Article Text

Abstract
Background and aims: Intraductal papillary mucinous tumours (IPMT) of the pancreas constitute a unique pathological entity with an overall incidence of associated invasive malignancy of 20%. The malignant potential of an individual IPMT cannot be accurately predicted. Preoperative estimation of the risk of associated invasive malignancy with IPMT would be of significant clinical benefit. As aberrations in cell cycle regulatory genes are associated with the progression of precursor pancreatic ductal lesions to invasive adenocarcinoma, we examined expression of key cell cycle regulatory genes in the cyclin D1/retinoblastoma pathway and the transforming growth factor β/Smad4 signalling pathway in a cohort of patients with surgically resected IPMT.
Methods: Sections of formalin fixed paraffin embedded pancreatic tissue from a cohort of 18 patients with IPMT were examined using immunohistochemistry for protein expression of cell cycle regulatory genes p16INK4A, p21CIP1, p27KIP1, cyclin D1, pRb, and p53, as well as the cell signalling molecule Smad4. A comparison of expression levels was made between adenoma/borderline IPMT (10 patients) and intraductal papillary mucinous carcinoma (IPMC) (eight patients, four of whom harboured invasive carcinoma). Statistical analysis was performed using the χ2 and Fisher's exact tests.
Results: Aberrant expression of the proteins examined increased in frequency from adenoma/borderline IPMT to IPMC. Specifically, there was a significantly greater incidence of loss of p16INK4A expression in IPMC: 8/8 lesions (100%) compared with 1/10 (10%) adenoma/borderline IPMT (p<0.001). Similarly, loss of Smad4 expression was associated with IPMC: 3/8 (38%) versus adenoma/borderline IPMT 0/10 (p<0.03). Loss of Smad4 expression within the IPMT was the best marker for the presence of invasive carcinoma (p<0.001).
Conclusions: These data indicate that loss of p16INK4A and Smad4 expression occur more frequently in IPMC alone, or with associated invasive carcinoma, compared with adenoma/borderline IPMT. Aberrant protein expression of these cell cycle regulatory genes in IPMT and pancreatic intraepithelial neoplasia in the current model of pancreatic cancer progression suggest similarities in their development and may also represent the subsequent risk of invasive carcinoma.
- intraductal papillary mucinous tumour
- pancreatic adenocarcinoma
- DPC4
- Smad4
- p53
- p16
- IPMT, intraductal papillary mucinous tumour
- IPMC, intraductal papillary mucinous carcinoma
- ERCP, endoscopic retrograde cholangiopancreatography
- TGF-β, transforming growth factor β
- Cdk, cyclin dependent kinase
- pRb, retinoblastoma protein
- PanIN, pancreatic intraepithelial neoplasia
Statistics from Altmetric.com
- IPMT, intraductal papillary mucinous tumour
- IPMC, intraductal papillary mucinous carcinoma
- ERCP, endoscopic retrograde cholangiopancreatography
- TGF-β, transforming growth factor β
- Cdk, cyclin dependent kinase
- pRb, retinoblastoma protein
- PanIN, pancreatic intraepithelial neoplasia
Intraductal papillary mucinous tumours (IPMT) of the pancreas are a distinct group of lesions that arise within pancreatic ducts. The overall incidence of invasive carcinoma associated with an IPMT is 10–20%.1 There are no effective means of determining preoperatively the presence or risk of invasive carcinoma associated with IPMT.1,2 As patients with IPMT commonly have significant comorbidities, identifying the risk of invasive malignancy preoperatively for an individual patient with IPMT would optimise treatment for those at high risk for invasive carcinoma while avoiding radical treatment in those in whom it may not be necessary. Attempts have been made to identify preoperatively invasive carcinoma in the presence of IPMT using computed tomographic appearances,3 magnetic resonance imaging,4 and endoscopic retrograde cholangiopancreatography (ERCP)5 but these techniques lack sufficient accuracy.
The World Health Organisation (WHO) has defined IPMT as an intraductal papillary growth of neoplastic columnar cells producing mucin. IPMT are subdivided into three groups based on increasing nuclear atypia and mitotic rate: adenoma (no significant atypia and mitoses almost absent), borderline (more irregularly structured papillae, frequent mitoses and moderate dysplasia consisting of some nuclear pleomorphism, and occasional distinct nucleoli), and intraductal papillary mucinous carcinoma (IPMC, severe epithelial dysplasia with irregular epithelial budding and bridging). According to the absence or presence of neoplastic glandular structures invading the pancreatic tissue surrounding the involved ducts, IPMC are separated into invasive and non-invasive types.6 Differences in clinical and histological characteristics have resulted in IPMT also being classified according to their distribution within the pancreas, differentiating main duct tumours from branch duct tumours.7–10
Evidence supporting IPMT as precursor lesions of invasive carcinoma is limited. IPMT share similar clinicopathological characteristics with pancreatic ductal adenocarcinoma including sex and age distribution and location within the pancreas. In addition, previous studies have identified similarities in the expression of some oncogenes and tumour suppressor genes in IPMT and invasive ductal adenocarcinoma. In particular, activating K-ras mutations,11 p53 protein accumulation,11,12c-erbB-2 overexpression, and oncofetal antigens such as CEA, CA19-9, and DUPAN-213 have been identified in a similar proportion of IPMT and invasive carcinomas. Chromosomal aberrations associated with IPMT and invasive carcinoma of the pancreas also share a similar profile.14 Heterogeneity of allelic loss within IPMT lesions has been identified14 suggesting progression within the IPMT. Studies comparing the proliferative indices of IPMT with normal ductal cells and invasive cancer, using immunohistochemistry for Ki-67 and PCNA, have reported a progressive increase from histologically benign IPMT to borderline lesions, to IPMC, and through to invasive carcinoma.12
Dysregulation of the normal cell cycle regulatory machinery is integral to the neoplastic process and there is compelling evidence implicating loss of cell cycle control in the development and progression of most human cancers.15 Abnormalities in the retinoblastoma pathway that controls G1 to S phase progression in the cell cycle and the transforming growth factor β (TGF-β) signalling pathway occur at high frequencies in pancreatic ductal adenocarcinomas.16 The tumour suppressor gene INK4A maps to chromosome 9p21 and encodes the cyclin dependent kinase (Cdk) inhibitor p16INK4A, a key regulator of cell cycle progression at the G1/S phase transition point.17 p16INK4A binds to Cdk4 and Cdk6 to inhibit their catalytic activity leading to reduced phosphorylation of the retinoblastoma protein (pRb) and hence G1 cell cycle arrest.17 p16INK4A is frequently inactivated in human cancer,17 and in pancreatic cancer p16INK4A is inactivated in more than 90% of ductal adenocarcinomas.18 The mechanisms of p16INK4A inactivation include homozygous deletion, mutation, and promoter methylation.19 Mutations of the tumour suppressor gene p53 occur in approximately half of all human malignancies.20 Inactivation of p53 has been consistently reported in 50–75% of pancreatic carcinomas.18 p53 protein has many biological functions, including regulation of programmed cell death and inhibition of cell proliferation by increasing intracellular levels of p21CIP1.20 The Cdk inhibitors p21CIP1 and p27KIP1 also act to prevent pRb phosphorylation by binding and inhibiting cyclin E/Cdk2 enzyme complexes.15 Cyclin D1 overexpression occurs in 68% of pancreatic cancers and is associated with a poor prognosis.21
The TGF-β growth inhibitory pathway signals through Smad proteins 1–18. Characterisation of Smad functions has defined three groups: receptor regulated Smads (Smad1, 2, 3, 5, and 8), common partner Smad (Smad4), and inhibitory Smads (Smad6 and 7).22 Normally, ligand induced TGF-β receptor activation results in the formation of heterodimeric complexes of Smad3 with Smad4 leading to the translocation of these complexes into the nucleus where Smad4 activates transcription of cell cycle inhibitory factors, particularly p21CIP1.23 Homozygous deletion or mutation of DPC4, the gene encoding Smad4 located on chromosome 18q, has been reported in 55% of pancreatic ductal adenocarcinomas.16 Loss of Smad4 may lead to upregulation of the retinoblastoma pathway and consequently progression from G1 to S phase of the cell cycle and hence increased cellular proliferation.
In an attempt to characterise the molecular pathology of IPMT and further define its natural history, we have examined expression of the cell cycle regulatory genes p16INK4A, p21CIP1, p27KIP1, cyclin D1, p53, pRb, and the cell signalling molecule Smad4 in a series of 18 patients with IPMT.
METHODS
Tissue
Archival formalin fixed paraffin embedded tissue was collected from Westmead Hospital, Royal North Shore Hospital, St Vincent's Hospital Campus, Royal Prince Alfred Hospital, and Concord Repatriation General Hospital, Sydney, Australia, from 18 patients with IPMT whose pancreata were resected between 1990 and February 2000. Ethical clearance was obtained from the participating institutions. Haematoxylin and eosin sections were examined and the IPMT were graded according to current WHO criteria by two of the pathologists (SAB and JGK). Tissue blocks that contained IPMT were selected for the study. Clinical information including presenting symptoms, ERCP appearances, and macroscopic pathology descriptions were collected retrospectively from patient records. Outcome data (survival and status of disease) were obtained by contacting the treating physician.
Immunohistochemistry
Haematoxylin and eosin staining and immunohistochemistry were performed on 4 μm serial sections of paraffin embedded formalin fixed tissue. For immunohistochemistry, sections were deparaffinised in xylene and rehydrated through a series of alcohols. Antigen retrieval was achieved by microwave heating in citrate buffer at pH 6.0. Endogenous peroxidase activity was quenched in 3% hydrogen peroxide in methanol and non-specific binding of secondary antibody blocked by incubation with normal horse serum. Individual sections were incubated with mouse monoclonal antibodies to either p21CIP1 (clone 70; Transduction Laboratories, Lexington, Kentucky, USA), p27KIP1 (clone 57; Transduction Laboratories), p53 (clone DO-7; Dako Corporation, Carpinteria, California, USA), p16INK4A (clone ZJ11; NeoMarkers, Fremont, California, USA), cyclin D1 (clone DCS-6; Novocastra, Newcastle upon Tyne, UK), pRb (clone G3-245; Pharmingen, San Diego, California, USA), or Smad4 (clone B-8; Santa Cruz, Santa Cruz, California, USA). A streptavidin-biotin peroxidase detection system was used in accordance with the manufacturer's instructions (Vectastain Elite Kit; Vector Laboratories, Inc., Burlingame, California, USA) and then developed using 3,3`-diaminobenzidine as substrate. Sections were counterstained with haematoxylin and light green. Formalin fixed paraffin embedded cell lines, where the status of the genes examined had been determined by other authors, were used as positive and negative controls (table 1).
Cell line controls for immunohistochemistry
Analysis
For each patient 2–5 separate samples of the lesion were examined. Staining was independently assessed by two observers (SAB and JGK) and scored as a percentage of nuclei staining positive within each tumour. Cytoplasmic staining was also assessed for Smad4. Scoring was performed by examining the full section for assessment of staining adequacy, then counting 500 nuclei/cells per lesion from representative high power fields, and expressing the result as a percentage. The criteria to achieve a positive score for each of the antigens studied were based on published criteria: pRb, a homogeneous pattern with >10% of nuclei staining positive; p53, a homogeneous staining pattern with >10% demonstrating nuclear p53 protein accumulation24; cyclin D1 was recorded as overexpressed when a homogeneous staining pattern with >5% of nuclei staining was observed25; p27KIP1, a homogeneous staining pattern with >50% of nuclei staining26; p16INK4A was only scored as absent when no nuclei were observed to stain in the IPMT or invasive carcinoma in the presence of islet cell and stromal cell nuclear positivity27; and Smad4 was scored as absent when <5% cytoplasmic or nuclear staining was evident. Immunohistochemical detection of Smad4 with this antibody is reported to be accurate in determining DPC4 gene status with high sensitivity and specificity.28 For p21CIP1, a homogeneous staining pattern with >50% of nuclei staining was designated as normal expression as there are no established conventions for p21CIP1 scoring and this molecule has similar functional attributes to p27KIP1. This cut off provided the greatest differential between adenoma/borderline IPMT and IPMC. Any discrepancies between the two observers were resolved by conference using a multiviewer microscope.
Statistical evaluation of differences in proportions between groups of IPMT was performed using χ2 analysis and Fisher's exact test with Statview 4.5 Software (Abacus Systems, Berkeley, California, USA). A p value of <0.05 was accepted as statistically significant.
RESULTS
Clinical and pathological data
The 18 IPMT were divided into two groups based on their nuclear atypia/degree of dysplasia, as defined by WHO criteria: IPMC (carcinoma in situ) was diagnosed in eight patients, four of whom had concomitant invasive carcinoma, and adenoma/borderline IPMT (mild/moderate dysplasia) in 10 patients (one adenoma and nine borderline lesions). Demographic and histopathological details of each patient are summarised in table 2. All patients had pancreatic resections for their lesions (17 Whipple pancreaticoduodenectomy, one total pancreatectomy).
Demographic, histopathological data, and aberrant protein expression in 18 patients with IPMT
The majority of tumours (16/18) formed a discrete lesion but all were associated with diffuse ductal changes of variable extent. Only two patients had true diffuse disease of the duct system without evidence of a significant focal lesion. Microscopically, only a mild degree of heterogeneity was seen, with less than 10% of any IPMT demonstrating a different level of dysplasia. No differential immunohistochemical staining was seen in those instances. The histological appearance of the surrounding ducts did not always reflect that of the IPMT but resembled ductal premalignant lesions described in the proposed progression model for pancreatic ductal adenocarcinoma.29 These ductal lesions were predominantly less dysplastic than the associated IPMT but they occasionally demonstrated more severe atypia than that seen in the IPMT. There was no association between location within the ductal system of the pancreas (main duct tumours versus branch duct tumours), the degree of IPMT dysplasia, or survival (data not shown). Of the four associated invasive ductal adenocarcinomas in the cohort, three had the usual tubular pattern and one corresponded to the mucinous non-cystic variant.
Mean age for the 18 patients was 65 years (range 47–80). Overall median follow up was 21 months (range 4–78). The majority were males (72%). At last follow up, three patients had died within 21 months (all of whom had associated invasive carcinoma), one of whom had negative resection margins. The fourth patient with invasive carcinoma had evidence of recurrent disease at 10 months after resection. Three of the remaining patients with IPMT and no invasive carcinoma had intraductal tumour involving the resection margin of the pancreas. Two are alive with no evidence of disease after 40 months and one was lost to follow up at 10 months.
Molecular pathology
Concordance was high between the scores of the two observers, with less than 10% variability. In all cases this difference did not affect the expression status with the cut offs used for positivity. A summary of the expression states of the cell cycle regulatory proteins and Smad4 is presented in table 3.
IPMT: proportions of lesions exhibiting abnormalities in protein expression
p16INK4A protein expression
There was loss of p16INK4A protein expression in 100% (8/8) of IPMC but only in 10% (1/10) of adenoma/borderline IPMT. This lesion was a borderline lesion (case No 10). The difference was independently significant (p<0.001). p16INK4A staining of islet cell, stromal cell, and duct cell nuclei served as internal positive controls. When p16INK4A protein was detectable within the IPMT, ≥5% of IPMT nuclei demonstrated positive staining (fig 1). Of note, one of the IPMC which demonstrated no staining in the intraductal component showed intense p16INK4A expression within the associated invasive adenocarcinoma. The remaining three invasive cancers showed loss of p16INK4A expression, as demonstrated in their associated IPMC.

(A) p16INK4A protein expression in a borderline intraductal papillary mucinous tumour (magnification approximately ×100). (B) Magnified view of a different region of the same section as (A) (magnification approximately ×200). Note stromal cell nuclear staining.
Smad4 protein expression
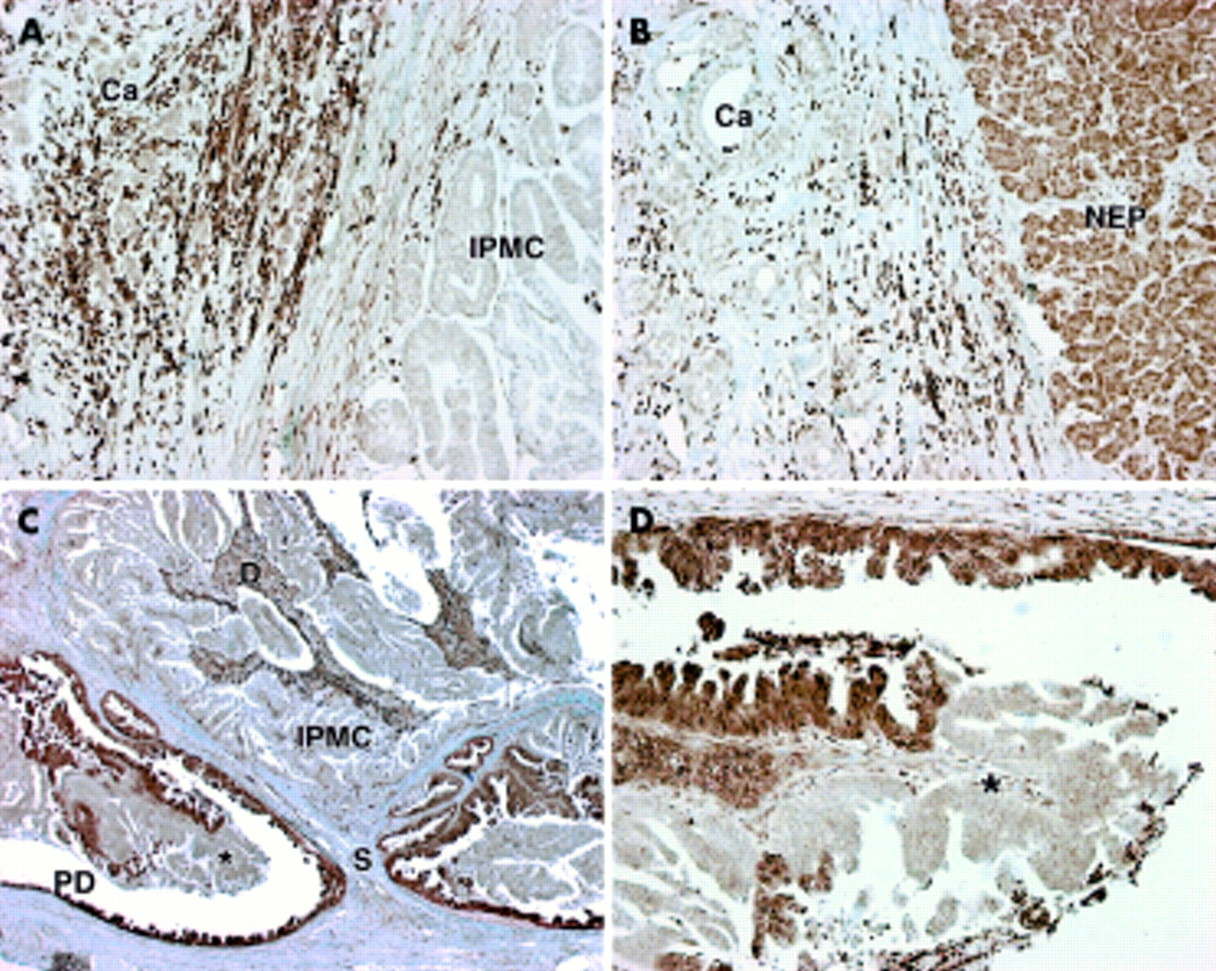
Smad4 expression was seen in all normal cells of the pancreas, including islet cells, acinar cells, stromal cells, and duct cells (fig 2B). Smad4 was either expressed in all cells of the lesion or lost altogether with no detectable expression in either the cytoplasm or nucleus. The cytoplasm stained intensely in all cells that were positive and nuclear staining was also seen in more than 80% of all Smad4 positive cells. All 10 adenoma/borderline IPMT expressed Smad4 while 3/8 (38%) IPMC lost Smad4 expression (fig 2A); these three cases all had associated invasive carcinoma. Smad4 expression of the invasive cancers was identical to expression within the intraductal component of the IPMC of all four cases. All three tubular invasive carcinomas lost Smad4 expression whereas the single mucinous non-cystic invasive carcinoma retained expression of Smad4. In two cases where the IPMC was sectioned at its base there was an abrupt loss of expression from the epithelial lining of the duct, which itself demonstrated severe dysplasia, to the IPMC which filled the lumen of the duct (fig 2C, 2D). Loss of Smad4 significantly differentiated IPMC from adenoma/borderline IPMT (p<0.03), and to a greater degree the presence of invasive cancer (p<0.001). Loss of Smad4 expression was only seen in those IPMC where there was associated invasive carcinoma.

(A) Loss of Smad4 protein expression in intraductal papillary mucinous carcinoma (IPMC) and invasive carcinoma (Ca) (magnification approximately ×100). Note stromal cell positive staining. (B) Loss of Smad4 protein expression in invasive carcinoma (Ca) with adjacent exocrine pancreas expressing Smad4 (magnification approximately ×100) (NEP, normal exocrine pancreas). (C) Abrupt loss of Smad4 protein expression from the walls of the main pancreatic duct to the IPMC. Macroscopically the IPMC filled and distended the main pancreatic duct (magnification approximately ×25) (PD, main pancreatic duct; S, stalk of IPMC; D, debris). (D) Magnified view of (C) demonstrating abrupt loss of Smad4 protein expression with no visible alteration in cellular appearance (magnification approximately ×200) (*location marker for comparison with (C)).
p53 protein expression
Homogeneous p53 protein accumulation was present in a greater but not statistically significant proportion of IPMC (63%) versus adenoma/borderline IPMT (30%) (fig 3A) (p<0.07). Staining of papillae was not uniform for p53. Staining at the bases of the papillae was greater in terms of both the number of positive cells as well as the intensity of the staining compared with cells at the tips of the papillae. This was the reverse of the pattern seen for p21CIP1 expression where nuclei of epithelial cells at the tips of the papillae stained in greater numbers and intensity than at the bases (fig 3B). In all cases of invasive carcinoma, expression status reflected that of the intraductal component of the IPMC.
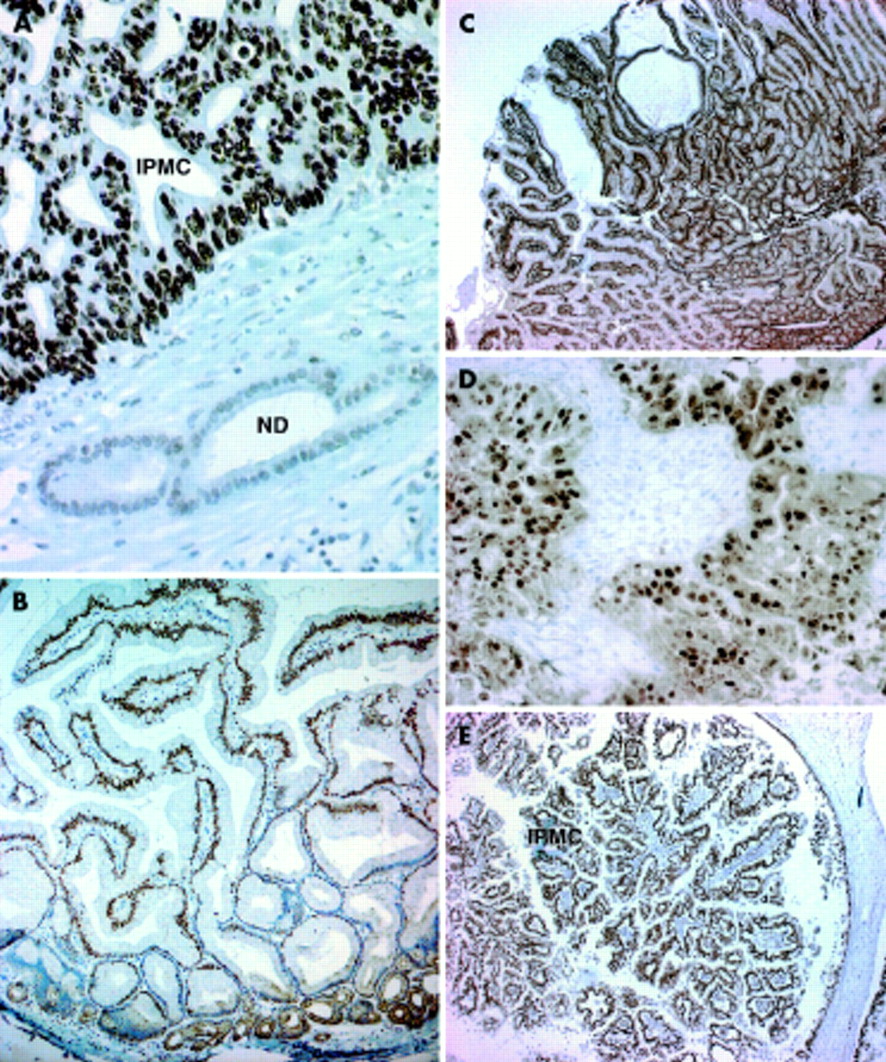
(A) p53 protein overexpression in intraductal papillary mucinous carcinoma (IPMC) with adjacent normal ducts (magnification approximately ×400) (ND, normal pancreatic duct). (B) p21CIP1 protein expression in borderline intraductal papillary mucinous tumour (IPMT). Note differential staining between the bases and tips of the papillae (magnification approximately ×100) (Base, base of papillae of IPMT). (C) p27KIP1 protein expression in borderline IPMT (magnification approximately ×25). (D) Cyclin D1 overexpression in IPMC (magnification approximately ×400). Note the negative stroma providing an internal negative control. (E) Retinoblastoma protein expression in IPMC (magnification approximately ×25).
p21CIP1/p27KIP1 protein expression
p21CIP1 was homogeneously expressed in greater than 50% of nuclei in 8/10 adenoma/borderline IPMT (80%) but only in 3/8 (12%) IPMC (p<0.06). Low p27KIP1 expression (<50% of nuclei) was observed in 3/10 adenoma/borderline IPMT (30%) versus 0/8 IPMC (p<0.09). Of the four IPMC with invasive carcinoma, all had low expression of p21CIP1 within the IPMC but in two of the four invasive carcinomas the expression level increased to positivity. Expression of p27KIP1 in all but three borderline IPMT was positive (>50%) (fig 3C): in one case (case No 3), low p27KIP1 expression was the only aberration of all proteins examined. In contrast with p21CIP1, p27KIP1 expression decreased to less than 50% in two of the four invasive carcinomas from >50% expression in the IPMC. Positive nuclear staining for p21CIP1 in smooth muscle cells of blood vessels and duodenal epithelium, where present, served as internal positive controls. Significant cytoplasmic localisation of p27KIP1 staining was not seen.
Cyclin D1 expression
Cyclin D1 was expressed homogeneously in >5% of nuclei in 5/8 IPMC compared with 3/10 adenoma/borderline IPMT (fig 3D). This difference was not statistically significant (p<0.17). In all cases of invasive carcinoma, the expression status reflected that of the coexistent intraductal component of the IPMC. Expression status did not correlate with coexistent invasive cancer or either p53, p16INK4A, p21CIP1, p27KIP1, or Smad4 expression status (data not shown).
Retinoblastoma protein expression
pRb was homogeneously expressed, with greater than 80% of nuclei staining positively within all 18 IPMT (fig 3E) and the four associated invasive carcinomas. Acinar, islet, and stromal cells as well as duct cells also expressed pRb.
Cumulative analysis
Adenoma/borderline IPMT and IPMC
The prevalence of aberrations in protein expression examined in this study increased with increasing severity of dysplasia within the IPMT, as can be seen in table 2 which shows the aberrations of expression for each patient.
Loss of p16INK4A alone was the strongest marker in differentiating adenoma/borderline IPMT from IPMC (p<0.001). Loss of Smad4 expression also differentiated adenoma/borderline IPMT from IPMC (p<0.03). Although p53 accumulation occurred in more cases of IPMC than adenoma/borderline IPMT the difference was not statistically significant (p<0.16). The combination of loss of p16INK4A and Smad4 expression was also statistically significant in differentiating adenoma/borderline IPMT from IPMC (p<0.03) although not as strongly as p16INK4A alone and no better than Smad4.
Invasive carcinoma with IPMT
Loss of Smad4 alone was the best marker for the presence of invasive carcinoma (p<0.01). Loss of p16INK4A expression in combination with loss of Smad4 expression also cosegregated with invasive carcinoma but was of no advantage over Smad4 alone (p<0.01). Loss of p16INK4A protein expression was also statistically significant (p<0.03).
DISCUSSION
Sequential accumulation of aberrations in protein expression with increasing dysplasia demonstrates the potential importance of the retinoblastoma pathway, most notably p16INK4A, in IPMT progression. The increase in the frequency of cyclin D1 overexpression and p53 accumulation from adenoma/borderline IPMT to IPMC, although not statistically significant, is also supportive of this hypothesis. It is possible that these latter changes represent earlier events in IPMT development than loss of p16INK4A expression and were not elucidated here because our series contained only one adenoma IPMT. In contrast, loss of Smad4 expression was a later event and perhaps as a consequence was specific for the presence of invasive carcinoma. The proportions of genetic aberrations of p16INK4A, cyclin D1, p53, and DPC4/Smad4 presented here approach those reported in large series of invasive ductal adenocarcinomas.16,19,21 The relationship of p16INK4A, p53, and DPC4/Smad4 to dysplasia within IPMT is similar to that reported in pancreatic ducts of the current pancreatic cancer progression model.29 The similarities in incidence and timing of aberrations in the expression of p16INK4A, p53, cyclin D1, and Smad4 suggest similarities in the development of pancreatic intraepithelial neoplasia (PanIN), pancreatic cancer, and IPMT.30
Loss of p16INK4A function is associated with alteration of the histological appearance of neoplastic pancreatic duct cells31 and is represented by the difference in p16INK4A expression between IPMC and adenoma/borderline IPMT in this cohort. Allelic loss at chromosome 9p, the locus for INK4A, has also been reported at higher frequencies in IPMC than in less dysplastic IPMT.14 These results suggest that p16INK4A loss is necessary but not sufficient to induce progression of non-invasive IPMC to invasive carcinoma. A satisfactory model for the regulation of p16INK4A expression in pancreatic cancer has yet to be formulated and this is evident in one case where although expression of p16INK4A was absent in the non-invasive IPMC it was expressed in the associated invasive cancer. A possible explanation is that p16INK4A was inactivated by promoter methylation within the intraductal component but then reactivated in the invasive cancer where the replication rate of the cells is likely to be greater. It is also possible that the invasive tumour arose from tissue not of IPMT origin. Given that invasive ductal adenocarcinomas of the pancreas are known to lose p16INK4A expression in more than 90% of cases, and that the loss of p16INK4A most likely occurs in preinvasive stages of tumour development,27 the findings of this study suggest that loss of p16INK4A may play a significant role in IPMT progression to invasive carcinoma. Expression of pRb was equivalent in all IPMT within our cohort. Aberrant p16INK4A expression has been reported to be related to the pRb status in other cancers32 but the proportion of phosphorylated pRb was not assessed in this cohort. It is also interesting to note that all three invasive carcinomas with a tubular pattern demonstrated aberrant expression of p53, p16INK4A, Smad4, cyclin D1, and p21CIP1 but the single mucinous non-cystic type carcinoma only showed aberrations in p16INK4A and p21CIP1 expression (table 2).
In spite of the potential limitation of the size of the cohort, there was a highly significant difference in gene expression, particularly of p16INK4A, between IPMC and adenoma/borderline IPMT. As published series of IPMT are composed of relatively small numbers, we have compared the results of another significant series by Fujii and colleagues14 with our own data. Fujii et al identified significant allelic losses in IPMT which were also more common in IPMC than adenoma/borderline IPMT. If we assume that allelic loss on chromosomes 9p, 17p, and 18q corresponds to loss of functional p16INK4A, p53, and DPC4/Smad4, respectively, then these results parallel those of our study (table 4).
Comparison of allelic loss data from Fujii and colleagues14 with protein expression data in this study
The current pancreatic ductal adenocarcinoma progression model (fig 4) defines progression from normal cuboidal epithelium, to flat hyperplasia (columnar cells with some crowding), to ductal papillary hyperplasia (increased crowding of columnar cells with papillary projections containing a stromal core). These early changes are termed PanIN-1A and PanIN-1B. The next stage in the progression model known as PanIN-2 (ductal papillary hyperplasia with atypia) is characterised by similar architectural characteristics as PanIN-1B but with mild to moderate nuclear atypia (nuclear stratification, some pleomorphism, occasional mitoses, and prominent nucleoli). All three lesions may have varying degrees of mucinous cell hypertrophy. PanIN-3 or atypical ductal hyperplasia demonstrates severe atypia and has in the past been termed carcinoma in situ, and is the final preinvasive stage.29 Accumulation of aberrations in protein expression presented in this study demonstrate remarkable similarities to the findings in the above progression model when the cytological appearances of IPMT are correlated with PanIN lesions (fig 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic diagram representing genetic aberrations in common between pancreatic intraepithelial neoplasia (PanIN) in the current pancreatic cancer progression model and intraductal papillary mucinous tumours (IPMT). In the pancreatic cancer progression model, progression of normal ductal epithelium through PanIN to invasive cancer is shown from left to right (adapted from Hruban and colleagues29 with permission, artwork by Jennifer L Parsons). In IPMT, progressive cellular atypia is shown from left to right, as defined by the WHO.6 Black bars denote temporal occurrence of aberrations in common for both PanIN lesions and IPMT. Grey arrows indicate potential paths of IPMT development. K-ras: activating mutations. HER-2/neu: overexpression. p16: loss of expression. p53: protein accumulation or mutation. Cyclin D1: overexpression. DPC4/Smad4: loss of expression. LOH: loss of heterozygosity. IPMC, intraductal papillary mucinous carcinoma (data from this study, references quoted in the text, and unpublished data).
In contrast with colorectal cancer where most invasive carcinomas develop from adenomatous polyps rather than flat adenomas, pancreatic ductal adenocarcinomas appear to develop predominantly from non-polypoid duct lesions (PanIN) with a small proportion developing via a polypoid lesion. In addition, invasive ductal adenocarcinoma in association with IPMT is more commonly of the mucinous non-cystic (colloid) type (65%) with only 35% being of the tubular type, which constitute over 95% of invasive pancreatic carcinomas that are not associated with IPMT.1 Three of four invasive carcinomas in our study were of the tubular type, all of which demonstrated loss of Smad4 expression, the mucinous non-cystic type maintaining Smad4 expression. In a recent publication by Iacobuzio-Donahue and colleagues,33 19/29 invasive carcinomas in association with IPMT were of the colloid (mucinous non-cystic) type, all of which demonstrated strong Smad4 expression by immunohistochemistry. Five of 10 tubular carcinomas also stained strongly for Smad4 (similar to the 55% reported incidence of loss of Smad4 expression in non-IPMT associated pancreatic carcinoma by the same group16); one was negative, and the remaining four demonstrated only faint cytoplasmic positivity. Faint staining above background was not seen in our cohort. The significance of faint cytoplasmic immunohistochemical labelling with this antibody is not known and may not represent wild-type Smad4 expression.
CONCLUSION
This study reveals similarities in genetic aberrations between IPMT, especially those associated with invasive adenocarcinoma of the tubular type, and PanIN in the currently proposed progression model of pancreatic ductal adenocarcinoma. The progressive accumulation of these aberrations likely reflects the risk of invasive carcinoma. IPMT may represent a morphologically distinct premalignant lesion with similar potential for invasive malignancy as its equivalent PanIN counterparts.
Acknowledgments
This research was supported by grants from the National Health and Medical Research Council (NH & MRC) of Australia, the Cancer Council New South Wales and the RT Hall Trust. AVB was supported by the Sir Roy McCaughey Fellowship from the Royal Australasian College of Surgeons and the Di Boyd Cancer Research Award from the St Vincent's Clinic Foundation. Access to tissue was kindly facilitated by Professor Ross Smith and Dr Christopher Bambach (Royal North Shore Hospital), Dr Roy P Brancatisano, Dr Michael Hollands and Dr Arthur Richardson (Westmead Hospital), Dr Mark Richardson and Dr Betty Lin (Concord Repatriation General Hospital), and Dr David Storey (Royal Prince Alfred Hospital).