Article Text

Abstract
Background and aims: Pancreatic cancer remains a devastating diagnosis with only limited therapeutic options. Specific inhibition of expression of target genes has become possible using small interfering (si) RNAs. We therefore investigated how far siRNA specific for bcl-2 may serve as a therapeutic option for pancreatic cancer in vitro and in vivo.
Methods: siRNAs targeting two different regions in the bcl-2 gene were transfected to YAP C and DAN G pancreatic carcinoma cells and human foreskin fibroblasts. Permutations were generated by changing 3′ and 5′ overhangs and varying the length of the paired RNA duplex. Transfection efficacy was determined using FITC labelled siRNAs and fluorescence microscopy. Cell survival and apoptosis were quantified at 24–120 hours. Pancreatic cancer xenografts in male nude mice were treated intraperitoneally with siRNAs daily for 24 days. siRNA pharmacokinetics in vivo were assessed using radioactively labelled siRNAs. Total protein and RNA were extracted for western Blot analysis and quantitative polymerase chain reaction.
Results and conclusions: bcl-2 specific siRNAs specifically inhibited expression of the target gene in vitro and in vivo. Antiproliferative and proapoptotic effects were observed in tumour cells but not in fibroblasts or non-malignant tissues. siRNA permutations and diverse overhangs influenced gene silencing efficacy. siRNA was quickly distributed to all organs and excreted via the kidney and liver. bcl-2 specific siRNA is a promising adjunctive treatment for pancreatic carcinoma.
- siRNA, short interfering RNA
- RNAi, RNA interference
- ds, double stranded
- RISC, RNA induced silencing complex
- FCS, fetal calf serum
- PBS, phosphate buffered saline
- FACS, fluorescence activated cell sorter
- FITC, fluorescein isothiocyanate
- RT-PCR, reverse transcription-polymerase chain reaction
- short interfering RNA
- RNA interference
- bcl-2
- pancreatic cancer
- apoptosis
Statistics from Altmetric.com
- siRNA, short interfering RNA
- RNAi, RNA interference
- ds, double stranded
- RISC, RNA induced silencing complex
- FCS, fetal calf serum
- PBS, phosphate buffered saline
- FACS, fluorescence activated cell sorter
- FITC, fluorescein isothiocyanate
- RT-PCR, reverse transcription-polymerase chain reaction
Pancreatic cancer is one of the most common causes of cancer related deaths in Western countries and represents approximately 10% of all gastrointestinal malignancies.1 Ductal adenocarcinoma is responsible for 90% of all cases and is often diagnosed at an advanced stage, with a median survival time of less than six months and 12 month and five year survival rates of only 10% and less than 3%, respectively. Hence pancreatic cancer has the poorest prognosis of many neoplasms.2,3 Only 10–14% of patients are eligible for curative surgery which prolongs median survival to 10–20 months.4,5 Various adjuvant, neoadjuvant, locoregional, or radiochemotherapy strategies could not significantly improve overall survival, and still remain palliative.4,5
Several genetic risk factors have been identified to date.6 Among these, activating point mutations in codon 12 of the K-ras proto-oncogene can be found in up to 90% of tumour specimens.7,8 Other factors include mutations of p16INK4, the deleted in colon cancer gene (DCC), p53, and related cell cycle control genes.9,10 Furthermore, the antiapoptotic gene bcl-2 is highly expressed in most pancreatic cancers.11,12 The family of bcl-2 homologues consists of proapoptotic members such as bax, bad, bid, or bim, and anti-apoptotic members such as bcl-2 or bcl-xL which regulate central pathways in cell survival, apoptosis induction, and resistance to chemotherapy.13–16 While antiapoptotic bcl-2 stabilises mitochondrial membranes, proapoptotic bax or bak disturb the integrity of these membranes after oligomerisation, leading to release of proapoptotic cytochrome c from the mitochondrial intermembrane space which then activates procaspase 9, triggers activation of other effector caspases, and causes the execution of apoptosis.16 Several studies have shown that inhibiting expression of antiapoptotic bcl-2 inhibits growth and induces apoptosis, frequently enhancing the effects of conventional chemotherapy in various malignancies—for example, lymphoma,17 leukaemia,18 breast cancer,19 melanoma,20 or gastric cancer.21 Suppression of expression of antiapoptotic bcl-2 has so far been investigated using antisense oligonucleotides.22 However, preclinical and clinical studies did not demonstrate convincing antitumour efficacy due to unspecific effects (for example, complement activation, thrombocytopenia, cytokine release), hepatotoxicity, and the need to introduce chemical modifications (for example, locked nucleic acids) to stabilise these molecules.22–24
We therefore exploited the recently discovered mechanism of specific suppression of a target gene by short double stranded (ds) oligoribonucleotides utilising the RNA interference (RNAi) pathway. RNAi is a highly conserved pathway of cleaving dsRNA (for example, after viral infection) by generating short interfering RNA (siRNA) molecules, duplexes of 21–23 nt in length, which use the RNase III-like enzyme DICER.25–28 Intracellularly, DICER generated or chemically synthesised siRNAs are separated into single strands by an ATP dependent helicase and become incorporated into the RNA induced silencing complex (RISC). RISC binds the cognate endogenous mRNA and cleaves the target molecule if the siRNA derived antisense strand is incorporated.29–31 siRNAs and RNAi have been used to silence different target genes in vitro and in model organisms in vivo.32–34 As a therapeutic tool, siRNAs have been used to suppress the replication of several viruses (for example, hepatitis C virus35 or human immunodeficiency virus),36 and expression of inflammatory genes or oncogenes such as bcr-abl, for example.37
We therefore studied the pharmacokinetics of parenterally administered siRNA, and determined if, and to what extent, a tumour gene specific siRNA targeted to bcl-2 could contribute to the treatment of pancreatic cancer. We also investigated if structural variations in bcl-2-siRNA could influence RNAi efficacy in vitro and in vivo.
MATERIAL AND METHODS
Cell culture
YAP C and DAN G pancreatic adenocarcinoma cells were cultured on six well tissue culture plates (Becton Dickinson, Mannheim, Germany) in RPMI-1640 medium (Biochrom, Berlin, Germany) containing 10% fetal calf serum (FCS, Biochrom), penicillin (107 U/l), and streptomycin (10 mg/l) at 37°C with 5% CO2. Human foreskin fibroblasts served as non-malignant controls and were cultured in Dulbecco’s modified Eagle’s medium (Biochrom) with the same supplements. All cell lines were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany).
Transfection of siRNA
siRNA was provided by Ribopharma AG (Kulmbach, Germany) or purchased from Qiagen (Hilden, Germany). Sequences of the five bcl-2 specific and three control siRNAs used are given in table 1. bcl-2 specific sequences were numbered consecutively.
Sequences of short interfering RNAs (siRNAs)
Transfections were performed at approximately 70% confluency in six well plates using oligofectamine (Invitrogen, Carlsbad, California, USA) according to the manufacturer’s instructions. Briefly, 0.25×106 cells were seeded in complete growth medium the day before transfection. For each transfection reaction, siRNA-oligofectamine complexes were prepared by mixing oligonucleotides with oligofectamine diluted 1:5 (v/v) with medium directly before addition to the cells. Final concentrations of the siRNA were between 1 and 100 nM. Transfections were performed in 1 ml of serum free medium for four hours. Thereafter, 0.5 ml of medium containing 30% FCS (v/v) was added to achieve complete growth conditions (10% FCS). In each experiment, untreated controls and mock transfected cells, receiving only oligofectamine without siRNA, were included. Cells were assayed 24 to 120 hours after transfection.
Flow cytometric analysis of apoptosis and bcl-2 protein levels
For quantification of apoptosis, culture supernatants were collected and cells washed twice with phosphate buffered saline (PBS), trypsinised, and lysed in a hypotonic solution containing 0.1% sodium citrate, 0.1% Triton X-100, and 50 μg/ml propidium iodide (Sigma, Deisenhofen, Germany). Analysis of labelled nuclei was performed on a FACSCalibur fluorescence activated cell sorter (FACS) using CELLQuest software (both from Becton Dickinson). The percentage of apoptotic cells was determined by measuring the fraction of nuclei with a subdiploid DNA content.
To assess bcl-2 protein levels by flow cytometry, cells were transfected and harvested as described. Fixation and membrane permeabilisation were performed with Fix and Perm, as recommended by the manufacturer (Caltag Laboratories GmbH, Hamburg, Germany). A FITC conjugated bcl-2 specific antibody (10 μl F7053; Dako, Glostrup, Denmark) was added for 15 minutes at room temperature in the dark. Samples were washed with PBS, centrifuged at 300 g, resuspended in PBS, and kept on ice in the dark until FACS analysis with fluorescence detector FL1-H. Ten thousand events were collected for each sample analysed.
Determination of cell viability
Cell viability was assessed using the trypan blue exclusion test. Cells were harvested after the incubation period and stained with trypan blue (Biochrom). The number of unstained (intact) cells was counted in a Neubauer chamber and expressed as relative cell numbers compared with untreated controls ( = 100%).
Analysis of ΔΨm
Mitochondrial injury was assessed by JC-1 staining (MoBiTec, Goettingen, Germany). Cells were adjusted to a density of 0.2×106/ml, trypsinised, washed in PBS, resuspended in 1 ml of medium, stained with 5 μg/ml JC-1 for 15 minutes at 37°C with 5% CO2 in the dark, then washed twice in PBS and resuspended in 0.5 ml PBS. Analysis was performed by FACS scan, and mitochondrial function was assessed as JC-1 green (uncoupled mitochondria, detector FL-1, 537 nm) or red (intact mitochondria, detector FL-2, 597 nm) fluorescence, as described previously.38
Fluorescence microscopy of FITC labelled siRNA
siRNA was labelled with fluorescein isothiocyanate (FITC-siRNA, obtained from Ribopharma, Kulmbach, Germany or from the siRNA Starter Kit, Qiagen, Germany) and transfected to cells seeded on ChamberSlides, as described above. Twenty four hours after transfection, cells were washed in 1× PBS (Biochrom) and fixed for 30 minutes in ice cold methanol at −20°C. After an additional washing in PBS, nuclei were counterstained with 1 μg/ml propidium iodide (Sigma) for 20 minutes at room temperature in the dark. Slides were covered with fluorescent mounting medium (Dako). Fluorescence microscopy was performed using a Zeiss Axioplan microscope (Carl Zeiss, Göttingen, Germany) at 490 nm excitation (FITC-siRNA, 525 nm emission) and 536 nm excitation (propidium iodide, 617 nm emission), respectively. Digital image analysis was performed using OpenLab software (Improvision, Heidelberg, Germany).
Preparation of RNA, cDNA, and quantitative real time PCR
Total RNA was prepared from 0.25×106 untreated controls or transfected cells, 24–120 hours after transfection using peqGOLD RNA Pure (Peqlab, Erlangen, Germany) according to the manufacturer’s instructions and subsequent phenol-chloroform extraction. RNA integrity was checked electrophoretically and quantified photometrically. The concentration of total RNA was adjusted to 0.1 μg/μl in RNAse free water.
For reverse transcription (RT), 1 μl of a primer mix containing 100 pmol/μl oligo(dT)15 primer and 100 pmol/μl random hexamer primer (5′-NNN NNN-3′), both from Promega (Mannheim, Germany), were incubated with 1 μg RNA and denatured for 10 minutes at 70°C. An RT reaction mix consisting of 5× RT reaction buffer (Invitrogen, Karlsruhe, Germany), 0.1 M DTT (Invitrogen), and a dNTP mix with 10 mM of each dNTP was added together with 40 U RNAsin RNAse inhibitor (Promega) and 100 U Superscript II reverse transcriptase (Invitrogen) and placed on ice for two minutes. Total reaction volume was adjusted to 20 μl with RNAse free water and incubated for 10 minutes at 25°C and cDNA synthesised for one hour at 42°C. The reaction was stopped by 15 minutes heat inactivation at 70°C and cDNA stored at −20°C until use.
Relative transcript levels were quantified by real time RT-polymerase chain reaction (PCR). Sense and antisense primers (each at 0.5 μM) and 0.125 μM 5′-phosphorylated probe, labelled with the reporter dye FAM (carboxy-fluorescein) at the 5′-end and with the quencher molecule TAMRA (carboxy-tetramethylrhodamine) at the 3′-end, were synthesised at MWG Biotech (Ebersberg, Germany). PCR primers and probes (table 2) were designed to span exon-intron boundaries, reducing the risk of detecting genomic DNA. Real time RT-PCR was performed with 1.5 μl of template cDNA on a LightCycler system (Roche, Mannheim, Germany) in a total reaction volume of 15 μl, including TaqDNA polymerase, dNTP-mix, reaction buffer, and 3 mM MgCl2, provided by the LightCycler FastStart DNA Master Hybridisation Probes Kit (Roche Molecular Biochemicals, Mannheim, Germany) according to the manufacturer’s instructions. For amplification of bax and bcl-2, PCR was performed with a precycling step at 95°C for 10 minutes for activation of TaqDNA polymerase, followed by 55 cycles with a denaturation step at 95°C of less than one second, 15 seconds annealing at 55°C, and 15 seconds extension at 65°C. β2-Microglobulin was amplified as an internal standard using a precycling step at 95°C for 10 minutes, 45 cycles of less than one second at 95°C, 15 seconds at 53°C, and 25 seconds at 63°C. A cooling step to 40°C finalised all amplifications. For every measured transcript, a 1:2 to 1:32 dilution series of one sample was used as standard. Data were analysed with the LightCycler software using the proportional second derivative maximum option. To normalise for differences in RNA amounts and variable efficacy of the reverse transcription reactions, results for bax and bcl-2 were normalised to β2-microglobulin levels. All samples were analysed in duplicate.
Primers and probes for quantitative real time polymerase chain reaction
Xenograft model
Human pancreatic carcinomas established from Panc-1 cells were transplanted subcutaneously to 4–6 week old nude mice (Harlan Winkelmann GmbH, Germany).39 Animals were housed in a light and temperature controlled environment and provided with food and water ad libitum. Treatment was started when tumours reached 7 mm in diameter. Animals received intraperitoneal injections of 200 μg/kg siRNA dissolved in 120 μl RNAse free physiological NaCl. Control mice were treated with NaCl alone. Tumour size was determined daily by measurement with a calliper square. After 24 days, animals were sacrificed by cervical dislocation and specimens of tumour, liver, lung, kidney, spleen, and bone marrow were either fixed in 10% phosphate buffered formalin or snap frozen in liquid nitrogen for protein extraction, as described below. Ethics approval was obtained for all experiments (621-2531.31-20/01, Government of Lower Franconia, Würzburg, Germany).
Semiquantification of proteins involved in apoptosis
Snap frozen tumour specimens were lysed by adding 2× sample buffer (2 mM N-ethylmaleimide, 2 mM phenylmethyl-sulfonylfluoride, 4% sodium dodecyl sulfate, 4% dithiothreitol, 20% glycerol, 0.01% bromophenol blue, 2 M urea, 0.01 M sodium ethylenediaminetetra-acetic acid (Na-EDTA), 0.15 M Tris-HCl). DNA was sheared by pipetting up and down for three minutes at room temperature. Samples were boiled at 95°C for 15 minutes, centrifuged at 13 000 rpm for 10 minutes, and then subjected to 14% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (Invitrogen). After blocking overnight at room temperature in a buffer containing PBS, 0.1% Tween 20, and 4% low fat milk powder, nitrocellulose membranes were incubated for 90 minutes either with polyclonal rabbit antihuman Bcl-2 (1:400, sc-783) or polyclonal rabbit antihuman Bax (1:500, sc-493; both Santa Cruz Biotechnology, Santa Cruz, California, USA). Membranes were washed three times for 10 minutes in a buffer containing PBS, 0.1% Tween 20, and 4% low fat milk powder, and incubated with an antirabbit IgG coupled to peroxidase (1:1000; Sigma) for one hour at room temperature. Reactive bands were detected with the ECL chemiluminescence reagent (Amersham Pharmacia Biotech, Freiburg, Germany) and analysed using GelScan 5 software (BioSciTec, Frankfurt, Germany). Signals were standardised to β-actin (1:5000; Sigma) content.
Distribution of radioactive labelled siRNA in vivo
siRNA probe bcl5 was labelled radioactively with 33α-UTP and applied as a single bolus in physiological saline by tail vein injection into non-tumour bearing male nude mice at a concentration of 1 mg/kg, corresponding to 360.000 cpm/μl. Pure 33α-UTP (360 000 cpm/μl) dissolved in physiological saline was used as a control. Three animals per group were kept in metabolic cages to collect faeces and urine. Five minutes to 24 hours after injection, animals were sacrificed by heart puncture and total blood samples collected and fixed with RNAlater (Qiagen, Hilden, Germany). Whole bodies were fixed in formalin, as described above, and cut in sagittal and paramedian plains and 5 μm serial sections. Sections were stained with haematoxylin and eosin and analysed for 24 hours in a Fuji FLA-3000 phospho and fluorescence imager (Raytest GmbH, Straubenhardt, Germany).
Immunohistochemistry
Tumour tissue was fixed with phosphate buffered 10% formalin and embedded in paraffin. Sections (5 μm) were cut and stored at room temperature until use. Routine histology (haematoxylin-eosin staining) was performed in order to evaluate basic histomorphological features of the specimens and to determine toxic effects on organs, especially the liver, spleen, kidney, and haematopoietic system. For immunohistochemistry, sections were dewaxed, rehydrated, and processed by microwave heating in citrate buffer (pH 6.0), as described previously.40 Specimens were incubated with monoclonal primary antibodies (anti-Ki-67, 1:50; anti-bcl-2, 1:100; Dako) overnight at 4°C and visualised using streptavidin-biotin complex (Biogenex, San Ramon, California, USA) coupled to alkaline phosphatase and developed using 3-hydroxy-2-naphtylacide-2,4-dimethylanilide as substrate. Nuclei were counterstained with haematoxylin. Replacement of primary antibodies by non-immune mouse or rabbit serum (BioGenex) or Tris buffered saline (pH 7.2) served as negative controls. Slides were digitised and analysed with the Ce2001 Cell Explorer software (BioSciTec, Frankfurt Germany). Semiquantification was performed for four independent high power fields in each slide, using electronic filtering for respective signals.
RESULTS
siRNAs are effectively delivered into pancreatic cancer cells in vitro
To optimise the transfection protocol, siRNA labelled with FITC at either the 5′-end of the sense strand or the 5′-end of the antisense strand was used. Compared with untransfected or mock transfected cells without FITC signals, distinct intracellular fluorescence staining was observed in cells transfected with FITC labelled siRNA. These signals showed intracytoplasmic and perinuclear localisation, in accordance with the current notion of RNAi occurring as a post-transcriptional process. No qualitative difference was observed between sense or antisense labelled siRNA molecules. As this assay was used to confirm cellular uptake of siRNAs, cells were fixed 24 hours after transfection. At this early time point, no morphological signs of cellular apoptosis were found, indicating low toxicity of the transfection reagent used. The transfection protocol used was optimised for transfecting YAP C cells and human foreskin fibroblasts at comparable efficacy (fig 1).

Short interfering RNA (siRNA) transfection efficacy, as assessed by fluorescence microscopy. Slides were analysed for fluorescein isothiocyanate (FITC) and propidium iodide (PI) fluorescence. Digital images were acquired and merged with OpenLab software for each channel independently. PI stained nuclei were detectable in all analysed slides, while FITC signals were not present in untreated controls (not shown) or mock transfected cells (top row). Cells transfected with 100 nM FITC-siRNA exhibited distinct intracellular and perinuclear staining (both fibroblasts or YAP C cells). No qualitative difference was detectable between siRNA labelled on the sense or antisense strand (not shown).
Bcl-2 specific siRNAs selectively induce apoptosis in pancreatic cancer cells
At 10 nM, siRNAs bcl1, bcl3, and bcl11 induced significant levels of apoptosis in YAP C, reaching a maximum of 25.0%, 37.1%, and 15.4% after 120 hours, respectively (fig 2A), which did not increase further using 100 nM siRNA. Apoptosis remained low after transfection of bcl5 and bcl8, reaching only 8.6% and 4.5%, respectively, after 120 hours. No effect on cell death was observed in cells transfected with 1 nM of any siRNA (data not shown), and even the most effective bcl-2 specific siRNA, bcl3, could not induce apoptosis in non-transformed skin fibroblasts (fig 2B). Furthermore, control siRNAs with no corresponding endogenous target sequences also failed to activate cellular death signalling pathways in pancreatic cancer cells (fig 2C) or fibroblasts (not shown), except for control siRNA 1 which induced some apoptosis in tumour cells. After 120 hours, apoptosis rates of untreated or mock transfected YAP C cells or fibroblasts remained below 2.0%. These results were paralleled in DAN G cells, with untransfected or mock transfected controls reaching only 4.8% and 5.1% apoptosis after 120 hours, respectively, and all bcl-2 specific siRNAs inducing significant levels of cell death even at 10 nM, reaching 18.5%, 12.3%, 11.9%, 27.9%, and 15.8%, respectively (fig 2D).

Induction of apoptosis by bcl-2 specific short interfering RNAs (siRNAs) in pancreatic cancer cells but not in non-transformed foreskin fibroblasts. Shown are apoptosis rates measured as subdiploid events in flow cytometry by propidium iodide staining after transfection with 10 nM bcl-2 specific (A) or non-silencing (C) siRNA in YAP C pancreatic carcinoma cells 72, 96, and 120 hours after transfection. Results for bcl-2 specific siRNAs in foreskin fibroblasts are shown in (B). (D) Results of apoptosis induction by 10 nM bcl-2 specific siRNAs in DAN G pancreatic carcinoma cells 120 hours after transfection. Values are means (SEM) of three independent experiments. *p<0.05.
Transfection of bcl-2 specific siRNA reduces pancreatic carcinoma cell numbers
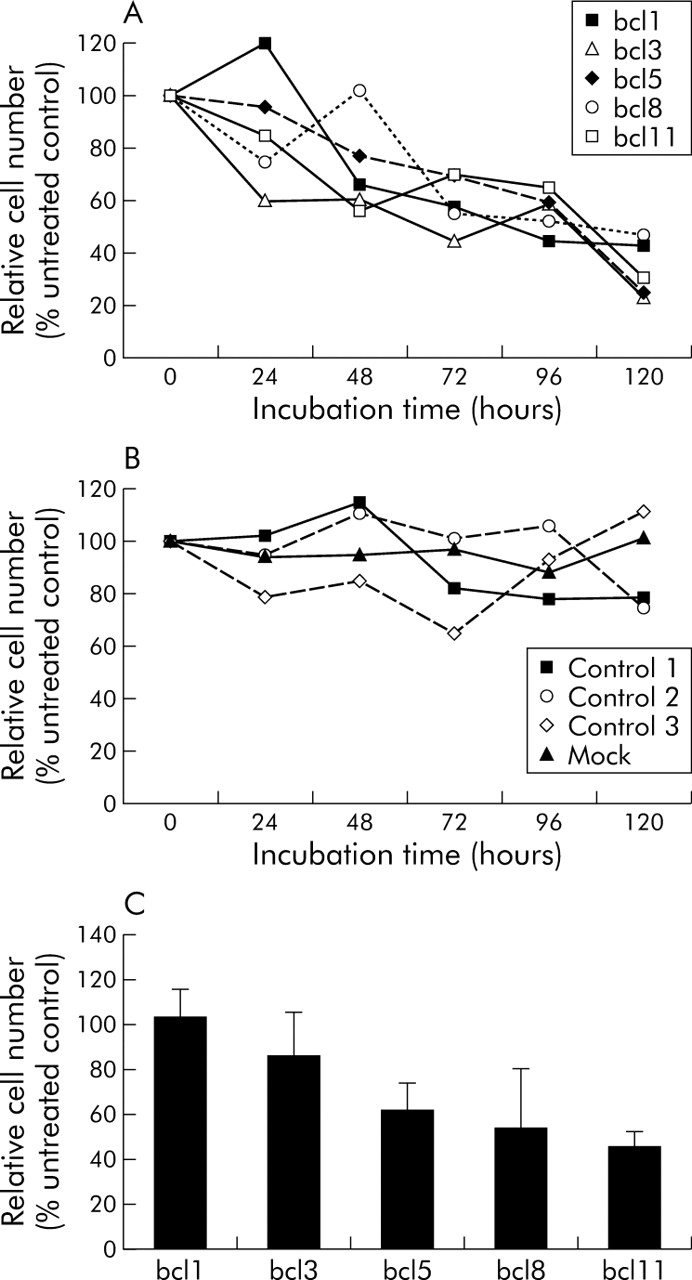
All bcl-2 specific siRNA sequences reduced the relative cell number of YAP C pancreatic carcinoma cells (fig 3A). This effect was most pronounced for bcl3, bcl5, and bcl11 (up to more than 70% after 120 hours), while bcl1 and bcl8 reduced cell proliferation by more than 50%. Reduced cell viability was detectable as early as 24 hours after transfection. In fibroblasts, cell viability was not significantly altered after transfecting either unspecific control siRNAs or bcl-2 specific siRNA sequences (not shown). Cell number remained stable in both cell types over this time course for mock transfected cells and was reduced slightly, but not significantly, after transfection of control siRNAs 1 and 2. The number of viable cells was slightly increased after transfection with control siRNA 3 (fig 3B). In contrast with YAP C, bcl1 did not reduce cell numbers in DAN G at 10 nM after 120 hours (103.2%), while the other bcl-2 specific siRNAs caused a reduction in viable tumour cells, reaching 86.7%, 62.5%, 54.6%, and 47.0%, respectively, after 120 hours (fig 3C).
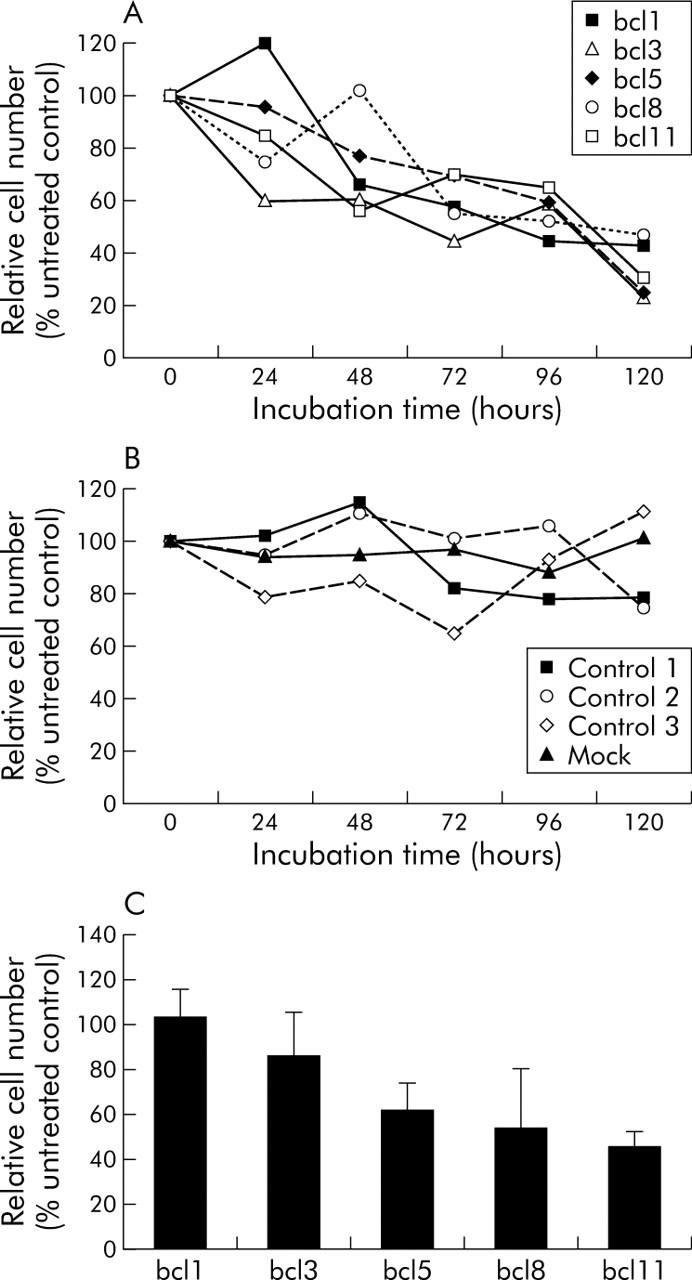
Relative cell number of YAP C cells, 24–120 hours after transfection with different short interfering RNAs (siRNAs). Absolute cell numbers of three independent experiments were determined by trypan blue staining in a Neubauer chamber. Average cell number of untreated controls was set at 100% and relative numbers of samples calculated accordingly. (A) Reduction of relative cell number after transfection of 10 nM bcl-2 specific siRNAs. (B) Stable cell viability after mock transfection or transfection of control siRNAs. (C) Reduced number of viable DAN G cells 120 hours after transfection with 10 nM bcl-2 specific siRNA.
Bcl-2 specific siRNA leads to rapid downregulation of the corresponding mRNA
Quantitative real time PCR revealed spontaneous decrease in bcl-2 mRNA levels in control or mock transfected cells over the time course of 120 hours while steady state levels of the housekeeping gene β2-microglobulin or the proapoptotic gene bax remained relatively stable (fig 4A, B), in accordance with cellular senescence during the time of the experiment.41 Transfection of bcl-2 specific siRNA led to a rapid but variable decrease in bcl-2 mRNA levels, with suppression persisting up to 120 hours. Here, bcl8 was most effective, while for bcl3 and bcl11 time and concentration dependent effects were observed. The proapoptotic counterpart of bcl-2, bax, remained stable in all experiments and showed an increase after transfection of bcl3 and bcl11. Thus, on the mRNA level, the bax/bcl-2 ratio was shifted towards apoptosis induction.

bcl-2 mRNA was rapidly downregulated by specific short interfering RNAs (siRNAs) in pancreatic cancer cells. Shown are mean values of two independent transfections of YAP C cells and two independent quantitative polymerase chain reaction amplifications of each transfection. Expression of bcl-2 (A) and bax (B) was normalised to β2-microglobulin mRNA levels. Results are expressed relative to normalised β2-microglobulin mRNA content of untreated controls ( = 100).
Expression of bcl-2 protein is diminished after transfection of bcl-2 specific siRNA
FACS analysis revealed a significant reduction in bcl-2 protein levels after transfection of bcl5 or bcl11 in a time and dose dependent manner in YAP C cells (fig 5). Untreated controls and mock transfected cells showed high and stable levels of bcl-2 staining, even after 120 hours (>70% of all detected events), while both bcl-2 specific siRNAs at 100 nM reduced bcl-2-FITC signals to 24.3% (bcl5) or 40.9% (bcl11). A minor but significant reduction in bcl-2 protein levels was observed after transfection of 10 nM bcl5 but not of bcl11 (p<0.05).

Reduction of bcl-2 protein levels in viable YAP C cells. (A) Protein expression of bcl-2 was determined by flow cytometry after transfection with bcl5 or bcl11 and staining with a monoclonal fluorescein isothiocyanate (FITC) labelled anti-bcl-2 antibody. Viable cells were gated by cell size (forward scatter) and fluorescence was analysed with fluorescence light detector FL1-H (530 nm). (B) Quadrant analysis revealed a time and dose dependent downregulation of bcl-2 protein positive cells 72–120 hours after transfection of corresponding short interfering RNAs (siRNAs). Mock transfected or untreated control cells showed no changes in bcl-2 protein expression. *p<0.05 versus mock transfected cells. Shown are means (SEM) of three independent experiments.
Downregulation of bcl-2 disturbs mitochondrial membrane potential
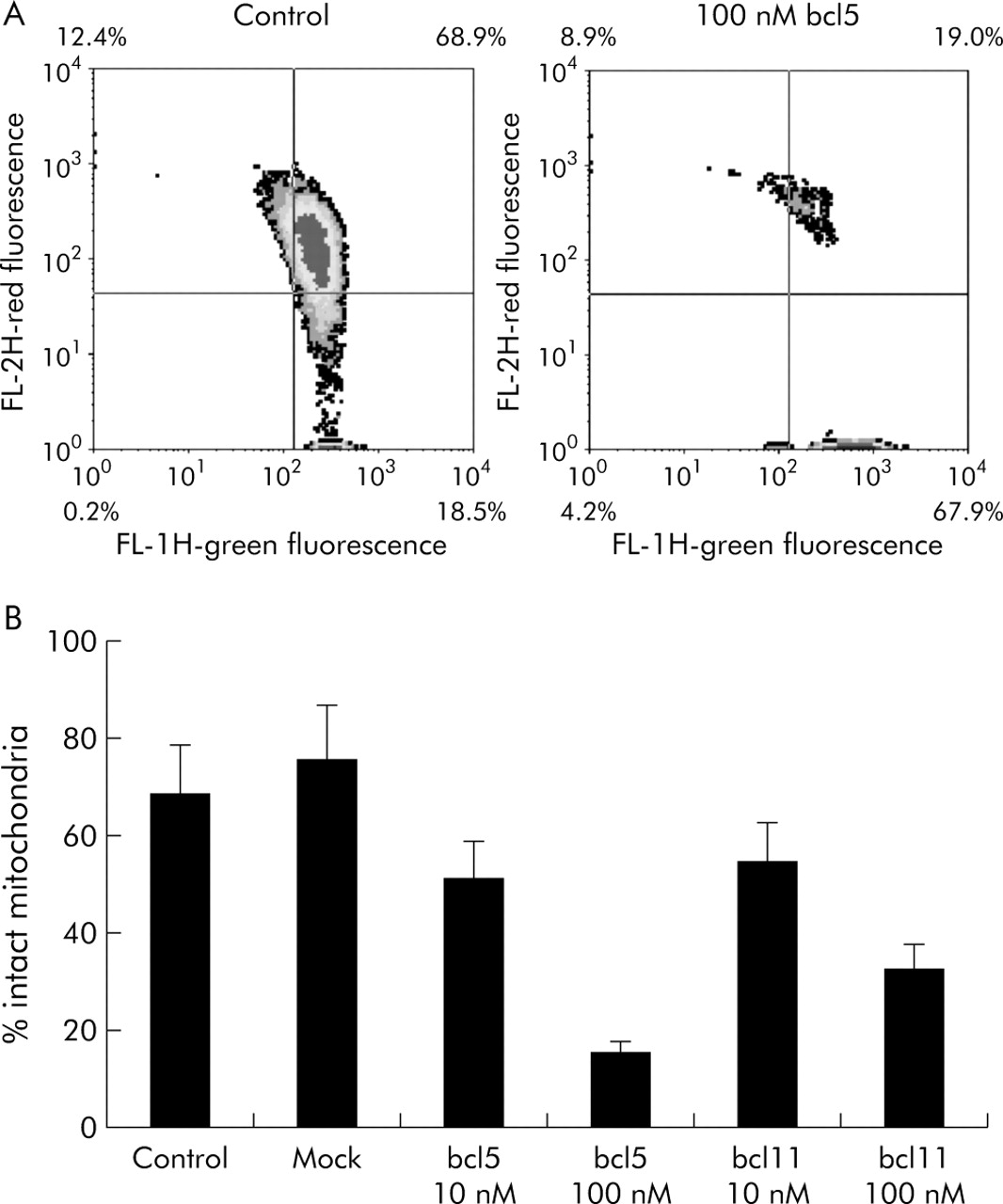
Paralleling the reduced expression of bcl-2 protein, the percentage of cells showing red fluorescence (that is, intact mitochondria) decreased compared with untreated or mock transfected cells (fig 6). With 10 nM of bcl5 or bcl11, JC-1 red fluorescence was reduced to 51% and 54.5%, respectively, after 120 hours, and was further reduced after transfection of 100 nM siRNA (15.1% and 32.5%, respectively). Red fluorescence correlated closely with levels of bcl-2 protein, as determined by flow cytometry (r2 = 0.9655). These findings indicate that bcl-2 is involved in maintaining mitochondrial membrane integrity and preventing induction of apoptosis.

Loss of mitochondrial membrane integrity after bcl-2-short interfering RNA (siRNA) transfection. (A) Representative density plots of fluorescence detector FL1-H (green fluorescence, breakdown of ΔΨm) versus FL2-H (red fluorescence, intact ΔΨm), depicting all acquired events of untreated YAP C cells (left) or 120 hours after transfection with 100 nM bcl5 (right). (B) Means (SEM) of three independent experiments. All bcl-2 specific siRNAs caused a rapid decrease in ΔΨm (p<0.05) while no significant change was observed for mock transfected cells.
siRNA rapidly distributes in vivo and is eliminated via the kidney and liver
Pharmacokinetic data using radioactively labelled siRNA were collected in male NMRI-mice. Ten minutes after a single bolus injection into the tail vein, radioactivity was quickly distributed by blood flow, showing intense signals in the heart and the retrocardial thoracic haematoma resulting from heart puncture (fig 7). Radioactivity was then redistributed to the kidney (two hours) and accumulated finally in the liver and gut wall (eight and 12 hours). In contrast, 33α-UTP alone showed signals predominantly in osseous tissues such as spine, skull, or the thoracic wall (fig 8, bottom row). These data indicate that a major proportion of siRNA is quickly excreted via the kidneys but is also metabolised and eliminated by the liver. While pancreata were clearly identified in haematoxylin-eosin stainings, unequivocal identification was not possible in phosphoimager analyses due to limitations in optical resolution. Overall, tissue distribution was homogenous, indicating that several target tissues can be reached with nude siRNA.
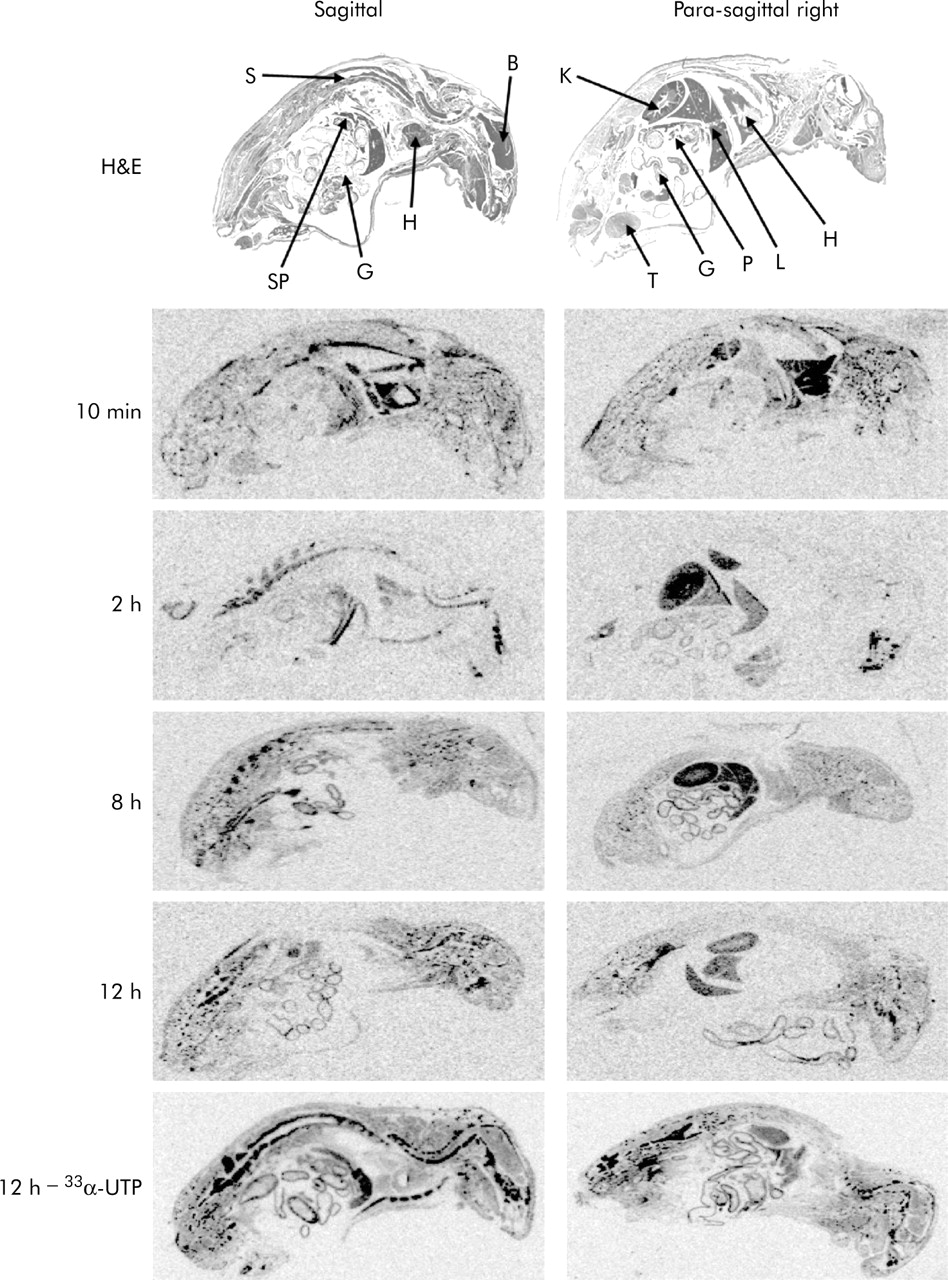
Distribution of 33P-labelled short interfering RNA (siRNA) in vivo. Representative phosphoimager analyses of sagittal (left column) and para-sagittal (right column) sections of animals sacrificed 10 minutes, and 2, 8, and 12 hours after intravenous bolus injection of labelled siRNA. Haematoxylin-eosin stainings of whole body sections are shown in the top row. Organs indicated by arrows are brain (B), gut (G), heart/thoracic haematoma (H), kidney (K), liver (L), pancreas (P), spine (S), spleen (SP), and testis (T). Bottom row shows representative sections of animals receiving pure 33α-UTP without nucleic acid as a control, sacrificed 12 hours after tail vein injection. Here, signals were more prominent in osseous tissues while in the siRNA treated animals, rapid redistribution from blood to kidney to liver and gut was visible.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
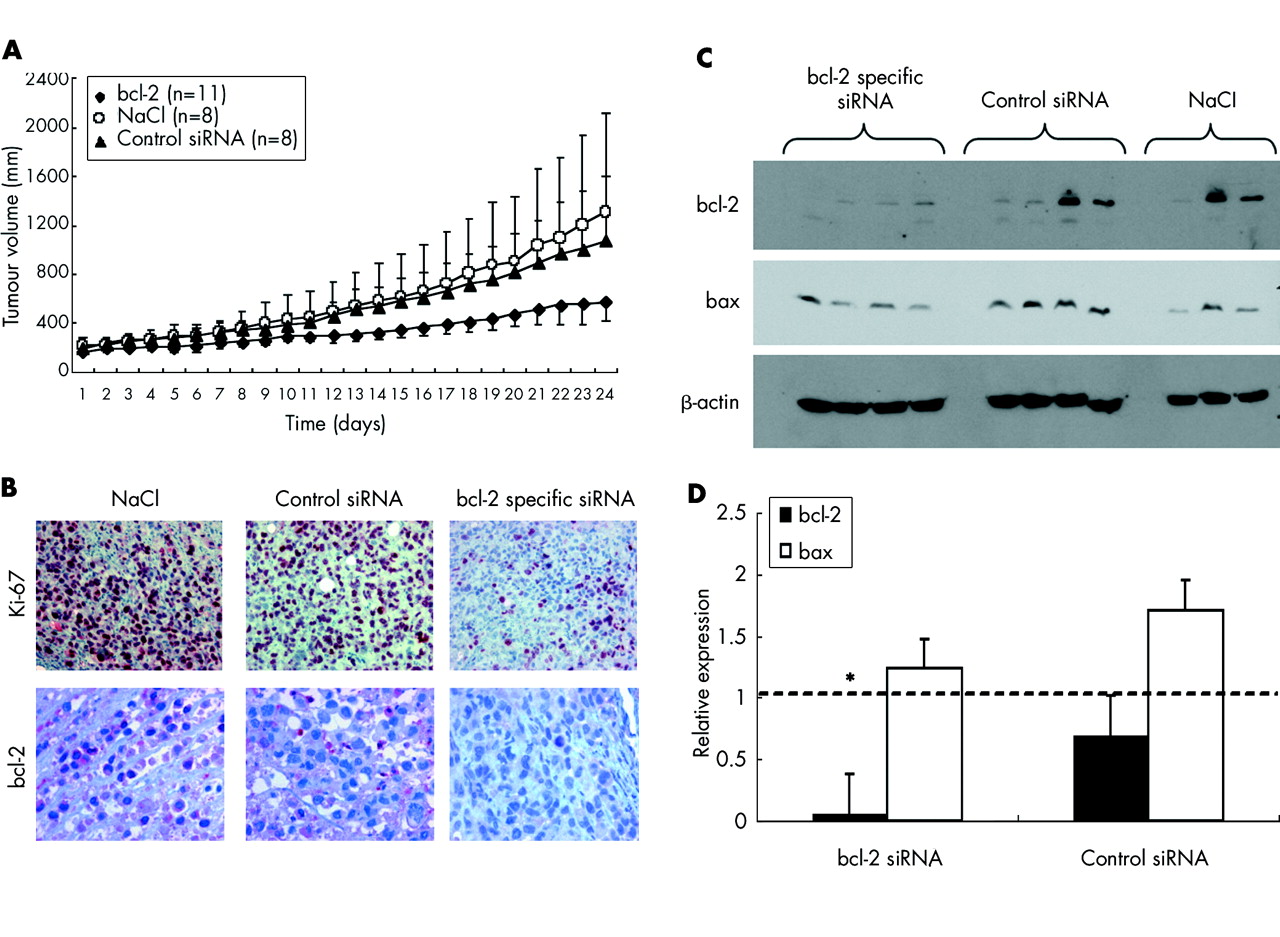
Growth inhibition of pancreatic cancer xenografts in nude mice by bcl-2 specific short interfering RNAs (siRNAs). (A) Size of subcutaneous pancreatic carcinoma xenografts in NMRI mice was determined by daily measurement with a standardised calliper square. Data represent mean (SD) tumour volume from animals treated with daily intraperitoneal injections of 200 μg/kg bcl-2 specific siRNA bcl5, control siRNA 1, or vehicle (physiological saline, NaCl), beginning at day 1. (B) Immunohistochemical analysis of formalin fixed paraffin embedded tumour specimens revealed decreased expression of Ki-67 in animals treated with siRNA against bcl-2 but not in control mice. Expression of bcl-2 was decreased by approximately 56% in animals treated with siRNA against the target gene versus physiological saline controls (NaCl). (C) Western blot analysis of xenograft samples revealed specific downregulation of bcl-2 protein in all animals treated with bcl5, in two of four animals treated with control siRNA 1, and in one of three mice from the NaCl group. Proapoptotic bax was detected in all samples; β-actin served as a loading control. (D) Semiquantification of western blot results. Mean (SEM) densitometry values per group relative to NaCl treated animals (broken line). *p<0.05 versus NaCl treated animals.
Bcl-2 specific siRNA delays growth of pancreatic cancer xenografts in vivo
To assess the efficacy of siRNA mediated gene silencing in vivo, mice (n = 11 for bcl-2, n = 8 for controls) bearing human pancreatic carcinoma xenografts were treated intraperitoneally with daily injections of either bcl5, control siRNA 1, or physiological saline. All mice survived the 24 days of treatment without any signs of organ toxicity, as shown by histological analysis (for example, no haematopoietic arrest in the bone marrow, no hepatic or kidney necrosis). No significant weight loss was observed in treated or untreated animals. Minor signs of fibrinous peritonitis were observed in all groups due to daily intraperitoneal injections. Mean relative tumour volume in the bcl5 group was 579.1 mm3 compared with 1078.3 mm3 in the control siRNA group and 1307.2 mm3 in the saline control group, representing a 56% reduction in tumour volume in mice treated with bcl-2 specific siRNA versus NaCl (fig 8A). Reduced tumour volumes were matched to decreased tumour cell proliferation, as demonstrated by immunohistological staining for Ki-67. In the bcl5 group, only 19.3% of tumour cells showed positive staining for Ki-67 (p<0.05) compared with 31.7% in animals receiving control siRNA or 43.4% in animals that received physiological saline (fig 8B, top row). Expression of bcl-2 (fig 8B, bottom row) was only decreased in animals treated with siRNA against bcl-2 (3.7% positive cells) but was highly expressed in animals that received NaCl solution or control siRNA (75.0% and 61.5%, respectively). These results indicate a high target specificity of RNAi in vivo. Semiquantitative western blotting from snap frozen xenograft specimens revealed specific downregulation of bcl-2 in three of four of the bcl5 treated animals while proapoptotic bax and the housekeeping protein β-actin remained unchanged (fig 8C). However, as yet unidentified factors may contribute to decreased tumour cell proliferation also. Thus in animals treated with control siRNA or with physiological saline, two of four also showed loss of bcl-2 whereas in others expression of the target gene remained elevated, indicating different endogenous expression of bcl-2 due to clonal expansion of low or high expressing tumour cells. Overall densitometric analysis showed a reduction in bcl-2 expression in the bcl5 treated group to a mean of 6.0% of expression in the NaCl group (p<0.05, fig 8D). In the control group, bcl-2 expression reached 70.0% of the saline control. Proapoptotic bax remained stable at a mean of 124% (bcl5) and 170% (control siRNA), shifting the bax/bcl-2 ratio to 20.7 in animals treated with bcl-2 specific siRNA compared with 2.5 in NaCl controls.
DISCUSSION
Efficient and well tolerated therapy for pancreatic cancer is urgently needed. Specific downregulation of genes that prevent apoptosis or chemotherapy resistance by RNAi is a promising treatment approach. Here, we have shown that siRNA molecules specifically targeting antiapoptotic bcl-2 lead to enhanced apoptosis and suppressed cell proliferation of pancreatic carcinoma in vitro and in vivo. Enhanced degradation of bcl-2 mRNA and protein was specific for the five bcl-2-siRNAs used, as several control siRNAs did not induce similar effects in pancreatic carcinoma cells while foreskin fibroblasts (used as control cells) remained largely unaffected.
Downregulation of bcl-2 by conventional antisense oligonucleotides has been shown to enhance apoptosis and chemotherapy sensitivity in a variety of human malignancies.20,21,42,43 The bcl-2 family consists of proapoptotic (for example, bax) and antiapoptotic (for example, bcl-2 or bcl-xL) members. While the exact mechanisms still remain elusive, the rheostat model suggests that the balance of pro and antiapoptotic bcl-2 proteins decides cell fate at the mitochondrial level.44,45 Downregulation of bcl-2 disturbs mitochondrial membranes leading to a breakdown of the mitochondrial membrane potential ΔΨm and subsequent release of cytochrome c from the mitochondrial intermembrane space which then activates the apoptosome and triggers the caspase cascade.44–46 Accordingly, we observed a breakdown of ΔΨm after transfection of bcl-2 specific siRNAs in YAP C cells but not after transfecting control siRNAs, as evidenced by FACS analysis. JC-1 staining of ΔΨm correlated with protein levels of bcl-2, indicating specific silencing by corresponding siRNAs. Importantly, applying bcl-2 specific siRNA in vivo leads to tumour reduction, accompanied by specific downregulation of bcl-2 mRNA and protein, as shown by RT-PCR, western blotting, and immunohistochemistry.
Furthermore, in accordance with cell culture results, cell proliferation (as shown by Ki-67 immunohistochemistry) was reduced in vivo. However, unspecific control siRNA also caused a slight decrease in Ki-67 staining, while proliferation was unaffected in the NaCl group. We also found low levels of bcl-2 expression in some control animals that did not receive anti-bcl-2 siRNA, as we did not pool protein extracts from all animals of one experimental group to perform semiquantitative western blotting. This is explained by expansion of different pancreatic cancer cell clones, some of which express bcl-2 only at low levels, in the experimental tumours (data not shown). However, overall, bcl-2 expression was downregulated significantly in the anti-bcl-2 siRNA treated group compared with the control groups. Furthermore, apart from the highly target specific effects of siRNAs, other activities appear to contribute to the observed growth inhibition of pancreatic cancer xenografts in vivo.
While it was shown that only long dsRNA molecules can trigger an interferon response and upregulate interferon dependent genes (for example, Janus family tyrosine kinase, JAK, and signal transducers and activators of transcription, Stats), recent reports have demonstrated that siRNAs can also induce changes in genes unrelated to the targeted sequence (for example, upregulation of p21 and p5347–49 which might induce bystander effects in untransfected cells). RNAi is a highly conserved biological response mechanism against genomic contamination by dsRNA, such as by viral infection, and several dsRNA binding proteins, such as wig-1, have been identified recently.50 These proteins have been shown to be involved in p53 signalling,51 inducing growth arrest for example,52 and therefore likely contributing to the observed growth inhibition after transfecting unspecific siRNA in vitro and in vivo. These factors, as well as clonal expansion of untransfected cells, may be responsible for the observed discrepancy between cell number and apoptosis rate.
Unpredictable interactions, toxicity, and variability in silencing different target genes, coupled with weak in vivo stability, have been major drawbacks for the routine administration of antisense oligonucleotides. This contrasts with the favourable in vivo stability of siRNA53 which was applied to numerous therapeutic genes in vitro and to mouse models in vivo.54,57 Variant silencing potency of siRNAs may be due to different transfection efficacies in several experimental settings. Importantly, it has been commonly observed that some siRNAs derived from and targeted to the same mRNA possess pronounced silencing activity, while others are barely active. We also observed variable proapoptotic effects of our five siRNAs targeting the bcl-2 gene but also of control siRNA 1. Our siRNAs varied in length for the paired duplex (21 or 22 nt) and the target region in the bcl-2 mRNA. Recent publications showed that the rational design of siRNAs can enhance their silencing activities.58–60 Thus separation of the siRNA duplex into sense and antisense strands is mediated by an ATP dependent helicase. Dependent on which end of the duplex is more accessible for the enzyme, either the desired antisense strand (unwinding from the 3′-end of the sense strand) or the unwanted sense strand (unwinding from the 5′-end of the sense-strand) is predominantly incorporated into RISC. When the sense strand is incorporated, no cleavage of the targeted mRNA is observed, which explains the ineffectiveness of some siRNAs. Strand separation from the 3′-end can be facilitated by selecting for A/U base pairs in positions 15–19, as has been done for all of our bcl-2 specific siRNAs (table 1). Further improvement in siRNA design (for example, by inserting mismatches in the nt 15–19 region of the duplex or chemical variations to the phosphate backbone) might further enhance strand separation and lead to increased silencing activity.55,56
siRNAs have been used to suppress expression of reporter genes under experimental conditions in mice.32–34,57 Ours is the first report on silencing of an endogenous target gene in a solid cancer after systemic administration of unmodified siRNAs in vivo, investigating the therapeutic utility of siRNA in the treatment of these disease entities. Furthermore, we showed that siRNAs are homogenously distributed in all organs of adult mice after intraperitoneal administration followed by renal clearance and a second hepatic elimination phase. Chemical modification of siRNA duplexes can further improve tissue targeting (for example, by tagging to RGD peptides for increased cellular uptake) and in vivo stability.53,61
More studies are needed to assess the pharmacokinetics and improve tissue specific targeting of certain genes with siRNA. Enhancing cleavage of bcl-2 mRNA by siRNA may contribute to the treatment of pancreatic cancer by improving chemotherapy sensitivity of this otherwise resistant carcinoma.
Acknowledgments
This work was supported by grants from the German Cancer Aid (10-2112-Oc1), the FUTUR-Programme of the State of Bavaria (project No. 263), and the ELAN-Programme of the Faculty of Medicine, University of Erlangen-Nuernberg (project 02.08.08.2).
We are indebted to Sandra Leitner, Catherina Kühl, and Gisela Weber for excellent technical assistance, and to Gabriele Krumholz for support in animal care and animal experiments. Professor Torsten Kuwert and Dr Olaf Prante, Department of Nuclear Medicine, are acknowledged for support in radioactive animal experiments. We thank Ribopharma AG, Kulmbach, Germany, for providing siRNAs and Dr Anke Geick, Ribopharma AG, for performing western blotting from in vivo material. We are grateful to Dr Hans-Peter Vornlocher, Ribopharma AG, for intense and fruitful discussions.
REFERENCES
Footnotes
-
↵* M Ocker and D Neureiter contributed equally to this work
-
Conflict of interest: None declared.