Experiments and data analysis were performed in part through the use of the Medical Research Core Facilities Center, Office of Research & Development at China medical University, Taichung, Taiwan.
Footnotes
↵* These Authors contributed equally to this study.
This article is freely accessible online.
- Received August 14, 2017.
- Revision received September 8, 2017.
- Accepted September 14, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Unlocking Synergistic Potential: Enhancing Regorafenib Efficacy in Hepatocellular Carcinoma Through Combination Therapy With 18{beta}-Glycyrrhetinic Acid
- A pipeline for natural small molecule inhibitors of endoplasmic reticulum stress
- Fisetin Suppresses Human Osteosarcoma U-2 OS Cell Migration and Invasion via Affecting FAK, uPA and NF-ĸB Signaling Pathway In Vitro
- Fisetin Enhances the Cytotoxicity of Gemcitabine by Down-regulating ERK-MYC in MiaPaca-2 Human Pancreatic Cancer Cells