Article Text

Abstract
Background and aims There are no chemopreventive strategies for pancreatic cancer or its precursor lesions, pancreatic intraepithelial neoplasia (PanINs). Recent evidence suggests that aspirin and inhibitors of angiotensin-I converting enzyme (ACE inhibitors) have potential chemopreventive properties. In this study, we used a genetically engineered mouse model of pancreatic cancer to evaluate the chemopreventive potential of these drugs.
Methods Drug treatment was initiated at the age of 5 weeks. LsL-KrasG12D; Pdx1-Cre or LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre transgenic mice were randomly assigned to receive either mock treatment, aspirin, enalapril, or a combination of both. After 3 and 5 months, animals were killed. The effect of aspirin and enalapril was evaluated by histopathological analyses, immunostaining, and real-time PCR.
Results After 3 and 5 months of treatment, enalapril and aspirin were able to significantly delay progression of mPanINs in LsL-KrasG12D; Pdx1-Cre mice. Furthermore, development of invasive pancreatic cancer in LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre transgenic mice was partially inhibited by enalapril and aspirin. Invasive pancreatic cancer was identified in 15 of 25 (60%) LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre untreated control mice, but in only three of 17 (17.6%, p=0.01) mice treated with aspirin, in four of 17 (23.5%, p=0.03) in mice treated with enalapril alone, and in five of 16 (31.2%, p=0.11) mice treated with a combination of both drugs. Using real-time PCR we found a significant downregulation of the target genes VEGF and RelA demonstrating our ability to achieve effective pharmacological levels of aspirin and enalapril during pancreatic cancer formation in vivo.
Conclusion Using a transgenic mouse model that imitates human pancreatic cancer, this study provides first evidence that aspirin and enalapril are effective chemopreventive agents by delaying the progression of PanINs and partially inhibiting the formation of murine pancreatic cancer. This study together supports the hypothesis that aspirin and ACE inhibitors might be a valid chemopreventive strategy.
- Pancreatic cancer
- PanINs
- chemoprevention
- ACE-inhibitor
- enalapril
- aspirin
Statistics from Altmetric.com
Introduction
Pancreatic cancer is a devastating and almost uniformly lethal malignancy that accounts for approximately 33 000 deaths in the USA every year, rendering it the fourth most common cause of cancer-related mortality.1 Although the past decades have seen intense research efforts aimed at a better understanding of the underlying aetiological and pathophysiological mechanisms, this increased knowledge could not so far be translated successfully into better clinical treatment strategies and improved patient survival. In fact, during the past 30 years the overall median 5-year survival rates for pancreatic cancer have improved only marginally and are currently around 5%.1 2 Notably, there is now strong evidence that invasive pancreatic adenocarcinoma proceeds through a morphological spectrum of non-invasive ductal lesions known as pancreatic intraepithelial neoplasia (PanIN), and that histological progression of these lesions towards invasive cancer is associated with the progressive accumulation of genetic abnormalities.3 The analyses of pancreatic carcinoma have unfortunately been hampered by a number of unique challenges. For instance, at the time of diagnosis, pancreatic cancer is usually at an advanced stage and has often metastasised. As a result, the stepwise tumorigenic progression has been inaccessible for study and the precursor cell types still remain an area of ongoing studies. The preclinical study of PanINs has recently been made possible by the generation of genetically modified animal models, which recapitulate human PanINs and invasive pancreatic cancer on a genetic and histomorphological level.4 5
Aspirin has been used to control pain and inflammation for over a century. Epidemiological studies associated a decreased incidence of colorectal cancer with the long-term use of aspirin in the early 1980s. In subsequent years, the use of other non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit cyclooxygenase (COX) enzymes, was linked to reduced cancer risk in multiple tissues including those of the breast, prostate and lung.6 7 As in many other types of malignant tissue, COX-2 is also over-expressed in pancreatic carcinoma. Maitra et al found an upregulation of COX-2 in a subset of PanINs which makes it a potential target for chemoprevention with selective COX-2 inhibitors.8 However, recent epidemiological studies investigating the preventive value of aspirin in pancreatic carcinoma reached conflicting findings, perhaps due to the substantial differences in patient populations, exposure information, and outcome information.9–12 Wang et al were the first to show that nuclear factor kappa B (NF-κB) activity is found constitutively in about 70% of pancreatic cancers, but not in normal pancreatic tissue.13 The findings by Sclabas et al14 showed that aspirin-mediated inhibition of NF-κB activation in response to inflammation is a possible mechanism for the cancer preventive effect of aspirin. They used an orthotopic mouse model with human pancreatic carcinoma cell lines to study the inhibitory effects of aspirin on pancreatic tumour formation. Animals given aspirin for 6 days before tumour cell injection showed a lower incidence of tumour formation compared with those receiving aspirin 2 weeks after inoculation. They suggested that aspirin-mediated anti-inflammatory approaches could be an effective strategy to prevent pancreatic carcinoma.
Inhibitors of angiotensin-I converting enzyme (ACE inhibitors) inhibit stimulation by angiotensin-II (Ang-II) by decreasing its conversion from Ang-I. ACE inhibitors (ACE-Is) were developed and applied clinically as first-line drugs for hypertension. Interestingly, much evidence has accumulated showing that ACE-Is suppress the growth of a wide variety of cultured cancer cells in vitro and inhibit tumorigenesis and angiogenesis induced in cancer animal models in vivo.15 16 Lever et al17 conducted a retrospective cohort study to assess the risk of cancer in hypertensive patients receiving ACE-Is or other antihypertensive drugs. Interestingly, the RR of cancer was lowest in women on ACE-Is. They concluded that long-term use of ACE-Is may protect against cancer. Ang-II is a multi-functional bioactive peptide and recent reports have suggested that it is a proangiogenic growth factor. It has been shown that Ang-II selectively increases blood flow, and ACE-Is decrease intratumoral blood flow without affecting blood flow in healthy organs. In experimental models, ACE-Is reduced the tumour cell growth rate and modulated gene expression in vitro. In vivo, captopril was shown to inhibit tumour growth and angiogenesis.18
In this study we show for the first time that the widely used drugs aspirin and ACE-Is both delay progression of PanINs and even cancer formation in a genetically engineered mouse model of pancreatic cancer. Our findings suggest that the drugs might be effective chemopreventive agents for pancreatic cancer.
Material and methods
Mice
Conditional LsL-Trp53R172H,19 LsL-KrasG12D and Pdx1-Cre4 strains were interbred to obtain LsL-KrasG12D; Pdx1-Cre double mutant animals or LsL-KrasG12D;LsL-Trp53R172H;Pdx1-Cre triple mutant animals on a mixed 129/SvJae/C57Bl/6 background as previously described.5 All mice were generated from the same initial stock. Animals were maintained in a climate-controlled room kept at 22°C, exposed to a 12:12-h light–dark cycle, fed standard laboratory chow, and given water ad libidum.
Genotyping
For genotyping, genomic DNA was extracted from tail cuttings using the REDExtract-N-Amp Tissue PCR kit (Sigma-Aldrich, St Louis, Missouri, USA). Three PCR reactions were carried out for each animal, to test for the presence of the oncogenic Kras (using LoxP primers), p53 and Pdx1-Cre transgene constructs (using Cre-specific primers along with Gabra as positive control), respectively.
Drug treatment
Drug treatment was initiated at the age of 5 weeks. Overall, 80 LsL-KrasG12D; Pdx1-Cre and 90 LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre transgenic mice were randomly assigned to receive either (1) mock treatment, (2) aspirin, (3) enalapril, or (4) a combination of aspirin and enalapril. The formation of the different study groups is explained in figure 1.

Study design of the LsL-KrasG12D; Pdx1-Cre and LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre transgenic mice.
Aspirin and enalapril were injected once a day by intraperitoneal injection. Aspirin was injected at a dosage of 20 mg/kg/body weight. This dosage is equivalent to a daily human dosage of 80–110 mg per day.20
Enalapril was injected at a dosage of 0.6 mg kg/body weight. This dosage was chosen after a report from Sakamoto et al.21
After 3 months and after 5 months of treatment, all mice that survived until these time points were killed. In cases where littermates were available for drug treatment only the first mouse was randomly assigned to one of the four given treatment groups; the second littermate was then assigned to the ‘matched’ control arm, and so forth. This scheme was chosen in order to obtain the highest possible degree of consistency and to avoid randomisation bias as far as possible.
Histological evaluation
After completion of drug treatment the mice were killed and the pancreas was removed and inspected for grossly visible tumours and either preserved in 10% formalin solution (Sigma-Aldrich) for histology or processed for RNA extraction (see below). Formalin-fixed, paraffin-embedded tissues were sectioned (4 μm) and stained with haematoxylin & eosin. Six sections (100 μm apart) of pancreatic tissues were histologically evaluated by an experienced gastrointestinal pathologist (AM) blinded to the experimental groups. MousePanIN (mPanINs) lesions were classified according to histopathological criteria as recommended elsewhere.22 23
To quantify the progression of mPanIN lesions in double transgenic LsL-KrasG12D; Pdx1-Cre mice in all pancreata from each animal, the total number of ductal lesions and their grades were determined. About 100–130 pancreatic ducts per mouse were detected in the entire fixed pancreas and analysed. The relative proportion of each mPanIN lesion to the overall number of analysed ducts was recorded for each animal.
In triple transgenic LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre mice the mice were classified by having developed pancreatic cancer or not.
Immunostaining
For immunolabelling, formalin-fixed and paraffin-embedded archived tumour samples and corresponding normal tissues were stained as previously described.24 Concentrations and sources of primary antibodies are listed in table 1. Briefly, slides were heated to 60°C for 1 h, deparaffinised using xylene, and hydrated by a graded series of ethanol washes. Antigen retrieval was accomplished by microwave heating in 10 mmol/l sodium citrate buffer, pH 6.0, for 10 min. For immunohistochemistry, endogenous peroxidase activity was quenched by 10 min incubation in 3% H2O2. Non-specific binding was blocked with 10% serum. Sections were then incubated with primary antibodies overnight at 4°C. For immunohistochemistry, bound antibodies were detected using the avidin–biotin-complex (ABC) peroxidase method (ABC Elite Kit; Vector Labs, Burlingame, California, USA). Final staining was developed with the Sigma FAST DAB peroxidase substrate kit (Sigma, Deisenhofen, Germany)
Concentrations and source of primary antibodies
RNA extraction and real-time RT-PCR
A portion of fresh tumour tissue was homogenised and lysed with 600 μl buffer RLT and whole RNA was extracted using the RNeasy kit (Qiagen, Hilden, Germany) with on-column DNA digestion following the standard protocol provided by the manufacturer. The mRNA was reverse transcribed into cDNA with oligo-dT primers using the Superscript 1st Strand System for RT-PCR (Invitrogen, Carlsbad, California, USA) at 42°C for 50 min. All PCRs were carried out on a 7500 Real Time PCR System (Applied Biosystems, Foster City, California, USA). Following an activation step at 95°C for 10 min, determination of mRNA expression was performed over 40 cycles with 15 s of denaturation at 95°C and annealing/extension/data acquisition at 60°C for 60 s using the Power SYBR® Green PCR kit (Applied Biosystems). Primer sequences are available on request. Relative fold mRNA expression levels were determined using the 2(−ΔΔCt) method.25 All reactions were performed in triplicates and results are presented as means and standard errors.
Statistical analysis
Survival curves were computed using the Kaplan–Meier method. The log-rank test was applied to identify significant differences. Differences in the mean of two samples were analysed by an unpaired t test. Comparisons of more than two groups were made by a one-way ANOVA with post hoc Holm–Sidak analysis for pair-wise comparisons and comparisons versus control and by Kruskal–Wallis one-way analysis of variance. Values of p <0.05 were considered statistically significant. Data were analysed using SPSS software (Version 14; SPSS, Inc.).
Results
Development of PanINs in LsL-KrasG12D; Pdx1-Cre and of pancreatic cancer in LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre mice
As previously described in the initial reports,4 5 we observed development of low grade (figure 2A, B) and high grade (figure 2C) mPanINs in LsL-KrasG12D; Pdx1-Cre (figure 2A,B) and fully invasive pancreatic cancers (figure 2D) in LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre transgenic mice. The histologies resembled ductal adenocarcinomas of the pancreas or its precursor lesions observed in humans.

(A) Low-grade mPanIN lesion (mPanIN-1A) in a LsL-KrasG12D; Pdx1-Cre mouse. (B) A second example of a low-grade mPanIN lesion in a LsL-KrasG12D; Pdx1-Cre mouse. Features of low-grade mPanIN lesions illustrated in A and B include the basally located nuclei, retained nuclear polarity, and absence of nuclear pleomorphism or mitoses. In addition, the abundant intracellular mucin is a characteristic feature. (C) High-grade PanIN lesion (mPanIN-3) after 3 months in a LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre mouse. Micropapillary architecture with loss nuclear polarity is discernible. (D) Invasive poorly differentiated ductal adenocarcinoma from 5-month-old LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre mouse. (E) AT1 expression was not detected in normal pancreatic tissue (asterisk). Note strong expression of AT1 in blood vessels (arrow). (F) Only trace levels of AT1 expression were observed in mPanIN-1A lesions, (G), while there was marked expression in higher-grade mPanIN-2. (H) In murine pancreatic cancer, AT1 positive cells were found homogenously distributed throughout the tumour. (I,J) nuclear factor kappa B (NF-κB) expression was not detected in normal pancreatic tissue (asterisk in I, J and K). In contrast, NF-κB expression was already detected in metaplastic lesions (I and J). (K) NF-κB was expressed in all grades of murine PanINs (arrows) and (L) was strongly expressed in pancreatic cancer tissue.
LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre-derived murine pancreatic cancer and its precursor lesions express angiotensin II type 1 receptor (AT1) in vivo
The pancreas was harvested from LsL-KrasG12D; Pdx1-Cre and LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre transgenic mice and stained for AT1 expression using immunohistochemistry. As illustrated in figure 2, AT1 expression was not detected in normal pancreatic tissue except in blood vessels (figure 2E). In neoplastic tissue, AT1 was hardly detected in mPanIN 1A (figure 2F), but expression became stronger in mPanIN 2 (figure 2G). AT1 positive cells were found in murine pancreatic cancer (figure 2H).
LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre-derived murine pancreatic cancer and its precursor lesions express NF-κB in vivo
After proving the expression of AT1, we sought to evaluate the expression of NF-κB in pancreatic cancer and PanINs in a genetically engineered mouse model.
NF-κB expression was not detected in normal pancreatic tissue (asterisk in figure 2I–K). In contrast, NF-κB was expressed in metaplastic tissue (figure 2I,J), mPanINs (figure 2K), and was strongly expressed in pancreatic cancer tissue (figure 2L).
These observations strongly supported the hypothesis that ACE inhibitors or aspirin might be useful chemoprevention agents for pancreatic cancer. Therefore, we determined the effect of these drugs in LsL-KrasG12D; Pdx1-Cre and LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre transgenic mice.
The angiotensin-I-converting enzyme inhibitor enalapril and aspirin delay progression of mPanINs in LsL-KrasG12D; Pdx1-Cre transgenic mice
After 3 and 5 months of treatment, enalapril and aspirin were able to delay progression of mPanINs in LsL-KrasG12D; Pdx1-Cre mice significantly (figure 3A,B). After 3 months, 54.1% of the ducts evaluated in the control mice showed normal pancreatic ducts, 28.6% were classified as mPanIN-1A, 16.1% as mPan-1B and 0.6% as mPanIN-2 and mPanIN-3, respectively. In contrast, mice treated with enalapril alone had 73.9% normal ducts (p=0.046), 14.9% mPanIN-1A (p=0.01), 10.7% mPanIN-1B (p=0.74), and 0.5% mPanIN-2 (p>0.05). We found similar results in LsL-KrasG12D; Pdx1-Cre mice treated with aspirin alone. These mice had 82.4% normal ducts (p=0.005), but only 13.1% mPanIN-1A (p=0.14), 4.5% mPanIN-1B (p=0.267), and no higher grade mPanINs. In mice receiving a combination of both drugs, we detected 76.4% normal ducts (p=0.009), 16.3% mPanIN-1A (p=0.01), 7.3% mPanIN-1B (p=0.46), and no higher grade mPanINs.

The angiotensin-I-converting enzyme inhibitor enalapril and aspirin delay progression of PanINs in LsL-KrasG12D; Pdx1-Cre transgenic mice. Quantitative analysis of mPanINs in experimental groups after (A) 3 months and (B) 5 months of treatment. Percentages of pancreatic ducts with no pathology (normal ducts), mPanIN-1A (1A), mPanIN-1B (1B), mPanIN-2 (2), and mPanIN-3 (3) lesions in control mice (n=10) and treated mice with either aspirin (ASS) (n=10); enalapril alone (ACE) (n=10) or combination of treatment (ASS+ACE) (n=10).
After 5 months of treatment, LsL-KrasG12D; Pdx1-Cre control mice had 58% normal ducts, 28% were classified as mPanIN-1A, 17.6% as mPanIN-1B and 0.4% as mPanIN-2. In contrast, mice treated with enalapril alone had 74.3% normal ducts (p=0.001), 15.2% mPanIN-1A (p=0.001), 10.5% mPanIN-1B (p=0.03), and no higher grade mPanINs. Mice treated with aspirin alone had 79.6% normal ducts (p=0.002), but only 11.6% mPanIN-1A (p=0.001), 8.4% mPanIN-1B (p=0.006), no mPanIN-2 and 0.4% mPanIN-3 (p>0.05). The combination of both drugs led to 65% normal ducts (p=0.02), 16.1% mPanIN-1A (p=0.03), 18.7% mPanIN-1B (p=0.37), and 0.2% mPanIn-2 (p>0.05) compared with the control group.
Formation of invasive murine pancreatic cancer in LsL-KrasG12D; LsL-Trp53R172H; Pdx1-C.re transgenic mice is partially inhibited by enalapril and aspirin
After 3 and 5 months of treatment, respectively, all LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre that survived until these time points were killed and the abdomens were opened and inspected for cancer formation (figure 4A,B). Invasive murine pancreatic cancer was identified in 15 of 25 (60%) LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre untreated control mice (figure 4A). In contrast, only three of 17 (17.6%, p=0.01) transgenic mice treated with aspirin developed murine pancreatic cancer during the study (figure 4B). In mice treated with enalapril alone, four of 17 (23.5%, p=0.03) developed murine pancreatic cancer. In mice that were treated with the combination of aspirin and enalapril, we found invasive murine pancreatic cancer in five of 16 (31.2%, p=0.11) mice (figure 4C). We did not find any distant metastases in liver or other organs of treated or untreated mice.

Formation of invasive pancreatic cancer in LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre transgenic mice is inhibited by enalapril and aspirin. (A) In more than 50% of control LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre mice invasive pancreatic cancer was identified (arrows). (B) In a large subset of aspirin or enalapril treated LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre mice, we found no invasive cancer after 3 or 5 months of treatment (arrows). (C) Pancreatic cancer formation in LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre was significantly reduced in aspirin and enalapril.
The target genes VEGF and NF-κB (RelA) are downregulated in invasive pancreatic cancer by enalapril and aspirin
Given the clinical and histopathological evidence that aspirin and enalapril inhibited the formation of pancreatic cancer, we next explored two typical target genes of aspirin and enalapril in the tumour tissue. It is well known that ACE-Is is able to downregulate vascular endotherlial growth factor (VEGF).26 Aspirin inhibits NF-κB, which can be measured by analysing the expression of RelA. RelA (p65) functions as the critical transactivating component of the heterodimeric p50–p65 NF-κB complex. Quantitative real-time PCR for both of these genes demonstrated significant downregulation in the tumour of LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre mice treated with aspirin or enalapril (figure 5). These experiments confirmed our ability to achieve effective pharmacological levels of aspirin and enalapril during pancreatic cancer formation in vivo.
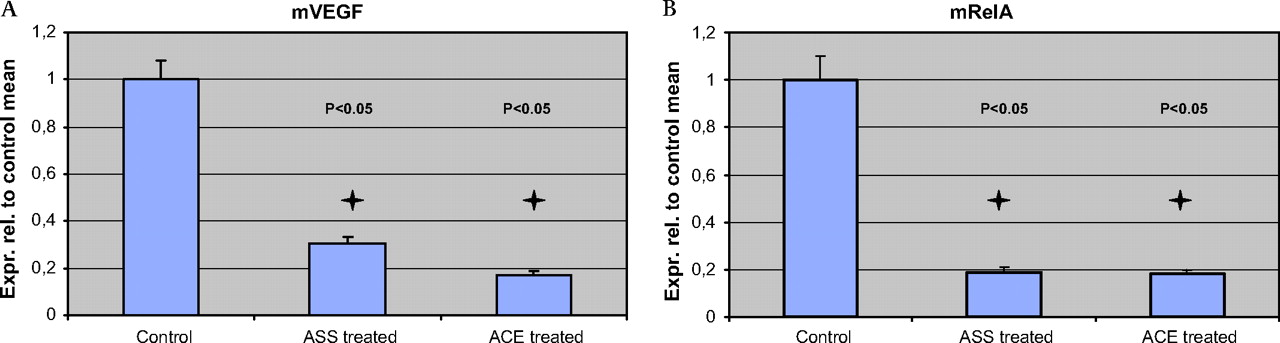
The target genes vascular endothelial growth factor (VEGF) and nuclear factor kappa B (NF-κB) (RelA) are downregulated in invasive pancreatic cancer by enalapril and aspirin. (A,B) Quantitative real-time PCR on RNA obtained from aspirin- and enalapril-treated mice demonstrates profound downregulation of the VEGF and RelA (p65) in the tumour. Error bars indicate SE of the mean for each measurement.
Expression of amylase in PanINs and pancreatic cancer of LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre transgenic mice suggests an exocrine origin of mPanINs
Recently, our group showed that Ela-CreERT2; LSL-KrasG12D mice spontaneously developed mPanIN, demonstrating that acinar/centroacinar cells can be spontaneously induced along a path towards pancreatic neoplasia by a single initiating genetic event.27 Therefore we stained mPanINs for amylase to evaluate an acinar origin. Indeed, we found scattered amylase expressing acinar cells within the metaplastic epithelium (figure 6). In low-grade mPanIN lesions, immunohistochemical studies demonstrated that occasional single cells expressing amylase were also present, confirming phenotypic exocrine differentiation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression of amylase in PanINs and pancreatic cancer of LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre transgenic mice suggests an exocrine origin of PanINs. (A,B) We found scattered amylase-expressing acinar cells within the metaplastic epithelium. (C,D) In mPanIN lesions, amylase positive cells were also present (arrows), confirming phenotypic exocrine differentiation.
Discussion
Since for pancreatic cancer the mortality rate approaches the incidence rate with only 1–4% of all patients surviving 5 years,2 it would be would be of great value to provide chemopreventive treatment for high-risk individuals. This holds especially true for individuals with an inherited predisposition to pancreatic cancer, such as members of families with familial pancreatic cancer, hereditary pancreatitis, familial atypical mole melanoma, Peutz–Jeghers syndrome and other tumour syndromes.28
The conditional LsL-KrasG12D; Pdx1-Cre mice,4 is considered a very valuable tool to study PanIN biology, since it mimics rather slow progression from PanIN 1 over PanIN 2 and 3 lesions to invasive cancer in around 12–15 months. Furthermore, LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre mice manifest widely metastatic pancreatic ductal adenocarcinoma that recapitulates the human spectrum. The use of these models provides now the opportunity to conduct chemopreventive studies on pancreatic cancer.
We show for the first time, that both aspirin and the ACE-I enalapril delay the progression of mPanINs and partially inhibited the formation of invasive murine pancreatic cancer formation in this genetically engineered mouse model. Just recently, Funahashi et al found that the selective COX-2inhibitor nimesulide delays the progression of pancreatic cancer precursor lesions in the KrasG12D mouse.29 In contrast to our study they tested the effect of nimesulide only in the LsL-KrasG12D; Pdx1-Cre mice but not in LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre mice. The delay of mPanIN progression in the LsL-KrasG12D; Pdx1-Cre mice shows the potential of aspirin to be an effective chemopreventive agent. Because the activation of an oncogenic Kras is an early event in PanIN formation our data now show that aspirin delays the progression from normal ducts to mPanIn1A and to mPanIN1B (figure 3). Because only PanIN3 are classified as carcinoma in situ, delaying the progression towards PanIN3 would have a major impact on cancer development. Therefore, our study strengthens the conclusion of Funahasi et al29 that aspirin or a COX-2 inhibitor might be an effective chemopreventive agent for pancreatic cancer probably by inhibition of NF-κB-driven COX-2 expression by blocking the MAPK and Akt/protein kinase B signalling.30 Our in vivo results are in line with several epidemiological studies.10 31 32 Recently, in a prospective study conducted by Anderson et al, the use of aspirin was associated with a reduced risk of pancreatic cancer among 28 232 post-menopausal women.10
Furthermore, we show for the first time that aspirin partially inhibits the formation of invasive murine pancreatic cancer in a significant amount of treated mice (figure 4). Invasive murine pancreatic cancer was identified in 60% untreated LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre control mice, but only 17% of aspirin treated mice developed murine pancreatic cancer during the study (figure 4).
NF-κB orchestrates the expression of genes that encode key determinants in inflammation, tumorigenesis and apoptosis, and thus promotes the cardinal clinical features of pancreatic carcinoma of locally aggressive growth and metastasis.33 Constitutive activation of NF-κB is a frequent molecular alteration in pancreatic carcinoma and is also found in human pancreatic carcinoma cell lines but not in immortalised, non-tumorigenic pancreatic epithelial cells.34 Aspirin inhibits NF-κB activation through specific inhibition of IKK-2 activity by binding to IKK-2 and reducing ATP binding.35 Furthermore, it is well known that also ACE inhibitors reduce the expression of NF-κB.36 As shown in the present study, both aspirin and enalapril were able to downregulate NF-κB in tumour cells by intraperitoneal injection (figure 5). This could be an important step in pancreatic cancer, because NF-κB is a critical downstream mediator of cell transformation mediated by oncogenic ras.37 In this context, the transcriptional activity of RelA is significantly increased in Ras transformed cells as compared to non-transformed cells.38 It is thus reasonable to expect that activation of the Ras pathway in human tumours may yet be another means to induce constitutive NF-κB activity and oncogenesis. In agreement with this hypothesis, RelA has been observed to be constitutively activated in 67% of pancreatic adenocarcinomas and the human K-ras oncogene to be frequently mutated in pancreatic cancer.13
In the current study, we employed an ACE-I as a chemopreventive agent. A retrospective cohort study on 5207 patients receiving ACE-I or other anti-hypertensive drugs with a 10-year follow-up demonstrated that ACE-I treatment may decrease the incidence of adult cancer cancer.17 Furthermore, a study of 483 733 US veterans has found that ACE-Is can significantly reduce risk of pancreatic cancer (OR 0.48, p<0.01).39 In the present study we show for the first time in a genetically engineered mouse model that enalapril is an effective chemopreventive agent for pancreatic cancer. These results are in line with several in vitro15 40–42 and in vivo findings.
Recently, Arafat et al26 analysed the expression and localisation of ACE and AngII type 1 receptor (AT1) in relation to VEGF in invasive human pancreatic cancer. VEGF is a crucial pro-angiogenic component in pancreatic ductal adenocarcinoma, and its high expression levels have been correlated with poor prognosis and early postoperative recurrence. They found an upregulation of ACE and AT1R in 75% of analysed cancers, when compared with matching controls. VEGF expression was significantly higher in tissues that expressed high levels of AT1 and ACE. The same group explored the signalling mechanisms involved in the AngII-mediated VEGF induction and correlated AT1 and VEGF expression in noninvasive precursor lesions. An AT1 antagonist inhibited the AngII-mediated induction of VEGF messenger RNA and protein in all PDA cell lines.43 These findings, together with the striking data we present now in this study, suggest the involvement of AT1 and Ang-II in tumour progression and angiogenesis and therefore valuable chemopreventive targets. Although a protective effect of a combination therapy with NSAIDs and ACE-Is has been reported in colon cancer,44 we did not reach a synergistic effect in the group of mice treated with both drugs.
Recently, we demonstrated that acinar expression of a mutant KrasG12D allele from its endogenous promoter results in the spontaneous induction of mPanIN lesions in adult mice,27 showing that differentiated acinar cells in adult mice are capable of generating the entire histological spectrum of mPanIN lesions including high-grade mPanIN-3, without the requirement for exocrine injury. In the present study we found the presence of scattered amylase-expressing acinar cells within the metaplastic epithelium (figure 6) also in mice with targeting of mutant KrasG12D to Pdx1-expressing cells. This underscores the spontaneous ability of mature acinar cells to transition to a ductal precursor phenotype in the appropriate oncogenic context.
In conclusion, we identified two drugs, aspirin and enalapril, with the potential to delay the formation of pancreatic cancer and its precursor lesions. The drugs we used are prescribed for millions of patients annually. While fundamental differences in biology suggest the need for caution in equating mouse tumours with their human counterparts,45 mouse models nevertheless represent an important source of insight regarding human neoplasia. Further studies are necessary to confirm the hypothesis that aspirin and ACE-Is might be a valid chemopreventive strategy for pancreatic cancer, which could be particularly effective in delaying PanIN progression in high-risk individuals.
References
Footnotes
Linked articles 200733.
Funding VF was supported by a Research Grant of the University Medical Center Giessen and Marburg. GF was supported by a fellowship grant within the postdoctorate programme of the German Academic Exchange Service (DAAD).
Competing interests None.
Ethics approval All experiments using mice were approved by the local committees for animal care and use.
Provenance and peer review Not commissioned; externally peer reviewed.