Article Text

Abstract
Background: Tumour-specific cytotoxic T lymphocytes (CTLs) can be activated in vivo by vaccination with dendritic cells (DCs). However, clinical responses to DC-based vaccination have only been observed in a minority of patients with solid cancer. Combination with other treatment modalities such as chemotherapy may overcome immunoresistance of cancer cells. It has been shown previously that gemcitabine sensitises human pancreatic carcinoma cells against CTL-mediated lysis. Here, a murine pancreatic carcinoma model was used to investigate whether combination with gemcitabine increases therapeutic efficacy of DC-based vaccination.
Methods: Bone marrow-derived DCs from C57BL/6 mice were loaded with UV-irradiated, syngeneic Panc02 carcinoma cells and were administered subcutaneously. For prophylactic vaccination, mice were vaccinated three times at weekly intervals prior to tumour challenge with Panc02 cells. Therapeutic vaccination was started when tumours formed a palpable nodule. Gemcitabine was administered intraperitoneally twice weekly.
Results: Prophylactic DC-based vaccination completely prevented subcutaneous and orthotopic tumour development and induced immunological memory as well as tumour antigen-specific CTLs. In the subcutaneous tumour model, therapeutic DC-based vaccination was equally effective as gemcitabine (14% vs 17% survival at day 58 after tumour challenge; controls, 0%). Combination of the two strategies significantly increased survival of tumour-bearing mice (50% at day 58 after tumour challenge). DC-based vaccination also prevented death from pulmonary metastatisation after intravenous injection of Panc02 cells.
Conclusion: DC-based immunotherapy may not only be successfully combined with gemcitabine for the treatment of advanced pancreatic carcinoma, but may also be effective in preventing local recurrence or metastatisation in tumour-free patients.
- CD, cluster of differentiation
- CFSE, carboxyfluorescein diacetate-succinimidyl ester
- CTL, cytotoxic T lymphocyte
- DC, dendritic cell
- DMEM, Dulbecco’s modified Eagle’s medium
- FACS, fluorescence-activated cell sorting
- FCS, fetal calf serum
- 5-FU, 5-fluorouracil
- GM-CSF, granulocyte–macrophage colony-stimulating factor
- IFN, interferon
- IL, interleukin
- LPS, lipopolysaccharide
- mAb, monoclonal antibody
- MHC, major histocompatibility complex
- MuLV, murine leukaemia virus
- NK, natural killer
- TGF, transforming growth factor
- Th1, T helper type 1
- TNF, tumour necrosis factor
Statistics from Altmetric.com
- CD, cluster of differentiation
- CFSE, carboxyfluorescein diacetate-succinimidyl ester
- CTL, cytotoxic T lymphocyte
- DC, dendritic cell
- DMEM, Dulbecco’s modified Eagle’s medium
- FACS, fluorescence-activated cell sorting
- FCS, fetal calf serum
- 5-FU, 5-fluorouracil
- GM-CSF, granulocyte–macrophage colony-stimulating factor
- IFN, interferon
- IL, interleukin
- LPS, lipopolysaccharide
- mAb, monoclonal antibody
- MHC, major histocompatibility complex
- MuLV, murine leukaemia virus
- NK, natural killer
- TGF, transforming growth factor
- Th1, T helper type 1
- TNF, tumour necrosis factor
Pancreatic carcinoma represents the fifth leading cause of cancer death in the Western world. The 5-year overall survival rate is <5%.1 Pancreaticoduodenectomy represents the only curative form of treatment. However, only 10–15% of patients diagnosed are eligible for surgical treatment. Chemotherapy with gemcitabine is currently the most effective treatment for advanced pancreatic carcinoma, resulting in a moderate increase in survival compared with 5-fluorouracil (5-FU) treatment.2,3 Recent trials have failed to show an improvement in survival when gemcitabine is combined with other chemotherapeutic drugs.4 Therefore, novel strategies are required for the treatment of advanced pancreatic cancer. Pancreatic carcinoma cells can be recognised by tumour-specific T cells.5,6 Moreover, tumour infiltration with cytotoxic T lymphocytes (CTLs) and T helper cells represents a favourable prognostic factor for patients with pancreatic adenocarcinoma.7 Tumour-reactive T cells capable of tumour rejection can also be isolated from blood of pancreatic carcinoma patients.8
Tumour-specific T cells can be activated by vaccination with dendritic cells (DCs).9,10 DCs are specialised antigen-presenting cells with the unique capacity to establish primary immune responses.11 DCs can deliver exogenous antigens into the major histocompatibility complex (MHC) class I processing pathway to activate CTLs, a process termed “cross-presentation”.12 Contact with microbial or inflammatory “danger signals” as well as T cell-derived activation signals induces DC maturation and interleukin (IL)12 secretion.13,14 IL12 plays a central role in regulating tumour-directed immune responses and stimulates natural killer (NK) cells, CTLs and interferon (IFN)γ-producing T helper type 1 (Th1) cells. Vaccination with tumour antigen-loaded DCs has been shown to elicit antitumoural CTL responses in vivo and to induce tumour regression in cancer patients.9,15–17 Pancreatic carcinoma cells can be rejected in vivo after vaccination with tumour antigen-loaded DCs.18,19 However, tumours are capable of forming an immunosuppressive environment rendering them insensitive to CTLs. Combination with other treatment strategies such as radiation or chemotherapy may break the immunoresistance of cancer cells and enhance the therapeutic efficacy of DC-based vaccination.
Gemcitabine not only exerts direct antitumoural activity, but also mediates immunological effects relevant for tumour immunotherapy.20–22 Antitumoural CTL responses can be induced by DCs cross-presenting antigens of tumour cells treated with a multidrug regime including gemcitabine.23 Enhanced cross-presentation of tumour antigens by DCs after gemcitabine treatment also leads to increased tumour recognition by CTLs in vivo.24 Recently, we were able to demonstrate that gemcitabine sensitises human pancreatic carcinoma cells to DC-induced tumour-specific CTL responses.25 Here, we used a murine pancreatic carcinoma model to investigate whether combination with gemcitabine augments therapeutic efficacy of DC-based vaccination in vivo.
MATERIALS AND METHODS
Mice, cell lines and media
Animal studies were approved by the Regierung von Oberbayern, Munich, Germany. C57BL/6 mice were from Harlan Winkelmann (Borchen, Germany). Transgenic OT-1 animals from the C57BL/6 background were provided by Professor Brocker (Department of Immunology, University of Munich). OT-1 T cells were grown in RPMI (Biochrome, Berlin, Germany) with 10% fetal calf serum (FCS; Gibco, Invitrogen Cooperation, Paisley, UK), penicillin and streptomycin. The H2-b-positive cell line Panc02 is derived from a methylcholanthrene-induced pancreatic adenocarcinoma in C57BL/6 mice.26,27 Panc02 cells were maintained in RPMI with 10% FCS, 2 mM l-glutamine (Life Technologies, Paisley, UK), 100 U/ml penicillin (Sigma, Munich, Germany) and 100 μg/ml streptomycin (Seromed, Jülich, Germany). Prior to co-incubation with DCs, Panc02 cells were irradiated using UV-B light with 0.75 J/cm2. The viability of UV-irradiated tumour cells was excluded by light microscopy and thymidine incorporation. The H2-Kb-positive lymphoma cell line EL4 (kindly provided by Professor Enders, Institut für Chirurgische Forschung, University of Munich) was maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FCS, penicillin and streptomycin.
mRNA extraction and reverse transcription-PCR
Samples were cleaned in phosphate-buffered saline (PBS), snap-frozen in liquid nitrogen and stored at –70°C. Total RNA was isolated using the Roche Total RNA Tissue Extraction Kit (Roche, Mannheim, Germany) after homogenising tissue with an Ultra Turrax instrument (Janke und Kunkel, Staufen, Germany). RNA was stored with an equal volume of ethanol at –80°C. The yield and purity of the RNA were determined by spectroscopic analysis. For reverse transcription, M-MLV Reverse Transcriptase (Gibco Life Technologies, Paisley, UK), RNase Inhibitor (Roche, Mannheim, Germany), oligo(dT) primer for cDNA synthesis (Roche, Mannheim, Germany) and dNTP (Promega, Madison, WI, USA) were used. PCR was performed with Taq DNA polymerase as recommended by the supplier (Roche, Mannheim, Germany). Primers were manufactured by Applied Biosystems (Weiterstadt, Germany) based on sequences from the literature.28
Monoclonal antibodies and flow cytometry
For phenotypic analysis, DCs were incubated for 30 min at 4°C with 5 μg/2×105 cells anti-mouse CD86-FITC monoclonal antibody (mAb; clone GL1), CD11b-PerCP mAb (clone M1/70), CD11c-APC mAb (clone HL3; all from BD Biosciences, San Jose, CA, USA), CD80-PE mAb (clone RMMP1, Caltag, Burlingame, CA, USA), MHC ll-PE mAb (clone NIMR-4, Southern Biotech, Birmingham, AL, USA) and appropriate isotype controls. Cells were examined by flow cytometry (FACSCalibur, BD Biosciences, Heidelberg, Germany). Data were analysed using CellQuest software (BD Biosciences, Heidelberg, Germany). MHC class I expression was examined using anti-mouse mAb (H2-Kb, clone AF6-88.5; and H2-Db, clone KH95, both from BD Biosciences, San Jose, CA, USA).
Peptides
The H2-Kb restricted peptides OVA257–264, TRP2181–188 and p15E604–611 were from Jerini Peptide Technologies (Berlin, Germany).29–31
Reagents
Gemcitabine (Gemzar®) was from Lilly (Indianapolis, IN, USA). Midazolam (Roche, Basel, Switzerland), medetomidin (Orion Corporation, Espoo, Finland), fentanyl (Janssen-Cilag, Neuss, Germany) and naloxon (Deltaselect, Dreieich, Germany) were used for anaesthesia.
Cytokines, growth factors, stimulating agents and ELISAs
Granulocyte–macrophage colony-stimulating factor (GM-CSF), IFNγ, IL1β, IL2 and IL4 were from PeproTech (London, UK) and IFNα was from HyCult Biotechnology (Uden, The Netherlands). Lipopolysaccharide (LPS), p:IC, R848 and tumour necrosis factorα (TNFα) were from Sigma-Aldrich (Steinheim, Germany). Murine ELISA kits for IL12 p70, IL12 p40 and IFNγ were from BD Biosciences (San Diego, CA, USA).
Generation and antigen loading of bone marrow-derived DCs
Bone marrow-derived DCs were prepared as described.32 Bone marrow cells were harvested from murine femur and tibia. Erythrocytes were lysed with ammonium chloride buffer (BD Biosciences, Heidelberg, Germany). Cells were cultured at 5×105 cells/ml in RPMI 1640 medium supplemented with 10% FCS, 2 mM l-glutamine, 100 μg/ml streptomycin, 1 IU/ml penicillin, 20 ng/ml GM-CSF and 20 ng/ml IL4 (DC medium). After 7 days, loosely adherent cells were harvested, and expression of DC markers was quantified by fluorescence-activated cell sorting (FACS). DCs represented 70% of the preparation. Co-incubation with Panc02 cells (referred to as “pulsing” in the following) was carried out at a DC/tumour cell ratio of 10:1. LPS (250 ng/ml) and IFNγ (100 ng/ml) were added for the last 24 h. At day 9, loosely adherent cells were harvested, washed and resuspended in RPMI, and analysed by FACS.
Tumour induction and vaccination strategies
For immunisation, 3×105 DCs were injected subcutaneously into the right flank in a final volume of 200 μl RPMI. Mice were challenged with Panc02 cells by contralateral subcutaneous injection (1×106 in 200 μl of RPMI). Therapeutic vaccination started after tumours had formed a palpable nodule. Gemcitabine was administered intraperitoneally twice weekly in a concentration of 25 or 50 mg/kg body weight. Tumour growth was expressed as the product of perpendicular diameters. Animals were killed when the tumour size exceeded 400 mm2 or when there were signs of animal distress observed twice in 48 h. For orthotopic tumour development, the left flank of anaesthesised mice was opened and the spleen mobilised to access the pancreas. A total of 2×105 Panc02 cells were injected into the pancreas. Tumour growth was monitored by a LaTheta™ in vivo CT scanner for small animals (Zinsser Analytic). Animal CT scans were performed in cooperation with Dr Heinz-Peter Scheuber (TierschutzInformationsZentrum for Biomedical Research, University of Munich) and Adam Glowalla (Zinsser Analytic, Germany).
In vivo cytotoxicity assay
The assay was performed by transferring carboxyfluorescein diacetate-succinimidyl ester (CFSE)high (2 μM)- and CFSElow (0.2 μM)-labelled C57BL/6 splenocytes into vaccinated or non-vaccinated mice. The CFSEhigh-labelled cells were pulsed with the p15E peptide (2 μg/ml), whereas the CFSElow-labelled cells were left unpulsed. A 1:1 ratio of CFSElow- to CFSEhigh-labelled cells was injected intravenously 20 h after injection of the target cells, spleens were removed and the ratio of CFSElow to CFSEhigh cells was determined by flow cytometry. Cytotoxicity was calculated by the formula: % cytotoxicity in a given mouse = ((% CFSEhigh vac/%CFSElow vac))/((% CFSEhigh control/% CFSElow control)).
In vitro cytotoxicity assay
The 51chromium-release assay was performed in cooperation with Professor R Wank, Department of Immunology, University of Munich. Target cells were incubated with Na251CrO4 (100 μCi/106 target cells) at 37°C for 1 h. Cells (5×103/well) were washed three times and co-cultured with effector cells in 96-well round-bottomed plates in a final volume of 200 μl. After 4 h of incubation at 37°C, 50 μl of supernatant were harvested and radioactivity determined by a gamma counter (Wallac Oy, Turku, Finland). Maximum release was assessed by incubation with Triton X-100 (Sigma, Munich, Germany) at a final concentration of 2.5%. Spontaneous release was determined in the absence of effector cells. Specific lysis was calculated according to the formula: specific 51Cr release = ((experimental counts – spontaneous counts)/(maximal counts – spontaneous counts))×100%.
Statistics
Student’s t test was applied to reveal significant differences in tumour protection. We compared the mean tumour size of controls and all treated animals (tumour-free and tumour-positive animals). A value of p<0.05 was accepted as the level of significance. Kaplan–Meier survival curves were analysed using the Cox proportional hazards model.
RESULTS
Vaccination with Panc02-pulsed DCs prevents subcutaneous tumour development
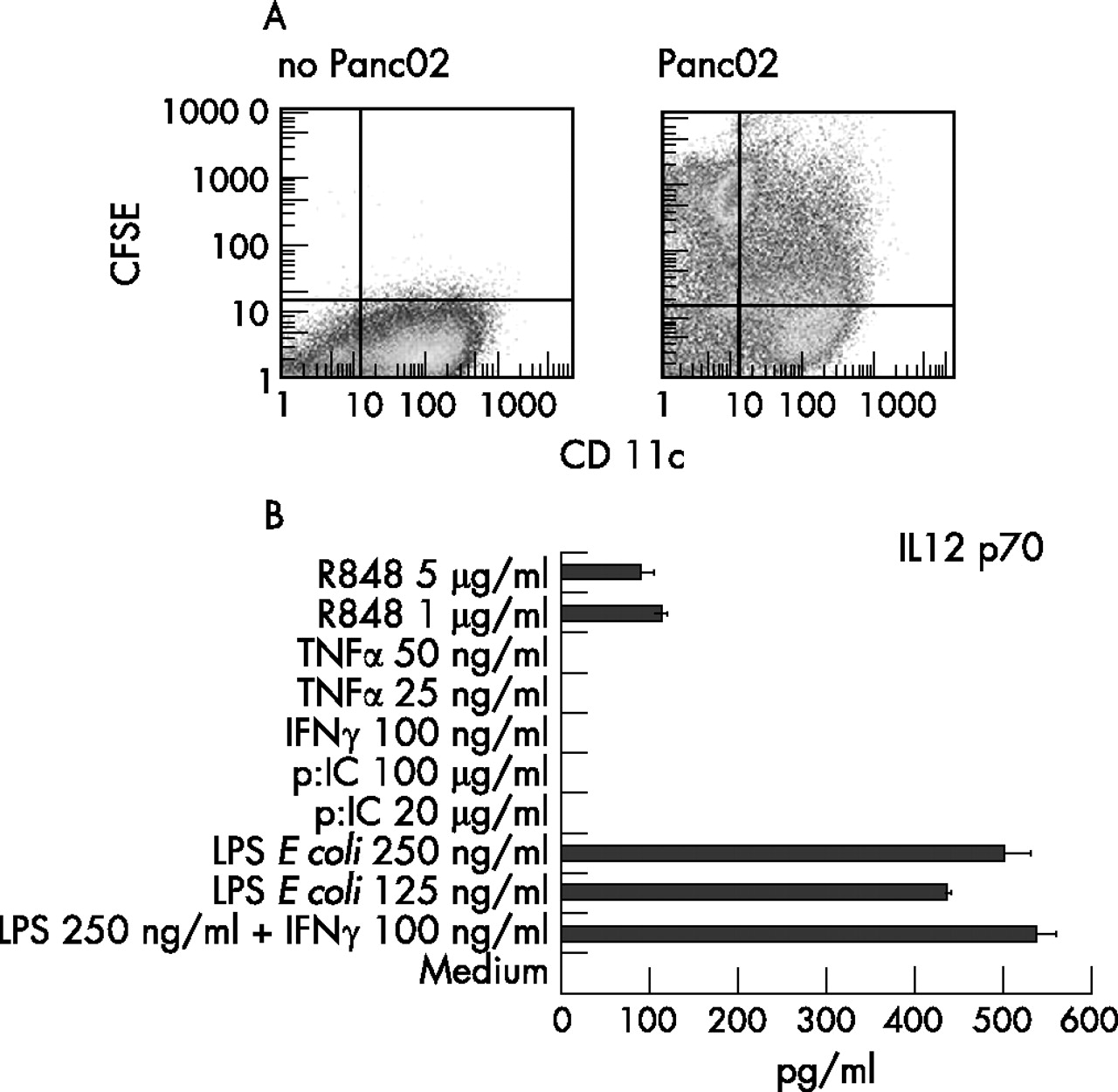
We have previously shown that DCs pulsed with UV-irradiated, apoptotic tumour cells are superior to tumour lysate-pulsed DCs in priming CTLs directed against human pancreatic cancer cells.33 This finding has recently been confirmed in the Panc02 tumour model.34 Thus, we chose UV-irradiated Panc02 cells as a source of tumour antigen. Bone marrow-derived DCs were capable of taking up dead Panc02-cells and secreted IL12 p70 upon stimulation with LPS and IFNγ (fig 1). Mice were completely protected from tumour development after three vaccinations with Panc02-pulsed DCs (fig 2A). In the untreated animals, tumours with a mean size of 110 mm2 developed within 35 days (fig 2A). Tumour size was only moderately reduced in mice receiving UV-irradiated Panc02 cells only or unloaded DCs (mean tumour size after 35 days approximately 40 mm2). Tumour protection in mice vaccinated with Panc02-pulsed DCs was associated with higher levels of IFNγ secretion in splenocytes isolated after two vaccinations (fig 2B).

Bone marrow-derived dendritic cells (DCs) effectively take up tumour antigen and secrete high levels of interleukin (IL)12 p70 after stimulation with lipopolysaccharide (LPS) and interferon (IFN)γ. DCs were derived from bone marrow of C57BL/6 mice by culture with granulocyte–macrophage colony-stimulating factor and IL4. On day 6 of culture, DCs were incubated with UV-irradiated, carboxyfluorescein diacetate-succinimidyl ester (CFSE)-labelled Panc02 cells at a ratio of 10:1. Uptake of tumour cells by CD11c+ DCs was determined by fluorescence-activated cell sorting (FACS) analysis (A; the results of one representative FACS analysis out of five performed are shown). At day 6 of culture, DCs were stimulated for another 24 h with different stimuli. Culture supernatants were harvested and analysed for IL12 production by ELISA (B; data are the mean (SEM) of two independent experiments).

Prophylactic vaccination with Panc02-pulsed dendritic cells (DCs) completely prevents development of subcutaneous Panc02 tumours. Female 6- to 10-week old C57BL/6 mice were vaccinated subcutaneously with either Panc02-pulsed or unpulsed syngeneic DCs (3×105) or UV-irradiated Panc02 cells (1×105) three times at weekly intervals, or were left untreated. Seven days after the last vaccination, mice were challenged subcutaneously with 1×106 Panc02 cells. Subcutaneous tumour growth was monitored at regular intervals and tumour size determined by multiplication of perpendicular diameters (A; data are the mean (SEM) of three animals in each group). In parallel experiments, mice from each group were sacrificed after two vaccinations, and in vitro interferon (IFN)γ production of splenocytes determined by ELISA (B, data are the mean (SEM) of two animals in each group). AG, antigen.
Vaccination with Panc02-pulsed DCs establishes immunological memory and long-term tumour protection
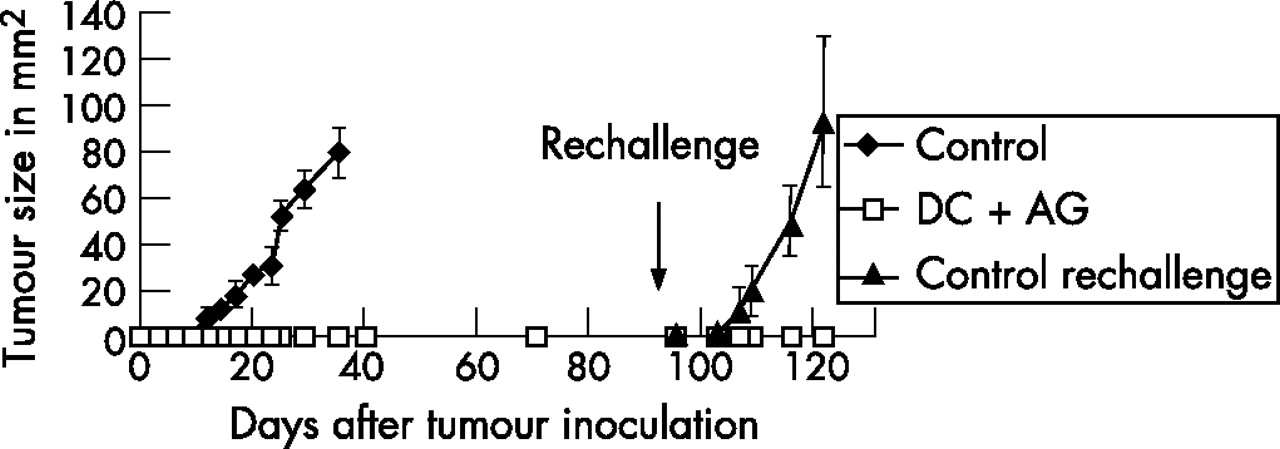
While control animals challenged with Panc02 cells died after 40–50 days, mice that had received prophylactic vaccination with Panc02-pulsed DCs survived more than 3 months without developing a tumour (data not shown). These mice were rechallenged with Panc02 cells at day 95 after the first tumour challenge. As shown in fig 3, they were still protected from tumour development, as opposed to controls that rapidly developed large tumours (mean tumour size 91 mm2 at day 26). Vaccinated mice that were rechallenged with Panc02 at day 95 showed long-term survival and were euthanised months later without signs of local or systemic tumour growth (data not shown).

Prophylactic vaccination with Panc02-pulsed dendritic cells (DCs) induces immunological memory associated with long-term tumour protection. Female 6- to 10-week old C57BL/6 mice were either vaccinated subcutaneously with Panc02-pulsed DCs (3×105) three times at weekly intervals or left untreated and challenged subcutaneously with 1×106 Panc02 cells 7 days after the last vaccination. Tumour size was determined at regular intervals by multiplication of perpendicular diameters. Mice receiving DC vaccination remained tumour free and were rechallenged subcutaneously with 1×106 Panc02 tumour cells at day 95 after initial tumour inoculation. Previously untreated and unchallenged mice served as a control group (data are the mean (SEM) of three animals in each group). AG, antigen
Vaccination with Panc02-pulsed DCs induces tumour-specific CTLs
Although mice could be protected from tumour development, it remained to be shown that DC vaccination induced a tumour-specific CTL response. The virus-associated murine leukaemia virus (MuLV) env-protein is encoded by the gp70 gene and selectively expressed by murine cancer cells. Figure 4A shows gp70 mRNA expression in Panc02 and C26 colon carcinoma cells derived from Balb/c mice, but not in normal tissue. CFSEhigh-labelled splenocytes from C57BL/6 mice loaded with p15E peptide derived from the MuLV env-protein and CFSElow-labelled, unloaded splenocytes were injected into the tail vein of mice either vaccinated three times with Panc02-pulsed DCs or left untreated. By quantifying the relative decrease of p15E-loaded, CFSEhigh splenocytes in vaccinated animals as compared with untreated animals, a specific lysis rate of approximately 12% could be calculated (fig 4B). Thus, Panc02-loaded DCs not only protect animals from tumour development, but also induce immunological memory and a tumour-specific CTL response.

Vaccination with Panc02-pulsed dendritic cells (DCs) induces tumour antigen-specific cytotoxic T lymphocytes (CTLs). The H2-Kb-restricted peptide p15E is derived from the murine leukaemia virus env-protein encoded by the virus-associated gp70 gene. Expression of gp70 mRNA was determined in Panc02 cells and C26 colon carcinoma cells (derived from the Balb/c mouse strain) as well as in samples from liver, spleen, pancreas and lymph nodes of C57BL/6 mice (A). For detection of p15E-specific CTLs in vivo, carboxyfluorescein diacetate-succinimidyl ester (CFSE)high-labelled splenocytes from C57BL/6 mice loaded with p15E and CFSElow-labelled, unloaded splenocytes were injected into the tail vein of mice either vaccinated three times with Panc02-pulsed DCs or left untreated. After 20 h, spleens were removed and the frequencies of the differently labelled splenocytes were analysed by flow cytometry. By quantifying the relative decrease of p15E-loaded, CFSEhigh splenocytes in vaccinated animals as compared with untreated animals, specific lysis could be determined (B; data are the mean (SEM) of four independent experiments).
Panc02 cells are recognised and killed by tumour-specific CTLs
Panc02-derived tumours are poorly immunogenic, similar to human pancreatic adenocarcinoma. Moreover, Panc02 cells express only low levels of MHC class I required for classical CTL-mediated lysis (data not shown). Thus, we analysed whether Panc02 cells could be recognised in vitro by antigen-specific CTLs prior to their use in the therapeutic in vivo vaccination model. CTLs derived from transgenic OT-1 animals specific for the OVA-epitope SIINFEKL were used.35,36 Panc02 cells were loaded with SIINFEKL peptide or an irrelevant control peptide (TRP2) and co-cultured with splenocytes from either wild-type mice or OT-1 mice in a 51Cr-release assay. EL4 cells loaded with SIINFEKL or an irrelevant peptide were used as control targets. Panc02 cells loaded with SIINFEKL were specifically lysed by splenocytes from transgenic OT-1 mice (fig 5).

Panc02 cells can be recognised and killed by antigen-specific cytotoxic T lymphocytes in a major histocompatibility complex class I-restricted manner. Panc02 cells were loaded with SIINFEKL or irrelevant TRP2 peptide and co-cultured with splenocytes derived either from wild-type C57BL/6 mice or from transgenic OT-1 mice in a 51Cr-release assay at effector:target ratios from 3.125:1 to 100:1. EL4 cells loaded with SIINFEKL or irrelevant TRP2 peptide and co-cultured with splenocytes from transgenic OT-1 mice served as positive controls in the 51Cr-release assay.
Combination of DC-based vaccination with gemcitabine increases survival in the Panc02 pancreatic carcinoma model
Four experimental groups with comparable tumour size (approximately 13 mm2) were formed 14 days after challenge with Panc02. Mice received either no treatment (control), Panc02-pulsed DCs (DC), gemcitabine (Gem) or Panc02-pulsed DCs plus gemcitabine (DC + Gem). Gemcitabine was given twice weekly, in a dosage of 50 mg/kg, in the combined treatment arm 2 days prior to and 2 days after vaccination with DCs. Therapy was continued for 5 weeks. As shown in fig 6A, animals in the control group had all died 58 days after initiation of therapy. DC-based vaccination alone was almost as equally effective as gemcitabine treatment in preventing death (14% survival at day 58 of therapy vs 17%; p = 0.13 and p = 0.08 for DC vaccine vs control and gemcitabine vs control). In contrast to each treatment strategy applied alone, survival could be significantly increased in the combined treatment arm (50% survival at day 58 of therapy; p<0.005 for combined treatment vs control). Local tumour growth could be reduced in all treatment groups: 32 days after the start of therapy, mean tumour sizes were 123 mm2 in the control group, 91 mm2 in the DC vaccination group and 85 mm2 in the gemcitabine group. In the combined treatment arm, mean tumour size was 67 mm2. However, none of these results reached statisitical significance when compared with the control group (fig 6B). The increase in survival of animals treated with DC-based vaccination plus gemcitabine exceeded a solely additive effect. Moreover, gemcitabine treatment alone could not significantly augment survival or reduce local tumour growth. Thus, we hypothesised a synergistic effect of gemcitabine treatment and DC-based vaccination independent from a direct cytotoxic effect of the drug. To test this hypothesis, we reduced the gemcitabine dosage by 50% and repeated the experiment. As shown in fig 7, combination of the DC-based vaccination with gemcitabine increased survival of Panc02 tumour-bearing mice at both dosages of 50 and 25 mg/kg (p = 0.01 and p = 0.001 for DC plus gemcitabine 50 mg/kg vs control, and for DC plus gemcitabine 25 mg/kg vs control). As before, no significant effect on survival could be observed in the groups treated with only one of the two strategies (p = 0.98 and p = 0.18 for DC vaccination alone vs control, and gemcitabine alone vs control).

Combination of dendritic cell (DC)-based vaccination with gemcitabine treatment increases survival in the subcutaneous Panc02 murine pancreatic carcinoma model. Female 6- to 10-week old C57BL/6 mice were challenged subcutaneously with 0.5×106 Panc02 cells. Fourteen days after tumour inoculation, mice were either left untreated (n = 5; control), or were vaccinated with Panc02-pulsed DCs at weekly intervals (n = 7; DC), treated with intraperitoneal gemcitabine (50 mg/kg body weight) twice weekly (n = 6; Gem) or treated with the combination of DC vaccine and gemcitabine (n = 6; DC + Gem). Therapy was continued for 5 weeks. Survival rates in the different treatment groups are depicted as Kaplan–Meier graphs (A). Tumour size was determined at regular intervals by multiplication of perpendicular diameters (B). Data are the mean (SEM) for the number of animals indicated for each group.

Synergistic effect of dendritic cell (DC)-based vaccination and gemcitabine is preserved after 50% dose reduction of chemotherapy. Female 6- to 10-week old C57BL/6 mice were challenged subcutaneously with 0.5×106 Panc02 cells. Fourteen days after tumour inoculation, mice were either left untreated (n = 7), vaccinated with Panc02-pulsed DCs at weekly intervals (n = 7) or treated with intraperitoneal gemcitabine (Gem 50 mg/kg body weight) twice weekly (n = 6). Mice treated with the combination of DC vaccine and chemotherapy received either 50 mg/kg body weight (n = 6) or 25 mg/kg body weight of gemcitabine (n = 8). Survival rates in the different treatment groups are depicted as Kaplan–Meier graphs.
Vaccination with Panc02-pulsed DCs prevents metastatisation and orthotopic tumour growth
Mice that were left untreated or vaccinated three times with Panc02-pulsed DCs were injected into the tail vein with 25×104 Panc02 cells 7 days after the last vaccination. As shown in fig 8A, untreated mice died within 60 days. In contrast, immunisation with Panc02-pulsed DCs prevented death in this model. Histopathological analysis confirmed the presence of tumour cells in the lungs of untreated mice (fig 8b). In an orthotopic tumour model, we tested the capacity of the DC vaccination to suppress local tumour growth. Mice were left untreated or vaccinated subcutaneously with 3×105 Panc02-loaded DCs. Seven days after the last vaccination, 2×105 Panc02 cells were injected into the pancreas as described in the Materials and methods section. Macroscopic tumours were removed 19 days after tumour induction, and tumour mass was assessed. In the control group, tumours reached a volume of approximately 400 mm2 (data not shown) and a mean weight of 400 mg (fig 9, left). Prophylactic vaccination with Panc02-loaded DCs completely prevented growth of orthotopic Panc02 tumours (p = 0.014 for DC vaccination vs control). An experimental small animal CT scanner was used to monitor orthotopic tumour growth in some animals. Three weeks after injection of Panc02 cells into the pancreas, tumours reached approximately 1 cm in diameter in untreated animals (fig 9; right, top).

Prophylactic vaccination with Panc02-loaded dendritic cells (DCs) prevents pulmonary metastatisation after intravenous injection of Panc02 cells. Mice were left untreated or vaccinated three times with Panc02-pulsed DCs. Seven days after the last vaccination, 25×104 Panc02 cells were injected into the tail vein. As depicted in the Kaplan–Meier analysis, non-vaccinated mice died within 60 days whereas vaccinated mice did not succumb to metastasis formation (A; n = 5). In untreated mice, lung metastases were found on histopathological analysis (B; one representative analysis of five performed).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Prophylactic vaccination with Panc02-loaded dendritic cells (DCs) prevents orthotopic tumour growth. Mice were left untreated (n = 6) or vaccinated subcutaneously with 3×105 Panc02-loaded DCs (n = 4). Seven days after the last vaccination, 2×105 Panc02 cells were injected into the pancreas after laparatomy and mobilisation of the spleen. Tumours were removed 19 days after surgery and tumour weight was determined (left; data are the mean (SEM) for each group). In some animals, orthotopic tumour growth was monitored by CT scanning. Representative abdominal CT scans of an untreated control animal (right, top) and of a vaccinated animal (right, bottom) 19 days after injection of Panc02 cells into the pancreas are shown.
DISCUSSION
Here, we show for the first time that DC-based vaccination can be combined with gemcitabine to increase survival in a mouse model of pancreatic cancer. Moreover, we were able to demonstrate that prophylactic DC vaccination can prevent metastatisation and orthotopic growth of murine Panc02 pancreatic carcinoma cells. We chose the Panc02 murine tumour model because its poor immunogenicity resembles one of the characteristic features of human pancreatic carcinoma.37 However, Panc02 cells can be recognised and killed by CTLs despite low levels of MHC class I expression, and immunotherapeutic approaches can be effective in this model.27,38 First, we demonstrated that Panc02-pulsed DCs can establish immunological memory associated with long-term tumour protection and are capable of inducing a tumour-specific CTL response. Of note, peptide-specific CTLs with in vivo cytolytic capacity could be induced by DCs loaded with a polyclonal, whole cell-based antigen preparation. Tumour infiltration by CTLs is associated with a better prognosis in pancreatic carcinoma and other solid cancers.7,39 We were unable to detect increased T-cell infiltration after DC-based vaccination by histopathology, and tumour size could not be significantly reduced in the therapeutic subcutaneous model. However, our data show that DC-based vaccination can prevent death from haematogenic tumour spread and inhibit formation of pulmonary metastases. This is in accordance with findings in the B16 murine melanoma model: although vaccination therapy is unable to prevent local growth of B16 murine melanomas, it is also very effective against pulmonary metastatic disease.30,40
Although all mice bearing heterotopic Panc02 tumours died from the disease, some animals tolerated very large subcutaneous tumours for a substantial period of time. In contrast, mice with orthotopic tumours rapidly succumbed to a wasting syndrome after formation of peritoneal metastases. Importantly, prophylactic subcutaneous DC vaccination completely prevented orthotopic tumour growth. Thus, the systemic immune response induced by the DC vaccine is capable of suppressing intraperitoneal tumour growth. Although Takigawa et al have previously shown that intraperitoneal injection of tumour lysate-pulsed DCs could inhibit peritoneal spread of orthotopic tumours in a hamster model of pancreatic cancer,41 this represents an important novel finding. Although not yet formally demonstrated, it appears that circulating tumour-specific CTLs induced by DC vaccination are capable of accessing the peritoneal cavity where tumour cells are recognised and killed. Thus, adjuvant DC-based vaccination may be effective in preventing recurrence from disseminated or local residual tumour cells after pancreaticoduodenectomy in patients with pancreatic carcinoma.
Although DC-based vaccination was equally effective as gemcitabine in the therapeutic setting, neither of the two strategies could prevent progression of established tumours nor significantly reduce mortality. While it is known that Panc02 cells are relatively resistant to chemotherapy, the ineffectiveness of the vaccine may be due to an immunosuppressive environment produced by the tumour (eg, by secretion of soluble factors detrimental to T cell-mediated immunity such as IL10 or transforming growth factor (TGF)β), to a limited access of effector cells to tumour tissue or inactivation of tumour-infiltrating lymphocytes, another feature that is similar to human pancreatic cancer.7,42 If the DC-based vaccine was combined with gemcitabine, the survival rate of tumour-bearing mice could be significantly increased. This is in accordance with previous findings showing that the effectiveness of immune stimulation by in vivo CD40 ligation can be augmented by gemcitabine.24 The exact mechanism by which gemcitabine enhances the effectiveness of the DC-based vaccine in our model remains unknown. In a human in vitro model, we were able to show that the drug can sensitise pancreatic carcinoma cells to CTL responses.25 Others have demonstrated that apoptosis induced by gemcitabine may increase cross-presentation of tumour antigens to CTLs by intratumoural DCs.24 Immunosuppression caused by the chemotherapy applied may limit combination with immunotherapeutic approaches to cancer. However, although inhibition of B cell-mediated immune responses has been described in animal models,43 gemcitabine can be administered to patients with pancreatic cancer without relevant loss of T cell and DC function.22 Moreover, preliminary data indicate that gemcitabine may even inhibit Th 2- and specifically augment Th 1-type immune responses in cancer patients.22 The effect of gemcitabine treatment on the local tumour environment and the frequency and function of tumour antigen-specific CTLs in our model is currently under investigation.
Although clinical trials have demonstrated that tumour-specific immunity can be regularly established by DC-based vaccines, it has also become evident that clinical responses to immunotherapy occur very rarely in patients with gastrointestinal malignancies.12 Today, researchers try to identify strategies that, on the one hand, help to overcome immunoresistance of tumours and, on the other hand, do not interfere with the activation of a tumour-directed immune response. Increasing evidence suggests that well-established treatment strategies such as radiation, surgical debulking or chemotherapy may be successfully combined with immunotherapeutic approaches.44–46 Gemcitabine is currently the treatment of choice for patients with irresectable pancreatic carcinoma and is not immunosuppressive. Here, we show that combination with gemcitabine augments therapeutic efficacy of a DC-based vaccine in a murine model of pancreatic cancer. These findings led to the initiation of a phase II trial at our institution investigating combined treatment of patients with advanced pancreatic adenocarcinoma with gemcitabine and an autologous DC-based vaccine.
Acknowledgments
C Bauer, F Bauernfeind and M Dauer were supported by grants from the University of Munich (FoeFoLe No. 481, Promotionsstudium Molekulare Medizin, Gravenhorst-Stiftung). We thank Professor Brocker from the Department of Immunology of the University of Munich for providing transgenic OT-1 animals.
REFERENCES
Footnotes
-
*These authors contributed equally. This work is part of the doctoral theses of F Bauernfeind and A Sterzik at the University of Munich, Munich, Germany.
-
Published Online First 29 March 2007
-
Competing interests: None.