


Abstract
At the cellular level, free radicals are tightly controlled by an inducible antioxidant program, since at low non-hazardous amounts they contribute to physiological signalling and homeostasis. However, high levels of oxidative stress promote the accumulation of damaged biomolecules, the impairment of cell signalling pathways and the increase of oncogenic hits. As the intracellular and extracellular levels of oxidative stress increase during ageing or in various diseases, so does the amount of damaged biomolecules, since the repair mechanisms are also targets of oxidative damage and thus become gradually ineffective over time. Depending on the severity of the biomolecular damage, the responses of normal human cells to oxidants may range from transient growth arrest to premature senescence, and even to cell death. Although some responses are clearly tumour suppressing (apoptosis), others may be potentially oncogenic as they combine damage accumulation with a retained ability for proliferation (transient growth arrest) or with inflammation (senescence, necrosis). This array of events significantly increases the likelihood of the appearance of tumour-initiating cells, which may then give rise to pre-neoplastic focal lesions and eventually to neoplasia. In the present manuscript, we will focus on the role of free radical-mediated biomolecular damage and inflammation in tumorigenesis.
Tumorigenesis is a multistage, multifactorial process that is generally described by three stages: initiation, promotion, and progression. Specifically, several factors disrupt the molecular signalling networks, providing tumour cells with sufficient proliferative advantage and diversity to evolve from healthy tissue and eventually to metastasize (1, 2). In 2000, Hanahan and Weinberg (3) proposed that six essential alterations in cell physiology (hallmarks) were required to promote malignant growth, leading to the development of a tumour. These alterations included self-sufficiency in growth signals, insensitivity to growth-inhibitory signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis; recently, this list has been expanded to also include the evasion of immune surveillance and the deregulation of cellular energetics (4, 5). Additional tumour-enabling characteristics include cancer-related inflammation, genome instability and mutation, as well as oxidative and proteotoxic stress (5, 6-8).
The stress phenotypes of cancer cells include (among others) DNA damage/replication stress, proteotoxic stress, mitotic stress, metabolic stress and oxidative stress. Although these features are not restricted to cancer cells, they certainly represent a common set of oncogenesis-associated cellular stresses with which the cell must cope (7).
During recent decades, the role of free radicals, mainly reactive nitrogen species (RNS) and reactive oxygen species (ROS), in cellular physiology has been studied extensively (9). Oxygen free radicals are produced by both endogenous and exogenous sources (see below) and are potential carcinogens because at high levels they cause significant biomolecular damage contributing to all three stages of tumour formation, namely mutagenesis, tumour promotion, and progression (10-12).
Inflammation has been linked to cancer since the end of the 19th century when Rudolph Virchow first noted leucocytes in neoplastic tissues and made a connection between inflammation and cancer. As was later shown, tumours also often develop at sites of chronic inflammation (13). Although inflammation, both related to increased endogenous oxidative stress and as a cause from oxidants exposure is linked with cancer, only recently have the mechanisms underlying this association started to be elucidated. For example, it has been demonstrated that inflammation down-regulates the mismatch repair (MMR) proteins by a variety of mechanisms leading to microsatellite instability (MSI) (6). Growth factors and chemokines produced by inflammatory cells also induce the overexpression of a number of transcription factors [e.g. nuclear factor-kappaB (NF-κB), signal transducer and activator of transcription 3 (STAT3), activating protein-1 (AP-1), hypoxia-inducible factor-1 (HIF-1)], as well as an altered expression of specific microRNAs in cancer cells that promote the expression of genes related to cell growth, apoptosis and invasion (14).
In the current review, we focus on the role of oxidative stress-mediated biomolecular damage and inflammation in tumorigenesis.
Signalling Pathways in Human Cells Affected by Oxidants
Exogenous sources of free radical production include UV light, X-rays, gamma rays, metal catalyzed reactions, and atmospheric pollutants. Endogenous oxygen free radicals may arise as second messengers during inflammatory neutrophil and macrophage cell activation, in various signal transduction pathways, or as by-products of normal aerobic metabolism [i.e. oxidative energy or P450 metabolism, peroxisome activity, or excessive stimulation of NAD(P)H oxidases] (11, 15, 16). The survival of an organism depends on the ability of its cells and tissues to adapt to or resist stress, and repair or remove damaged molecules or cells. The redox balance within the cell is achieved by the action of antioxidants (vitamins C and E, and thiol antioxidants such as glutathione and thioredoxin) and antioxidant enzymes [superoxide dismutase (SOD), catalase, glutathione peroxidase, glutathione reductase] (17).
The cellular responses to free radicals include a wide range of mechanisms that are rapidly activated in response to oxidative insults (11). At the heart of the antioxidant and cellular detoxification program lie the Kelch-like ECH-associated protein 1 (Keap1)–NF-E2-related factor-2 (NRF2) regulatory pathway, which plays a central role in the protection of cells against oxidative and xenobiotic damage (18). The actin-associated protein KEAP1 binds to and tethers NRF2 in the cytoplasm; upon stress, this association is disrupted resulting in the release of NRF2 which then translocates into the nucleus to perform its transcriptional activity (19). Besides NRF2, ROS are also involved in the activation of additional transcription factors including, NF-κB, heat shock factor-1 (HSF1), AP-1, p53 and STATs (15, 20). NF-κB transcription factor is a dimer in its active form, consisting of proteins from the REL family, namely p50, p52, c-REL, v-REL, p65, and REL B (21). NF-κB is involved in the regulation of a wide array of different genes involved in stress response, inflammation, immune function, differentiation, apoptosis, cell survival, and growth (21, 22). Similarly to NRF2, HSF1 is kept inactive in the cytosol in the absence of stress, mainly by binding to HSP90 (23). Upon stress, the inhibiting chaperones bind misfolded or damaged proteins, liberating HSF1, which can then translocate to the nucleus and activate several target genes (23, 24). The activation of AP-1 by cytokines, H2O2 or other physical and chemical stresses mostly results in increased cell proliferation (25, 26), while p53, the so-called guardian of the genome, is critically involved in the processes of many different stress responses (27). Besides the aforementioned nuclear transcription factors, free radicals can also regulate a wide panel of kinases or phosphatases, as well as the H-Ras oncogene and the transforming growth factor-beta (TGF-β) (11).
Among RNS, nitric oxide (NO) occupies a prominent position as it is involved in physiological processes, including vasodilatation, neurotransmission and host defence (14). Low levels of NO are involved in the formation of cGMP that, among other processes, ultimately regulate cell growth and differentiation (14, 28). The effects of ROS on the cell are also partly mediated by mitogen-activated protein kinases (MAPKs) (29). In mammalian systems, there are three subfamilies of MAPKs: extracellular signal-regulated kinases (ERKs) that mediate cell proliferation, and c-Jun N-terminal kinases (JNKs) with p38 MAPKs (also known as stress-activated protein kinases) that mediate stress responses and apoptosis (30).
Oxidative Stress-mediated Biomolecular Damage and Tumorigenesis
Increased oxidative stress is causally related to various diseases, including atherosclerosis, cardiovascular disease, diabetes, inflammatory joint disease, rheumatoid arthritis, neurological diseases and a wide range of tumour types (31). High levels of oxidative stress are established when the production of free radicals exceeds the cell's ability to eliminate them, resulting thus in the deregulation of redox-sensitive signalling pathways, as well as in the damage of most cellular biomolecules, including DNA, proteins, sugars and lipids (10, 32, 33).
ROS can cause DNA alterations, such as apurinic/apyrimidinic DNA sites, oxidized purines and pyrimidines, single- and double-strand DNA breaks (SSBs and DSBs), and oxidatively generated non-DSB clustered DNA lesions (OCDLs) (34, 35). DNA protein cross-linkages may also arise that cannot be effectively repaired, due to cumulative oxidative stress (36). The two most common DNA base modifications are 8-oxo-7,8-dihydroguanine (8-oxodG) and 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapydG). Both arise from the addition of a hydroxyl radical to the C8 position of the guanine ring, producing the 8-hydroxy-7,8-dihydroguanyl radical that can be oxidized to 8-oxodG or reduced to FapydG (37). The latter is an unstable open-ring structure and seems to be the prevalent guanine-derived lesion formed under low oxygen conditions (38). Hydroxyl radical may also interact with pyrimidines giving rise, among other products, to 5,6-dihydroxy-5,6-dihydrothymine (thymine glycol) and 5,6-dihydroxy-5,6-dihydrocytosine (cytosine glycol) (35). It has been estimated that in a given cell, about 105 oxidative lesions per day are formed (39). Of these, 8-oxodG and thymine glycol are used as markers of oxidative stress, due to their abundance (35). The apurinic/apyrimidinic DNA sites are mainly formed as intermediates during the repair process of oxidized bases and they have a high mutagenic potential as they are able to block DNA polymerases (40). The interaction between hydroxyl radicals and the deoxyribose backbone of DNA can lead to SSBs and DSBs of DNA (35). When SSBs and oxidized bases occur in a clustered formation (i.e. within two or more bases within few helical turns), they give rise to OCDLs which are complex, difficult to repair lesions that can give rise to chromosomal instability (41). In addition, ROS-mediated mutations in mitochondrial DNA (mtDNA) are emerging as important contributors to tumorigenesis (42). Sensitivity of mtDNA to ROS may relate to the fact that it is located in close proximity to the respiratory chain, it is not protected by histones and has a limited repair capacity (43, 44). Moreover, ROS can cause epigenetic alterations in particular, alterations in the DNA methylation pattern of genes. For example, exposure of hepatocellular carcinoma cells to hydrogen peroxide induces the expression of the transcription factor SNAIL which recruits histone deacetylase 1 and DNA methyltransferase 1 on E-cadherin gene promoter, resulting in gene silencing by hypermethylation (45). Down-regulation of E-cadherin has been correlated with epithelial-to-mesenchymal transition (EMT) and metastasis in a wide range of carcinomas (46). Interestingly, a number of tumour suppressor genes (e.g. p15INK4B and p16INK4A) have been identified as being silenced by oxidative-induced aberrant CpG island promoter methylation (47, 48). These latter observations suggest that there may be sites in the genome that are more susceptible to oxidative stress-related pro-tumorigenic effects.
Sustained oxidative stress also affects the proteome causing protein damage (e.g. via formation of peroxyl radicals or by oxidation of cysteine residues) that triggers conformational changes which ultimately lead to enhanced or diminished binding capacity to other proteins or to DNA (in the case of transcription factors), as well as to enzymatic inactivation (49, 50). These modifications may lead to loss of function, gain of function, or switch to a different function. For instance, in the case of inhibition of phosphatases, their inability to regulate kinase-mediated transduction pathways can lead to alterations of physiological functions such as aberrant cellular growth (51).
Apparently, the free radical-mediated biomolecular damage will eventually promote the deregulation of the various redox-sensitive signalling pathways. Deregulation of AP-1 protein signalling may participate in oncogenic transformation by interacting with activated oncogenes such as H-Ras (52), while p53 signalling is disrupted in the majority of human cancer types (53). Involvement of NF-κB in tumorigenesis mostly occurs in tissues in which cancer-related inflammation typically occurs (such as the gastrointestinal tract and the liver); recent studies have also shown that NF-κB signalling plays critical roles in EMT and cancer stem cell physiology (22). In non-stressed cells, the upstream regulator of p38 MAPKs (see above), apoptosis signal-regulating kinase 1 (ASK1), is associated with thioredoxin and thus, inhibited (54). When ROS levels increase, this complex dissociates and ASK1 exerts its function by activating p38 MAPKs (55) leading to growth arrest and/or apoptosis. On the other hand, in cancer cells, ROS induce the hyperphosphorylation of JNK that leads to AP-1 activation and to enhanced cellular proliferation (30). ROS-generated during hypoxia can prevent the hydroxylation (and thus, the degradation by the ubiquitin-proteasome system) of the transcription factor HIF-1α, allowing its translocation to the nucleus, where it dimerizes with HIF-1β, promoting the transcription of genes that mediate proliferation and survival in conditions of limited oxygen availability (56). Interestingly, recent studies revealed that RAS-driven proliferation requires ROS buffering of RAS-activated ERK1/2 activity (57), while the NRF2 antioxidant and cellular detoxification program may enhance oncogenesis in certain cellular settings (58). Finally, the ataxia-telangiectasia mutated (ATM) protein kinase, which is best known for its role in the DNA damage response, was recently found to function as a redox sensor that controls the levels of ROS in human cells (59).
Sustained oxidative stress or ROS-mediated deregulation of cell signalling in the early phases of tumorigenesis may trigger a wide panel of responses in normal human cells, depending on the level of damage, including reversible cell cycle arrest and repair, cellular senescence or death (apoptosis or necrosis) (11, 20, 60, 61). Although some of these responses are clearly tumour-suppressive (apoptosis), others may be potentially oncogenic as they combine damage accumulation with a retained ability for proliferation (transient growth arrest) or with inflammation (senescence, necrosis; see also below). It seems also that during tumorigenesis, cancer cells gradually acquire higher levels of oxidative stress. This property may relate to overproduction of ROS by mitochondria with impaired electron transport chain and mtDNA damage (62), or to inactivation of antioxidant enzymes. A decreased activity and expression of the mitochondrial form of manganese superoxide dismutase (Mn-SOD) has been reported in colorectal carcinomas (63) and pancreatic cancer cells (64). High endogenous levels of oxidative stress have also been found in various types of leukaemia (65), in human colorectal carcinoma (66), as well as in breast (67), stomach (68) and ovarian (69) cancer. Moreover, ROS levels in prostate cancer cells correlate positively to tumour aggressiveness (70).
These data clearly indicate that oxidative stress is at work throughout the stages of tumorigenesis (20), either directly by promoting biomolecular damage, or indirectly by deregulating the signalling pathways involved in proliferation, angiogenesis and metastasis. Cellular stress responses may not always result in cell death as it is becoming clear that transformed cells suppress stress signals in order to survive. During the early stages of tumorigenesis, this suppression is not complete and cells can still be sensitized to stress by anticancer drugs (71), but in those cells that progress towards a malignant phenotype this suppression is accomplished. This may explain why an anticancer therapy that is efficient in early stages of the disease loses efficacy as the tumour advances.
Chronic Inflammation, Oxidative Stress and Tumour Formation
Chronic inflammation is characterized by tissue destruction due to active inflammatory responses that induce oxidative stress and reduce the cellular antioxidant capacity. In some cases, inflammation precedes the malignant transformation, while in others, an oncogenic alteration induces an inflammatory microenvironment that accelerates the development of tumours. Two pathways linking inflammation and cancer have been proposed. In the intrinsic one, activation of different classes of oncogenes drives the expression of inflammation-related programs that lead to the establishment of an inflammatory milieu; while in the extrinsic one, inflammatory conditions promote cancer development (72).
A number of chronic inflammatory and infectious diseases that increase cancer risk have been described, including autoimmune diseases (e.g. inflammatory bowel disease for colon cancer) and microbial infections (e.g. Helicobacter pylori for gastric cancer and mucosal lymphoma) (72). There is also emerging evidence that prostatic inflammation may contribute to prostate cancer growth by inducing hyperplastic or neoplastic changes (73, 74), while pancreatic inflammation may play a key role in the early development of pancreatic malignancy (75). Epidemiological data have also demonstrated a positive relation between colorectal malignancy and obesity (76). B-Lymphocytes mediate chronic inflammatory states that promote de novo epithelial carcinogenesis (77), while the acidic microenvironment produced by the tumour cells inhibits the antitumor function of cytotoxic T-lymphocytes (CTL) (78) and natural killer (NK) cells (79). Interestingly, there are some notable exceptions; rheumatoid arthritis is an example of a chronic inflammatory disease without an increased cancer risk, whereas oncogenic human papilloma viruses are examples of cancer-prone chronic infections without inflammation (14).
The innate immune cells [neutrophils, monocytes/macrophages, mast cells, dendritic cells (DC) and NK cells] mediate the inflammatory response by releasing cytokines, chemokines, matrix-remodelling proteases and free radicals. Moreover, DC and NK cells activate, in principle via cytokine release, the adaptive immune response (80).
A prolonged generation of ROS and RNS contributes to the persistence and accordingly to the pathological consequences of chronic inflammation (81). As mentioned, depending on the level of ROS, different redox-sensitive transcription factors are activated and coordinate distinct biological responses. A low sustainable oxidative stress mainly induces NRF2, while an intermediate amount of ROS triggers an inflammatory response through the activation of NF-κB and AP-1. Finally, high levels of oxidative stress trigger the disruption of electron transfer, thereby resulting in apoptosis or necrosis (82). NF-κB is a transcription factor linking inflammation and cancer. NF-κB regulates several genes whose products inhibit apoptosis and enhance cell cycle progression, angiogenesis and metastasis (83). A number of NF-κB target genes encode mediators of the innate immune response and inflammation, which include cytokines, chemokines and cyclooxygenase 2 (COX2) (which leads to the production of prostaglandins), as well as NOS2 that is the inducible isoform of the nitric oxide synthase (84). However, NF-κB can protect cells from oxidative stress inducing the expression of ferritin heavy chain and superoxide dismutase 2 (SOD2) (85, 86). Interestingly, ROS seem to have an inhibitory effect on NF-κB activation in lung epithelial cells (87).
Besides immune inflammatory cells, it also seems that tumour-promoting inflammatory processes can be triggered by senescent or necrotic cells. Specifically, it has been found that young human cells, beyond telomere-dependent replicative senescence (88), can also become senescent (despite having long telomeres) if they are exposed to subcytotoxic oxidative stress (60); these prematurely senescent normal human cells accumulate during in vivo human ageing (60, 88). Recent studies have shown a striking increase in the secretion of pro-inflammatory proteins by senescent cells, rendering this cellular state an important additional contributor to chronic inflammation and tumorigenesis (89). Similarly, necrotic cell death by acute oxidative stress releases pro-inflammatory signals into the surrounding tissue microenvironment that recruit inflammatory cells of the immune system which, apart from surveying the extent of tissue damage and removing the associated necrotic debris, may also, in the context of neoplasia, be actively tumour promoting (90).
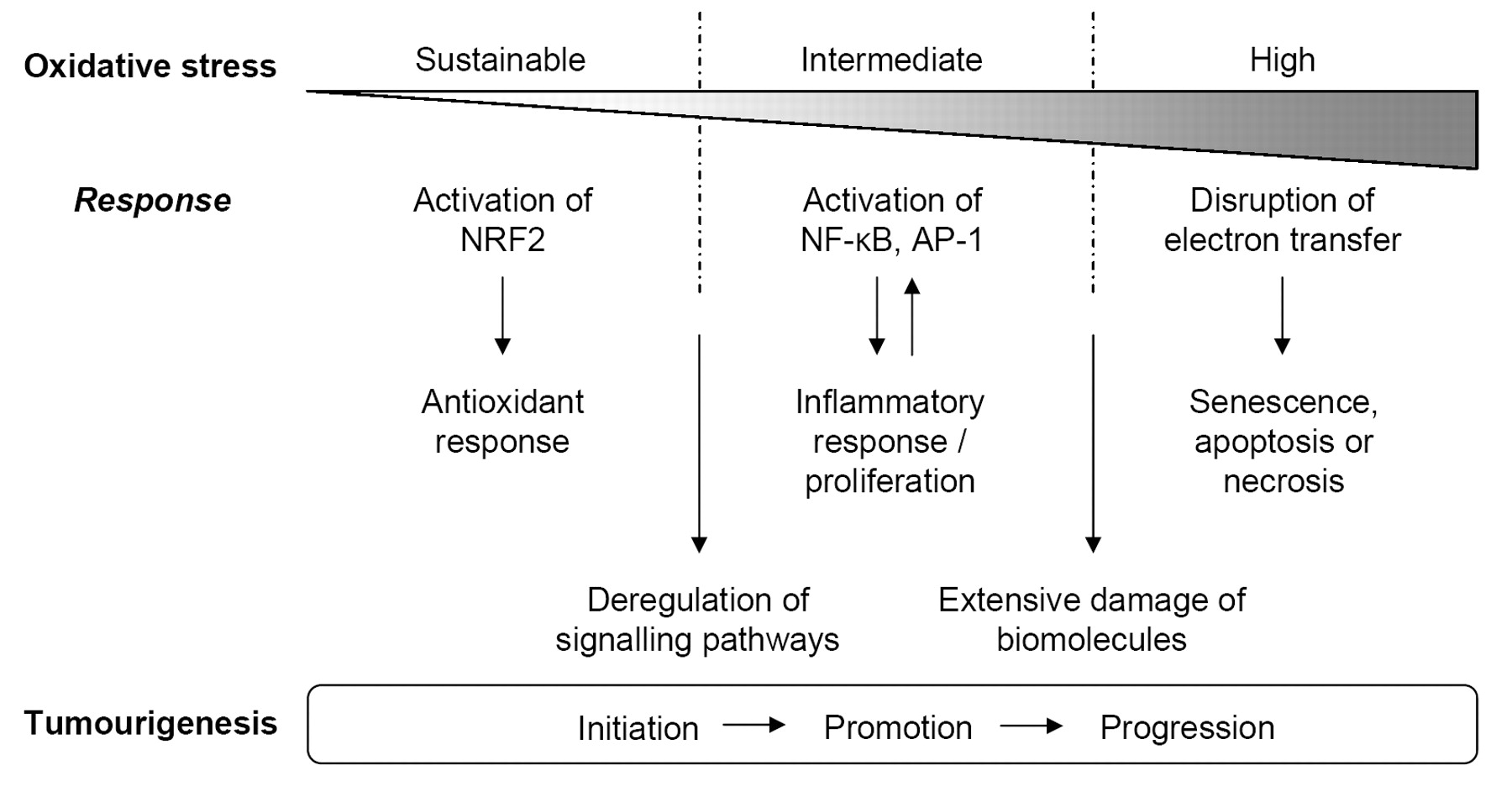
Gradual increase of cellular oxidative stress triggers a series of responses that may favour tumourigenesis. Pro-tumourigenic effects mostly relate to deregulation of signalling pathways and to extensive damage of all types of biomolecules.
Collectively, these data indicate that the inflammatory response triggered directly or indirectly by a prolonged state of oxidative stress can have potent pro-tumorigenic effects. Until recently, these effects were thought to affect only tumour-surrounding cells (91), but evidence has been provided that cytokines can also exert a detrimental function in tissues distant from non-metastatic early stage tumours (92). Indeed, an elevated serum level of chemokine (C-C) ligand 2 (CCL2), that correlated with DSBs and OCDLs in distant proliferative tissues of tumour-bearing mice has been observed; CCL2 is thought to be produced by the activated macrophages that were found to infiltrate not only the tumour, but also distant tissues (92). These interesting findings suggest that the onset of systemic aspects of cancer may arise earlier than previously believed (93).
Conclusion
As cancer research progresses, we are becoming increasingly aware that the abilities that cancer cells must acquire (self-sustained growth, limitless replicative potential, angiogenesis) or lose (insensitivity to growth-inhibitory signals and to apoptosis) during tumorigenesis are not gained through a single and unequivocal sequence of events (5). In this view, the state of the cells (e.g. redox state) acquires a fundamental importance as it can drive those changes by both altering the balance of pathways involved in cell proliferation at the proteome level, as well as by inducing direct genetic damage (20). The notion that ROS can also trigger an inflammatory response (81) and that, in turn, inflammation can contribute to the establishment of increased oxidative stress in cells (84), reveals that mutual reinforcement of certain, otherwise, physiological conditions can determine a state of imbalanced signalling which may lead to tumour initiation, promotion and progression (Figure 1). Therefore, inflammation and oxidative stress may lead to transformation of cells and tumour progression through sustained stimuli. Interestingly, metastasis has recently been proposed as a means of escape from oxidative stress (94). Of note, the cytoskeleton rearrangement underlying cell motility uses ROS as intermediates (95) and is therefore facilitated by oxidative stress.
The modulation of ROS, and more generally of redox homeostasis, holds promise as an effective cancer treatment of high specificity targeting, mainly cancer cells, due to the differences in ROS levels between normal and tumour cells (62). Indeed, it has recently been demonstrated that a natural compound, piperlongumine, was able to selectively kill cancer cells by inducing an increase in ROS levels and apoptotic cell death, both in vitro and in vivo (96); notably, this effect was evident, irrespective of the cell's p53 status. In a similar, and relatively unexplored approach, the increased reliance of transformed cells on systems that ameliorate the deleterious effects of proteotoxic stress (e.g. chaperones or the proteolytic systems) could be a promising strategy for the development of novel cancer-targeting therapeutic agents. Moreover, blocking a source of persistent inflammation can enhance cancer immunotherapy, as was recently demonstrated for CCL2 (97, 98). In conclusion, as more levels of complexity are added to our current notion of tumorigenesis, we are getting closer to unravelling the mechanisms that determine the various phases of human cell transformation. This knowledge will eventually lead to the development of novel combinatorial antitumour therapies.
Acknowledgements
We apologize to those investigators whose work, due to space constrains, was not cited. This work was supported by a European Union FP7 Capacities grant INsPiRE (REGPOT-CT-2011-284460).
- Received February 15, 2012.
- Revision received March 8, 2012.
- Accepted March 9, 2012.
- Copyright © 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- Changes in Serum Interleukin-8 and sRAGE Levels in Multiple Myeloma Patients
- Cholecystectomy Patients with High Plasma Level of Catalase Have Significantly Lower Analgesia Requirement: A Prospective Study of Two Different Cholecystectomy Techniques with Special Reference to Patients with Cancer
- Gallstone Patients with Enhanced Oxidative Stress Biomarker Superoxide Dismutase (SOD1) Plasma Levels Have Significantly Lower Number of Postoperative Analgesic Oxycodone Doses: A Prospective Study with Special Reference to Cancer Patients
- LINE-1 ORF1 Protein Is Up-regulated by Reactive Oxygen Species and Associated with Bladder Urothelial Carcinoma Progression
- Low Levels of Microsatellite Instability at Simple Repeated Sequences Commonly Occur in Human Hepatocellular Carcinoma
- Silibinin Induces Cell Death through Reactive Oxygen Species-Dependent Downregulation of Notch-1/ERK/Akt Signaling in Human Breast Cancer Cells