Article Text

Abstract
Background and aims: Emerging evidence suggests that highly treatment-resistant tumour-initiating cells (TICs) play a central role in the pathogenesis of pancreatic cancer. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) is considered to be a novel anticancer agent; however, recent studies have shown that many pancreatic cancer cells are resistant to apoptosis induction by TRAIL due to TRAIL-activated nuclear factor-κB (NF-κB) signalling. Several chemopreventive agents are able to inhibit NF-κB, and favourable results have been obtained—for example, for the broccoli compound sulforaphane—in preventing metastasis in clinical studies. The aim of the study was to identify TICs in pancreatic carcinoma for analysis of resistance mechanisms and for definition of sensitising agents.
Methods: TICs were defined by expression patterns of a CD44+/CD24−, CD44+/CD24+ or CD44+/CD133+ phenotype and correlation to growth in immunodeficient mice, differentiation grade, clonogenic growth, sphere formation, aldehyde dehydrogenase (ALDH) activity and therapy resistance.
Results: Mechanistically, specific binding of transcriptionally active cRel-containing NF-κB complexes in TICs was observed. Sulforaphane prevented NF-κB binding, downregulated apoptosis inhibitors and induced apoptosis, together with prevention of clonogenicity. Gemcitabine, the chemopreventive agents resveratrol and wogonin, and the death ligand TRAIL were less effective. In a xenograft model, sulforaphane strongly blocked tumour growth and angiogenesis, while combination with TRAIL had an additive effect without obvious cytotoxicity in normal cells. Freshly isolated patient tumour cells expressing markers for TICs could be sensitised by sulforaphane for TRAIL-induced cytotoxity.
Conclusion: The data provide new insights into resistance mechanisms of TICs and suggest the combination of sulforaphane with TRAIL as a promising strategy for targeting of pancreatic TICs.
Statistics from Altmetric.com
Pancreatic adenocarcinoma is an aggressive malignancy usually diagnosed when in an advanced state, for which there are few or no effective treatments. It has the worst prognosis of any major malignancy, and the vast majority of patients die within the first year after diagnosis, and <1% of patients are alive after 5 years.1 One of the major hallmarks of pancreatic cancer is its extensive local tumour invasion and early systemic dissemination. The molecular explanations for these characteristics of pancreatic cancer are incompletely understood. Emerging evidence has shown that the capacity of a tumour to grow and propagate is dependent on a small subset of distinct tumour-initiating cells (TICs).2 3 TICs have recently been identified in several tumour entities including pancreatic cancer.4–6 The current consensus definition describes a TIC as a cell within a tumour that is able to self-renew and to produce the heterogeneous lineages of cancer cells that comprise the tumour bulk.7 The implementation of this concept explains the use of alternative terms, such as “cancer stem cell” and “tumourigenic cell” to describe tumour-initiating cells.2 3 8–10 Common anticancer treatments such as radiation and chemotherapy do not eradicate the majority of highly resistant TICs,11 suggesting the need for new therapeutic options.
The human tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is considered to be a novel anticancer agent; however, recent studies have shown that many pancreatic cancer cells are resistant to the apoptosis-inducing effects of TRAIL due to constitutive and TRAIL-activated nuclear factor-κB (NF-κB) signalling.12 Homodimeric or heterodimeric NF-κB complexes consist of proteins present in the cytoplasm, namely p50, p52, RelA/p65, RelB and cRel. Dimers are kept in an inactive state by a family of IκB (inhibitor of NF-κB) proteins.13 A pivotal step in the activation of NF-κB is the phosphorylation of IκB molecules, which in turn leads to their proteasome-dependent degradation and allows the translocation of NF-κB dimers to the nucleus where they can bind to specific DNA response elements.14 Several chemopreventive agents are able to inhibit NF-κB and therefore are proapoptotic. Cruciferous vegetables, such as broccoli, have a high content of glucosinolate-derived compounds, the precursors of anticarcinogenic isothiocyanate sulforaphane (SF). SF protects from DNA damage, induces apoptosis and inhibits NF-κB, as well as blocking cell proliferation.15 Other anticarcinogenic plant substances with similar properties are the polyphenol resveratrol (RE), present in high levels in grapes and derived products such as red wine,16 and the flavonoid wogonin (WO), a popular herbal remedy in China and several other Asian countries.17
Why some pancreatic cancer cell lines have elevated NF-κB signalling, while others have not, is unknown. Therefore, it is tempting to speculate that this might be due to the presence of TIC populations within pancreatic tumours whose pronounced resistance might be due to activated NF-κB signalling. In the present study, we identified TIC-like populations in established pancreatic cancer cell lines, and in paraffin sections of patient tumours. The presence of TICs correlated with resistance towards gemcitabine or TRAIL due to binding of transactivation potent NF-κB dimers in TIChigh but not in TIClow pancreatic cancer cells. The chemopreventive agent SF was able to abrogate resistance by interfering with NF-κB binding. We propose that SF is a promising agent for TIC-targeted therapies in pancreatic carcinoma.
MATERIALS AND METHODS
Established cell lines
AsPC-1, BxPc-3, Capan-1 and MIA-PaCa2 pancreatic cell lines were obtained from the American Type Culture Collection (Manassas, Virginia, USA) and were cultured as described.18 The murine plasmocytoma cell line S107 has been described and was grown in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Karlsruhe, Germany) containing 10% heat-inactivated fetal calf serum (PAN Systems, Aidenbach, Germany).
Primary cell lines
Primary fibroblasts from skin were kindly provided by Dr H-J Stark (DKFZ, Heidelberg, Germany). Mesenchymal stem cells and fresh tumour cells from resected pancreatic tumours were isolated as described.18 19 Patient material was obtained with the approval of the ethical committee of the University of Heidelberg. Diagnoses were established by conventional clinical and histological criteria according to the World Health Organization (WHO). All surgical resections were indicated by the principles and practice of oncological therapy.
Nude mice and xenografts
MIA-PaCa2 cells (8×106 in 150 μl) were injected subcutaneously into the right anterior flank of 4- to 6-week-old NMRI (nu/nu) male mice (day 0). After the tumours had reached a mean diameter of about 8–10 mm, mice carrying MIA-PaCa2 xenografts were randomly divided into groups of six animals each and treatment was initiated. The mice were treated intraperitoneally with SF at a dose of 4.4 mg/kg and/or recombinant Super Killer TRAIL (25 ng/tumour) intratumourally on day 4, 5 and 6 after tumour transplantation. In detail, we diluted 1 μl of TRAIL stock solution (500 ng/μl) in 1 ml of phosphate-buffered saline (PBS) (500 ng TRAIL/ml) and injected 50 μl of this solution per tumour. Tumour growth was monitored by measuring two diameters daily with calipers, and tumour volumes (V) were calculated using the formula V = ½ (length×width2). Mice were euthanised at tumour sizes >1500 mm3. For evaluation of tumourigenicity, cells were trypsinised, counted, diluted to appropriate injection doses and mixed with Matrigel Matrix (BD Biosciences, Bedford, Massachusetts, USA) at a 1:1 ratio. TIChigh MIA-PaCa2 and TIClow BxPc-3 cells were injected subcutaneously (1×103 cells/flank, total volume 200 μl) at opposite flanks of the same animals. Animal experiments were carried out in the animal facilities of the DKFZ after approval by the authorities (Regierungspräsidium Karlsruhe).
Analysis of plasma liver enzymes
Peripheral blood was taken before liver perfusion under deep anaesthesia. One drop of heparin was added and, after centrifugation, supernatants were collected. The levels of plasma lactate dehydrogenase (LDH), glutamate-oxalacetate-transaminase (GOT/AST) and glutamate-pyruvate-transaminase (GPT/ALT) were measured using a DRY-CHEM FDC3500 (Fuji Medical, Tokyo, Japan) according to the manufacturer’s guidelines.
Cytotoxic agents
Gemcitabine (a kind gift from Eli Lilly, Indianapolis, Indiana, USA) was diluted in PBS to a 100 μM stock. Recombinant human TRAIL/Apo2 ligand produced in Escherichia coli was from Axxora (Lörrach, Germany). Stock solutions of SF and RE (both from Sigma, Deisenhofen, Germany) were prepared in ethanol and a stock solution of WO (Phytolab, Vestenbergsgreuth, Germany), phorbol 12-myristate 13-acetate (PMA; Calbiochem, San Diego, California, USA) and Gö6983 (Sigma-Aldrich Chemie, Steinheim, Germany) in dimethylsulfoxde (DMSO). Final concentrations of the solvents in the media were ⩽0.1%.
Measurement of apoptosis
Cells were stained with fluorescein isothiocyanate (FITC)-conjugated annexin V (BD Biosciences, Heidelberg, Germany) and externalisation of phosphatidylserine as well as the forward side scatter profile were identified by flow cytometry (FACScan, BD Biosciences). DNA fragmentation was detected by the Nicoletti method.18
MTT assay
Tumour cells were resuspended at a density of 3×104 to 105/ml in 96-well microplates, 100 μl per well. After treatment, the MTT (3-(4,5-dimethylthiazo-2-yl)-2,5-diphenyl tetrasodium bromide) assay was performed as described.18
Spheroid assay
Cells were cultured in NeuroCult NS-A basal serum-free medium (human) (StemCell Technologies, Vancouver, BC, Canada) supplemented with 2 μg/ml heparin (StemCell Technologies), 20 ng/ml human epidemal growth factor (hEGF; R&D Systems, Wiesbaden-Nordenstadt, Germany), 10 ng/ml human basic fibroblast growth factor (hFGF-b; PeproTech GmbH, Hamburg, Germany) and NeuroCult NS-A proliferation supplements (human) (StemCell Technologies.). Cells were seeded at low densities (1×104 cells/ml) in 12-well low adhesion plates, 1 ml per well. Upon formation of spheroids, cells were reseeded at 1×104/ml in order to evaluate the potential for formation of secondary spheroids.
Colony-forming assays
Cells were seeded at a density of 3×104 in 12-well tissue culture plates (BD Falcon, San José, California, USA). After 24 h the cultures were treated. Forty-eight hours after treatment, the cultures were trypsinised, plated at a density of either 400, 500, 600 or 800 cells per well in 6-well tissue culture plates in parallel and incubated for 10 days without changing the medium. For determination of colony formation, cultures were fixed (3.7% paraformaldehyde and 70% ethanol) and stained with 0.05% Coomassie blue. The number of colonies with >50 cells was counted under a dissecting microscope. The percentage cell survival was calculated (plating efficiency of non-treated cultures = 1, or relative survival rate).
Detection of aldehyde dehydrogenase (ALDH) activity
A total of 1×106 cells were treated with 5 μl/ml ALDEFLUOR substrate (Aldagen, Durham, North Carolina, USA), incubated for 30 min at 37°C and analysed by flow cytometry according to the manufacturer’s instructions.
Detection of caspase activity
A kit providing a fluorochrome inhibitor of caspases (FAM-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethylketone (FLICA)) was used according to the manufacturer’s protocol (Immunochemistry Technologies, Bloomington, Minnesota, USA). FLICA binds covalently to specific active caspases (thus the fluorochrome accumulates in cells which have active caspases). For red fluorescence, a sulforhodamine-labelled fluoromethyl ketone peptide inhibitor of caspases, and for green fluorescence carboxyfluorescein-labelled inhibitors were used. In short, FLICA specific for caspases 3, 7, 8 or 9 was added to cell media and incubated for 1 h at 37°C in 5% CO2. Cells were then washed in washing buffer and analysed by immunofluorescence microscopy.
Whole-cell extract preparation for electrophoretic mobility shift assay
Cells were washed twice in PBS and resuspended in three packed cell volumes of buffer C as described.20 The samples were subjected to three cycles of freezing in liquid nitrogen and subsequent thawing on ice. After centrifugation, the supernatant was used as whole-cell extract.
Electrophoretic mobility shift assay (EMSA)
Protein extracts (5 μg) were incubated for 30 min at room temperature with 3 μg of poly(dI/dC), 10 μg of bovine serum albumin in buffer containing 50 mM NaCl, 1 mM dithiothreitol, 10 mM Tris–HCl, 1 mM EDTA, 5% glycerol and radiolabelled double-stranded oligonucleotides containing an immunoglobulin (Ig)-κ enhancer consensus NF-κB site.21 The DNA–protein complexes formed were then separated from free oligonucleotides on a native 4% polyacrylamide gel. For supershift experiments, 2.5 μg of protein extract were preincubated for 30 min with specific antibody before being treated as described before. Antibodies used for supershift experiments were from Santa Cruz Biotechnology (Santa Cruz, California, USA).
Detection of stem cell markers by flow cytometry
The expression of surface markers was analysed with two-colour flow cytometry using a FACScan flow cytometer (Becton-Dickinson, Heidelberg, Germany). A total of 1×106 cells were incubated with Venimmun (Aventis-Behring, Marburg, Germany) at 4°C for 20 min to inhibit unspecific binding of antibodies. After washing with PBS/5% fetal calf serum (FCS), cells were incubated with unconjugated or with FITC- or phycoerythrin (PE)-conjugated primary antibody. After washing, cells were incubated with FITC- or PE-labelled secondary antibodies at 4°C for 30 min to detect unconjugated primary antibody. Specific antibodies were anti-CD44 monoclonal antibody (mAb) at a dilution of 1:50 (Clone G44-26, Pharmingen, Heidelberg, Germany), antiepithelial-specific antigen (ESA) mAb at a dilution of 1:50 (Biozol, Eching, Germany), anti-CD24 undiluted (ML5 or SWA11 from non-purified hybridoma supernatant)22 and anti-CD133 mAb diluted 1:10 (Miltenyi Biotec, Bergisch Gladbach, Germany). PE-conjugated goat antimouse IgG (BD Pharmingen, Heidelberg, Germany) or FITC-conjugated goat antimouse IgG (Jackson ImmunoResearch, Suffolk, UK) were used as secondary antibodies. The data were analysed using CELLQuest software (Becton-Dickinson, Heidelberg, Germany). PE- or FITC-labelled mouse IgG (BD Pharmingen) served as isotype controls. Gating was implemented based on negative control staining profiles.
Transfection of small interfering (siRNA)
siRNA oligonucleotides were obtained from Santa Cruz Biotechnology (Heidelberg, Germany). One day before transfection, 1×105 cells per well were seeded in 12-well tissue culture plates. One hour before transfection, medium was replaced by antibiotic-free DMEM and transfection of siRNA was performed with Lipofectamine 2000 according to the protocol provided by the manufacturer (Invitrogen, Karlsruhe, Germany).
Protein isolation and western blot analysis
Whole-cell extracts were prepared by a standard protocol and proteins were detected by western blot analysis using polyclonal rabbit antibodies anti-IκBα, anti-cRel (SC-371 and SC-70, Santa Cruz Biotechnology, Heidelberg, Germany), anti-cIAP1, anti-FLIP or mAb anti-XIAP (R&D Systems, Wiesbaden-Nordenstadt, Germany). Goat antirabbit or antimouse (Santa Cruz) secondary antibody conjugated to horseradish peroxidase (HRP) and an enhanced chemiluminescence detection system (Super Signal West Femto, Pierce, Rockford, Illinois, USA) were used for detection.
Immunohistochemistry and immunocytochemistry
Immunohistochemistry on 5 μm frozen or paraffin-embedded tissue sections was performed using the ABC Elite kit from Linaris (Wertheim-Bettingen, Germany), the DAB (3,3′ diaminobenzidine) staining kit from Invitrogen (Karlsruhe, Germany) or ZytoChem-Plus HRP polymer kit with AEC (3-amino-9-ethylcarbazole) as a chromogen (Zytomed Systems, Berlin, Germany) according to the manufacturers’ instructions. IκBα, RelA and cRel were detected using rabbit polyclonal antibody from Santa Cruz Biotechnology (Heidelberg, Germany) and goat antirabbit biotinylated IgG (Vector, Burlingame, California, USA) as a secondary antibody. The signal was amplified with the ABC kit, and DAB was used as a chromogen. Samples were counterstained with haematoxylin, dehydrated in graded alcohol, rinsed in xylene and mounted in Entellan (Merck, Darmstadt, Germany). CD133 in tumour tissues was detected by rabbit polyclonal anti-CD133 antibody from Biozol (Eching, Germany) and the colour was developed using the ZytoChem-Plus HRP polymer (mouse/rabbit) kit with AEC as a chromogen. Samples were directly mounted in water-soluble mounting medium. To detect CD133+/CD44+ cells, CD44 was detected by a mouse mAb (clone G44-26, Pharmingen, Heidelberg, Germany). Tissue sections were blocked with 10% normal goat serum and incubated with primary mouse anti-CD44 mAb followed by washing in PBS and incubation with secondary goat antimouse Alexa 488 IgG (Molecular Probes, Karlsruhe, Germany). After repeated washing, sections were incubated with rabbit anti-CD133 antibody followed by goat antirabbit Alexa 594 IgG (Molecular Probes), washed and mounted in Fluoromount G. CD24 was detected by immunofluorescence with mouse mAb from non-purified and undiluted hybridoma supernatant22 and goat anti-mouse Alexa 594 IgG (Molecular Probes). Omission of primary antibody served as a negative control. Randomly chosen fields were examined at ×400 magnification using a Leica DMRB microscope. Images were captured using a Kappa CF 20/4 DX digital colour camera (Kappa, Gleichen, Germany) and analysed with Kappa ImageBase 2.2 software. Similar staining protocols were used to detect RelA and cRel in pancreatic cancer cell lines and tissue samples of xenografts. Microvessel density in acetone-fixed frozen xenograft sections (5 μm) was deteced by staining with rat anti-mouse CD31 mAb (PharMingen, San Diego, Califonia, USA) as described previously.30 Randomly chosen areas of tumours were examined under ×400 magnification and counted. Any distinct area of positive staining for CD31 was counted as a single vessel. Results were expressed as the mean number of vessels (SE) per high-power field.
Statistical evaluations
For MTT and fluorescence-activated cell sorting (FACS) measurements, statistical evaluations are presented as the mean (SD). Data were analysed using the Student t test for statistical significance. p Values were considered significant if they were <0.05. For xenografts on nude mice, a distribution-free test for tumour growth curve analyses for treatment experiments with xenografted cancer cells was used as described by Koziol et al.23
RESULTS
TIC marker expression correlates with resistance, differentiation, sphere and colony formation, ALDH activity and growth on nude mice
TIC-like characteristics in four human pancreatic carcinoma cell lines were defined by expression of characteristic surface markers, the potential to grow in immunodeficient mice, apoptosis resistance, enrichment of a CD44+/CD24− phenotype by chemotherapy, degree of differentiation, colony- and spheroid-forming capacity, invasion pattern and differentiation potential (fig 1, table 1 and Supplementary fig 1).

A CD44+/CD24− phenotype represents a pancreatic tumour-initiating cell (TIC)-like population. (A) The expression of proposed surface markers for TICs was examined by fluorescence-activated cell sorting (FACS) analysis. AsPC-1, Capan-1, MIA-PaCa2 and BxPc-3 cells were double stained with fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)-conjugated anti-CD44 monoclonal antibody (mAb), PE-conjugated anti-CD133, FITC-conjugated antiepithelial-specific antigen (ESA) or with unconjugated anti-CD24 antibodies (Abs) as indicated. PE- or FITC-conjugated goat anti-mouse immunoglobulin G (IgG) was used as secondary Ab. The results shown in the upper panel are representative of three independent experiments. The lower panel shows means of three experiments including the SD. (B) Larger pictures: cells were seeded at a density of 1600 cells per well in a 6-well plate. Ten days later, colony formation was analysed by fixing the cells in paraformaldehyde and staining with Coomassie blue. Smaller pictures: spheroid formation was induced as described in the Materials and methods section and visualised by microscopy under ×200 magnification. (C) In the upper panel, cells were left untreated (CO) or were treated with gemcitabine at the indicated concentrations. After 72 h DNA fragmentation was measured by Nicoletti staining and FACS analysis. In the middle panel, cells were left untreated (CO) or were treated with recombinant tumour necrosis factor-related apoptosis-inducing ligand (TR: 5, 25, 50 ng/ml) and 24 h later mitochondrial activity/viability was measured by MTT assay. In the lower panel, human primary skin fibroblasts (Fibroblasts) or mesenchymal stem cells (MSC) were treated and analysed as described above for the middle panel. The experiments were performed three times with identical outcome and the means (SD) are shown. (D) MIA-PaCa2 and BxPc-3 cells were treated with ALDEFLUOR substrate and analysed by flow cytometry for detection of aldehyde dehydrogenase (ALDH) activity. (E) Parental BxPc-3 cells were repeatedly treated with 25 nM gemcitabine followed by treatment with 50 nM gemcitabine (BxPc-3 GEM). Four days later, expression of CD24 and CD44 was examined by FACS analysis of surviving cells. SSC, sideward scatter.
In this way we defined the CD44+/CD24− population as the TIC-like phenotype in pancreatic cancer cell lines. In detail, double staining with specific CD44 and CD24 antibodies revealed that AsPC-1, MIA-PaCa2 and Capan-1 cells contained >90% CD44+/CD24− cells, in contrast to BxPc-3 cells, which contained only 17.5% (fig 1A). All examined cells also expressed ESA, some of which were double positive for CD44 and some not, while expression of CD133 was undetectable. While an ESA single- or an ESA+/CD44+ double-positive phenotype did not correlate with any TIC property, the CD44+/CD24− phenotype correlated with colony- and spheroid-forming capacity (fig 1B), resistance towards gemcitabine and TRAIL (fig 1C), ALDH activity (fig 1D) and a poor to moderate differentiation grade (table 1) as well as with the documented ability to grow as xenografts in nude mice.24–26 It has been reported that BxPc-3 cells grow very slowly in nude mice,27 consistent with their lowest content of putative TICs. We confirmed this finding by transplanting 103 MIA-PaCa2 cells into the right flank and 103 BxPc-3 cells into the left flank of nude mice. While MIA-PaCa2 xenografts became visible 17 days after injection, BxPc-3 xenografts needed 28 days to form tumours (Supplementary fig 2). An additional hint of TIC features such as multipotency is the published finding that xenografted Capan-1 or AsPC-1 cells differentiate into phenotypically diverse populations which show morphological, biological and biochemical characteristics similar to those of the tumour of origin.28 29 The differentiation potential of TICs present in the cultured cell line is confirmed by our spheroid assays in which we observed that some cells, the putative TICs, acquired the ability to grow in primary and secondary spheres, while other cells still grow adherent. These features correlate with differentiation of TICs into functionally different cells. Since Capan-1, MIA-PaCa2 and AsPC-1 cells harbour >90% of putative TICs, we did not enrich them further but examined treatment resistance in these TIChigh cells compared with BxPc-3 TIClow cells. According to the recent suggestion that chemotherapy treatment of tumours may lead to enrichment of TICs,6 repeated treatment of BxPc-3 cells with 25 nM gemcitabine followed by treatment with 50 nM gemcitabine for 4 days enhanced the percentage of CD44+/CD24− cells from an initial 17% to 42% (fig 1E). Finally, we would like to point out that TRAIL resistance in TIChigh pancreatic cancer cell lines is not due to the absence of TRAIL receptor expression, since all cell lines have already been shown to express the TRAIL death receptor protein.30 31 In control experiments we confirmed that TRAIL exhibited no pronounced cytotoxicity to normal cells such as primary skin fibroblasts and to normal stem cells such as mesenchymal stem cells isolated from human bone marrow (fig 1C).

Sulforaphane (SF) sensitises tumour-initiating cells (TICs) with no pronounced toxicity to normal cells. MIA-PaCa2 and BxPc-3 cells were pretreated with resveratrol (RE), wogonin (WO) or SF in the concentrations indicated. After 24 h, tumour necrosis factor-related apoptosis-inducing ligand (TR; 5 ng/ml) was added (black bars) or not (white bars) and (A) cells were stained with Coomassie and photographed, or (B) mitochondrial activity/viability was analysed 24 h later by MTT assay. (C) AsPC-1 and MIA-PaCa2 cells were seeded at a density of 3× 104 cells per well in a 12-well plate and were treated with SF (10 μM), RE (50 μM) or WO (50 μM) in the presence or absence of TR (5 ng/ml) as described above. Ten days after treatment, clonogenic survival was examined by fixing the cells in paraformaldehyde and staining with Coomassie blue. The number of colonies with >50 cells was counted under a dissecting microscope. The numbers of the control (CO) groups were set to 1 and the survival fraction is shown. The experiments were performed three times with identical outcome and the means (SD) are shown. An asterisk marks significant difference from the corresponding control, p<0.05 (t test).
SF abrogates resistance of TIChigh cells without cytotoxicity to normal cells
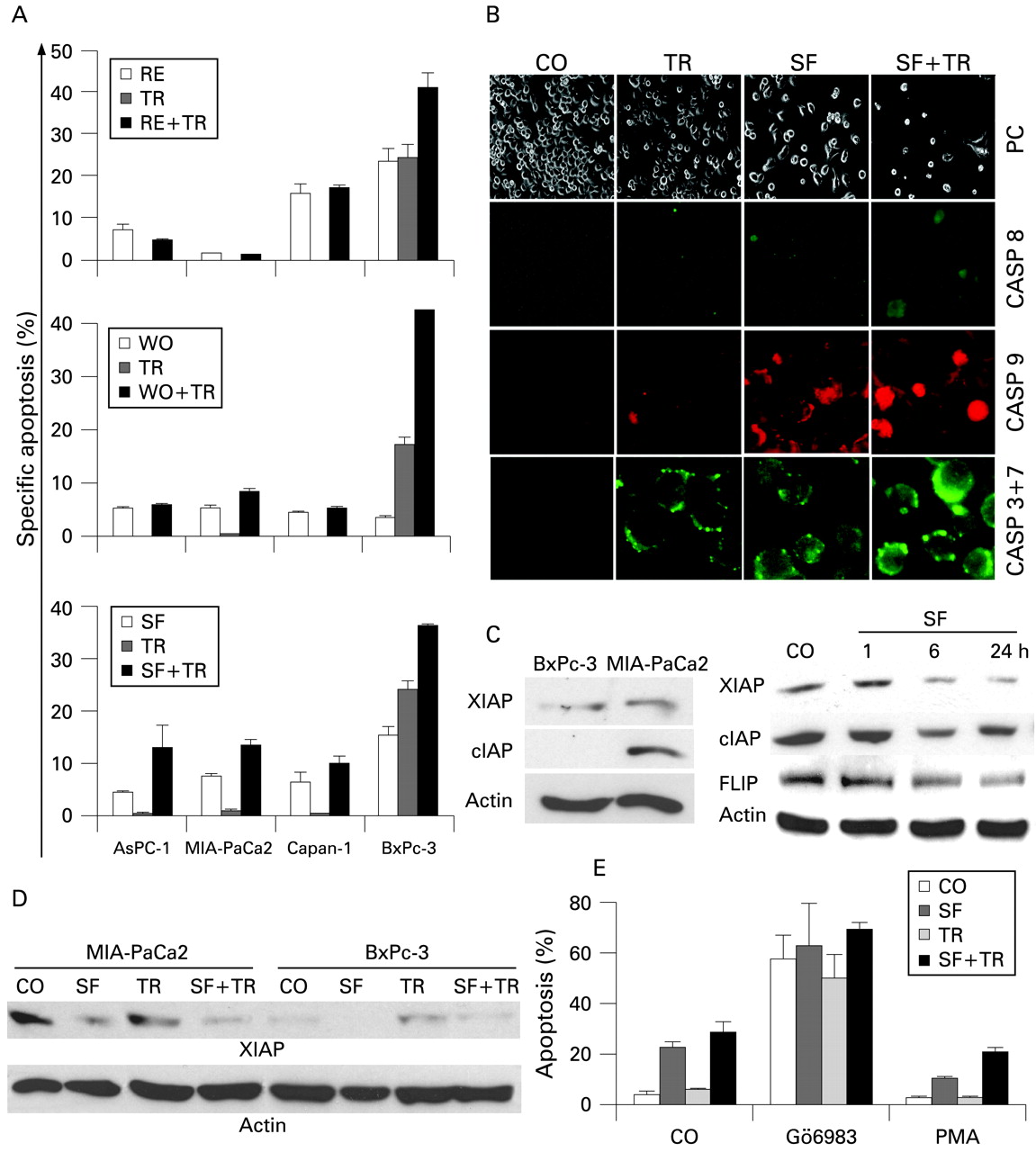
To test whether chemopreventive agents might overcome the resistance of TICs, MIA-PaCa2, AsPC-1, Capan-1 and BxPc-3 cells were treated with RE, WO or SF. After 48 h, viability was examined by MTT assay. While RE or WO alone had no pronounced effect on viability of TIChigh MIA-PaCa2 cells, SF strongly reduced viability already at low concentrations in MIA-PaCa2 and Capan-1 cells and in medium to high concentrations also in AsPC-1 cells (fig 2A,B, Supplementary Fig. 3). Most importantly, all chemopreventive agents used exhibited no pronounced toxicity in low and medium concentrations to normal cells as tested in primary skin fibroblasts and mesenchymal stem cells (Supplementary Fig. 4). When used in combination with recombinant TRAIL, an additive effect was observed in MIA-PaCa2, BxPc-3 and mesenchymal stem cells, but not in AsPC-1, Capan-1 cells and skin fibroblasts. Ten days after treatment, SF and, to a lesser extent, also RE and WO, strongly inhibited clonogenic survival, which was further reduced in combination with TRAIL (fig 2C). The results of the MTT and clonogenic assays are corroborated by apoptosis measurement using annexin staining combined with FACS analysis (fig 3A), detection of caspase activity (fig 3B) and downregulation of the apoptosis inhibitors XIAP, cIAP1 and FLIP (fig 3C,D). Since SF was most potent in inducing apoptosis even in resistant TIChigh cell lines, we examined whether this chemopreventive agent might influence the activity of protein kinase C (PKC), known to have antiapoptotic activity. Therefore, MIA-PaCa2 cells treated with SF, TRAIL or both agents together were incubated with a PKC inhibitor (Gö6983) or a PKC activator (PMA). While inhibition of PKC strongly increased apoptosis induction by SF, TRAIL and both agents together, activation of PKC strongly diminished it (fig 3E). This result suggests that regulation of the PKC pathway is involved in TRAIL- and SF-induced apoptosis.

Sulforaphane (SF) sensitises tumour-initiating cells (TICs) by inducing apoptosis and caspase activity. Pancreatic cancer cell lines were left untreated (CO) or were treated with (RE), wogonin (WO) or SF alone or in combination with tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) as described in fig 2C. (A) Apoptosis was measured by annexin staining and flow cytometry. The percentage of specific apoptosis was calculated as follows: 100×(experimental apoptosis (%)–spontaneous apoptosis in the control (%))/(100%–spontaneous apoptosis in the control (%)). The experiments were performed three times with identical outcome and the SD is shown. (B) MIA-PaCa2 cells were left untreated (CO), or were treated with SF (10 μM). After 24 h, TRAIL (TR, 5 ng/ml) was added to untreated (TR) or pre-treated cells (SF+TR) and the activity of caspase 8, 9 and 3+7 was analysed by fluorochrome-linked inhibitors of caspases (FLICA). Fluorescence was detected by fluorescence microscopy using an Olympus IX70 microscope at a magnification of ×400. The shape of the cells under phase contrast microscopy at a magnification of ×200 is shown. (C) XIAP, cIAP1 and FLIP expression was detected by western blot analysis in untreated MIA-PaCa2 and BxPc-3 cells or in MIA-PaCa2 cells treated with SF for the times indicated. (D) XIAP protein expression in MIA-PaCa2 and BxPc-3 cells upon treatment with SF (10 μM) for 48 h, TR (5 ng/ml) for 24 h or a combination of both agents together (SF+TR). (E) MIA-PaCa2 cell were pretreated with the protein kinase C (PKC) inhibitor Gö6983 (20 μM) for 45 min or with the PKC activator phorbol 12-myristate 13-acetate (PMA; 200 nM) for 15 min followed by treatment with SF, TR or SF plus TR as described in fig 2. Apoptosis was measured by annexin staining and flow cytometry. CASP, caspase.

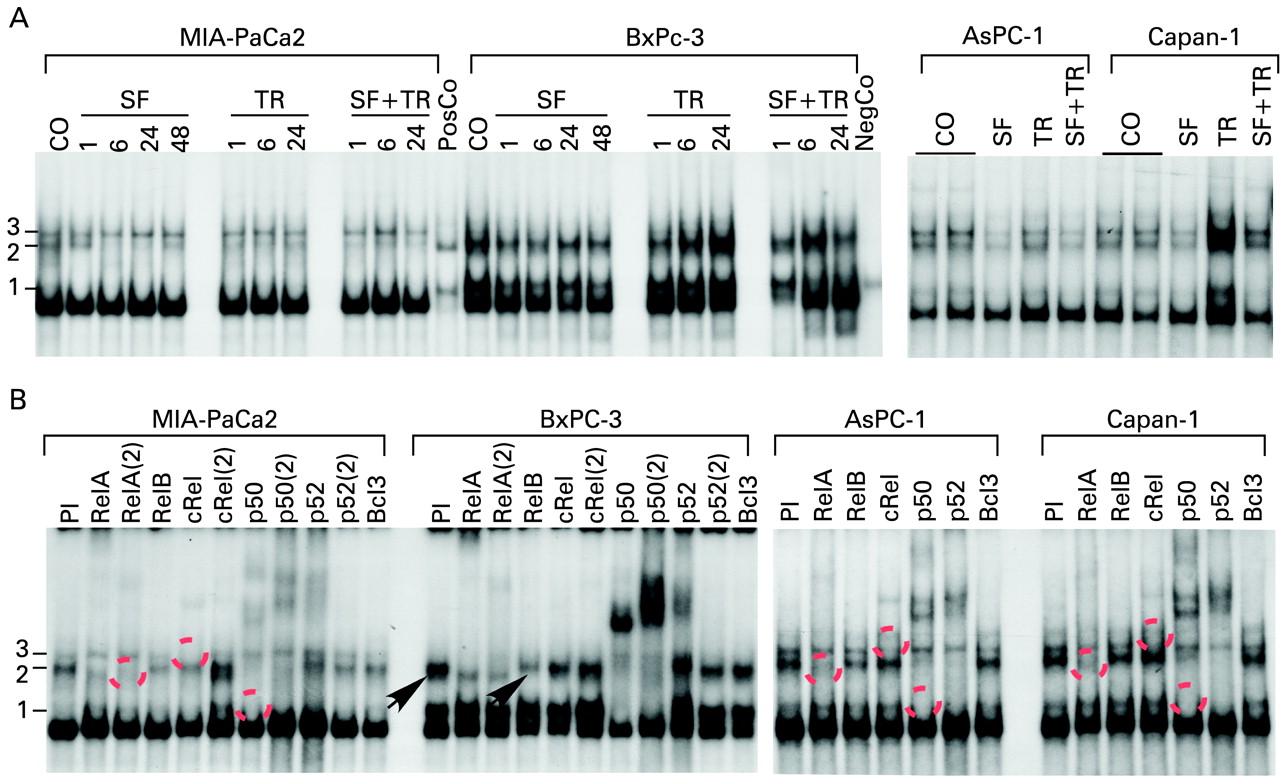
Sulforaphane (SF) inhibits binding of transactivation-competent nuclear factor-κB (NF-κB) dimers in tumour-initiating cell (TIC)high cells. (A) Pancreatic cancer cell lines were left untreated (CO) or were treated with SF (10 μM) for 24 h or for the times indicated. Twenty-four hours later tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) (TR, 5 ng/ml) was added to non SF-pretreated cells (TR) or to cells pretreated with SF (SF+TR) and incubated for an additional 24 h or for the times indicated. Untreated murine plasmocytoma S107 cells served as negative control (NegCo). S107 cells treated with recombinant tumour necrosis factor served as positive control (PosCo). After incubation, nuclear proteins were prepared and DNA binding was analysed by electrophorstic mobility shift assay (EMSA) using a specific 32P-labelled oligonucleotide probe for NF-κB. Three different bands became visible corresponding to three different NF-κB subunits binding to the oligonucleotide (1, 2 and 3). (B) Nuclear proteins derived from untreated control cells were preincubated for 30 min with specific antibodies followed by EMSA analysis. In some cases, two different antibodies for the same proteins were used: RelA/RelA(2), cRel/cRel(2), p50/p50(2), p52/p52(2). Broken circles mark the disappearance of shifted complexes due to specific interaction with co-incubated antibodies. The arrows mark a shift of RelB exclusively observed in BxPc-3 cells. PI, preimmune serum.
SF reduces binding of transactivation-competent NF-κB dimers in TIChigh cells
Since PKC may lead to NF-κB activity, we analysed the status of this transcription factor. EMSA experiments were performed with extracts of MIA-PaCa2, AsPC-1, Capan-1 and BxPc-3 cells, pretreated with SF and subsequently co-stimulated with TRAIL or incubated with TRAIL alone. EMSA with an NF-κB-specific probe revealed distinct basal DNA-binding activity in TIChigh and TIClow cell lines (fig 4A). Compared with the negative and positive control (unstimulated or TNF-induced S107 cells), binding was elevated, indicating an activated NF-κB system in all pancreatic tumour cell lines used. In TIChigh cells, binding of two complexes (3 and 2) was observed, of which in particular the faster migrating band 2 was diminished in MIA-PaCa2 cells upon SF treatment. In TIClow BxPc-3 cells, we found a prominent complex 2 while complex 3 was missing. TRAIL stimulation did not enhance NF-κB DNA-binding activity in MIA-PaCa2 and AsPC-1 cells but was able to induce NF-κB-binding in BxPc-3 (TIClow) and Capan-1 (TIChigh) cells. In all cells, SF pretreatment resulted in inhibition of NF-κB when cells were co-treated with TRAIL. Interestingly, we could detect a pronounced difference between TIChigh and TIClow cell lines. Only BxPc-3 cells showed a prominent faster migrating band 1 which was not affected by SF.
To characterice further the composition of complexes 1–3 we performed supershift assays with specific antibodies recognising the various NF-κB subunits (fig 4B, table 2).
Of the three complexes, the fastest migrating band was completely shifted by p50-specific antibodies in BxPc-3 (TIClow) cells, indicating that these cells have a high amount of p50/p50 homodimers. In all cell lines, complex 2 was composed of p50/RelA and p52/RelA heterodimers. Interestingly, complex 3 was only observed in TIChigh cells and was almost completely reduced by a cRel antibody, suggesting that this binding activity is composed of cRel/cRel homodimers. RelB-containing dimers could be detected only in BxPc-3 cells. Immunostaining with cRel- or RelA-specific antibody revealed nuclear translocation of these NF-κB subunits already in untreated MIA-PaCa2 cells, and neither TRAIL nor SF had an obvious effect on nuclear translocation as analysed by immunostaining and immunofluorescence microscopy (fig 5A) or enhanced expression of IκBα as demonstrated by western blot analysis (fig 5B). To elucidate further the mechanisms of SF-induced NF-κB inhibition, we employed siRNA specific for cRel in MIA-PaCa2 cells to prevent expression and thus binding of cRel. In the presence of cRel siRNA we found strong reduction of cRel expression, as examined by western blot analysis (fig 5C) while a nonsense siRNA oligonucleotide did not affect cRel expression. Transfected cells were analysed for apoptosis in the presence of SF, TRAIL and both agents together. In line with the results above, we observed in nonsense siRNA-transfected cells specific SF-mediated apoptosis and to a minor extent TRAIL-mediated death. Administration of both agents together resulted in the highest level of apoptosis. In cRel siRNA-transfected cells, SF, TRAIL or both agents together were able to induce apoptosis to an even greater extent, supporting our hypothesis that cRel binding is a critical event in elevating NF-κB activity and apoptosis resistance in TIChigh cells. As expected, the opposite result was obtained by inactivation of IκBα with siRNA (fig 5D). In summary, our data provide strong evidence that TIChigh, in contrast to TIClow, cells largely lack transactivation-deficient p50/p50 complexes and are enriched in transactivation-competent p50/RelA and p52/RelA dimers. Importantly, transactivation-competent cRel homodimers are specifically found in TIChigh cells. Thus, we conclude that the apoptosis resistance of TIChigh cells is most probably due to the enhanced expression of NF-κB target genes mediating apoptosis resistance. Furthermore, SF reduces the DNA binding of such transactivation-competent NF-κB dimers but not their nuclear localisation and thereby may impair the expression of NF-κB target genes XIAP, cIAP1 and FLIP with antiapoptotic effects (compare fig 3C,D).
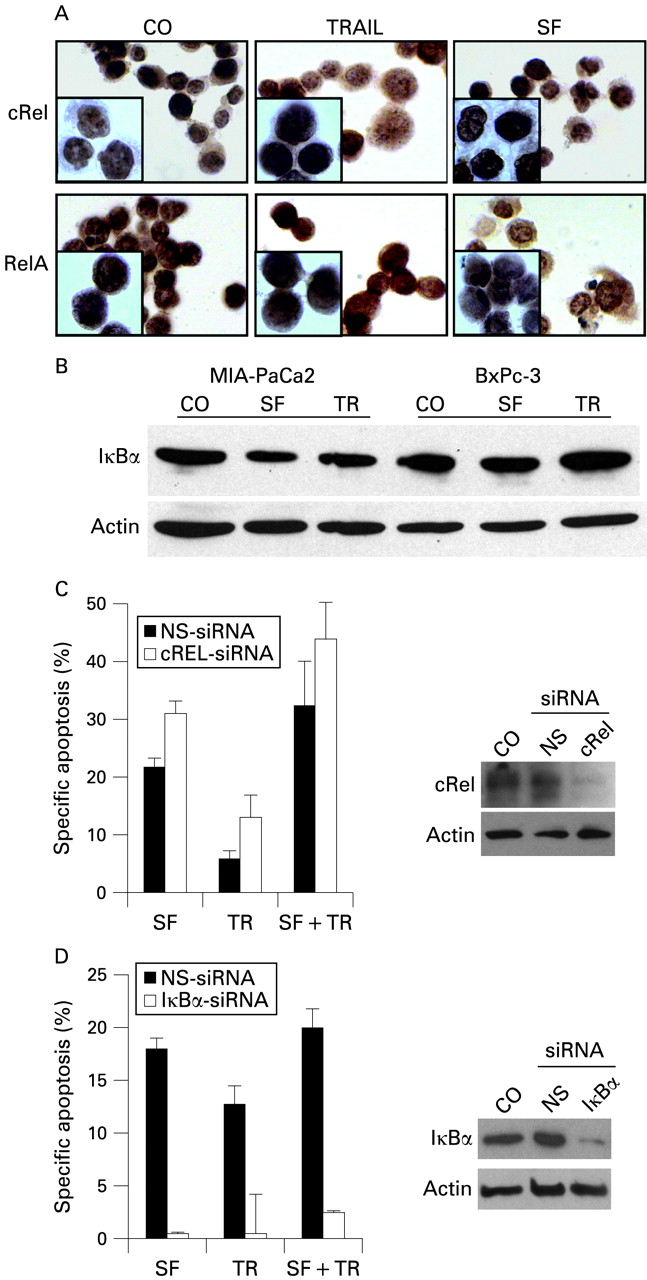
Sulforaphane (SF) does not influence translocation of nuclear factor-κB (NF-κB) and degradation of IκBα (inhibitor of NF-κB). (A) MIA-PaCa2 cells were left untreated (CO) or were treated with tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) (TR, 5 ng/ml) or SF (10 μM), and 6 h later expression of cRel and RelA was analysed by immunocytochemistry at a magnification of ×400 or ×1000 (inserts). (B) IκBα expression was detected in MIA-PaCa2 and BxPc-3 cells treated as described above by western blot analysis. (C and D) MIA-PaCa2 cells were transfected with non-specific (NS) or specific small interfering RNAs (siRNAs) directed towards IκBα or cRel. Five days later, cells were either treated with SF alone for 48 h or pretreated with SF for 24 h, followed by co-treatment with TRAIL for an additional 24 h. Specific apoptosis was determined as described in fig 3A. For detection of siRNA-mediated inhibition of protein expression, cellular proteins were harvested 5 days after transfection. Expression of IκBα and cRel was examined by western blot analysis, and β-actin served as internal standard.
SF overcomes resistance of TIChigh tumours xenografted to nude mice
To address whether SF might influence sensitivity of TIChigh tumours in vivo, we xenografted MIA-PaCa2 cells subcutaneously into nude mice and measured tumour growth during a period of 7 days in untreated mice or upon treatment with SF, TRAIL or both agents together (fig 6A). While administration of SF or TRAIL alone resulted in strong growth retardation, co-treatment with SF and TRAIL had an additive effect on the inhibition of tumour growth which was, however, not statistically significant compared with SF or TRAIL alone. However, SF in the presence of TRAIL totally repressed tumour growth during the days of treatment. Most importantly, we found that SF or TRAIL treatment were only minimally cytotoxic to mice since no change in body weight occurred (fig 6B) but only a transient elevation in plasma levels of LDH and GOT/AST 24 h after treatment with TRAIL but not after treatment with SF. LDH, GOT/AST and GPT/ALT were not elevated when measured in mice 5 days after three consecutive treatments (fig 6C). Since we found no necrotic liver tissue in paraffin sections of perfused mouse livers (data not shown), these data suggest that therapy with SF, TRAIL or both agents together is well tolerated in vivo.

Sulforaphane (SF) and tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) are effective and safe in vivo. (A) MIA-PaCa2 cells were injected subcutaneously into nude mice. After the tumours had reached a mean diameter of ∼8–10 mm, treatment was started. Mice received SF, TRAIL (TR) or both agents together at days 4–6 after implantation as described in the Materials and methods section. The tumour volumes were measured daily for the duration of the experiment. Relative increase in tumour size was calculated as follows: 100×(experimental tumour size–initial tumour size)/initial tumour size. Mice were euthanised on day 8. Data are presented as the mean (SE) of six animals. An asterisk marks a significant difference from control (CO), p<0.05 (t test). In addition, tumour growth curves were analysed according to Koziol et al23, and the p value for CO vs SF+TR was 0.007. (B) Mice were weighed during the experiment. The body weight of each individual mouse was set to 100% before treatment and body weight after treatment was correlated to initial weight. (C) Heparinised blood of mice was examined 24 h after a single treatment with TR, SF or both agents together. Likewise, blood from mice treated three times with TR, SF or both agents together was examined 5 days after the last treatment (120 h). The levels of plasma lactate dehydrogenase (LDH), glutamate-oxalacetate-transaminase (GOT/AST) and glutamate-pyruvate-transaminase (GPT/ALT) were measured using a DRY-CHEM FCD3500. (D) Twenty-four hours after single treatment of mice, xenograft tumour tissue samples were analysed by immunohistochemistry for expression of IκBα, cRel and RelA (×200 or ×400 (inserts)).
To examine, whether SF might influence the IκB status or nuclear translocation of RelA and cRel in xenografts in vivo, we performed immunohistochemistry on tumour tissue samples. No difference in expression or nuclear localisation of RelA, cRel or IκBα proteins was detectable between xenografts of untreated and treated mice (fig 6D). These results underline our in vitro finding and suggest that SF sensitises cells for TRAIL-induced apoptosis mainly by preventing promoter binding of transactivation-competent NF-κB complexes.
Our in vivo finding significantly differs from the in vitro results by the finding that TRAIL prevented growth of TIChigh MIA-PaCa2 tumour cells much more effectively in the mouse xenograft model. This may be due to an antiangiogenic effect of TRAIL, as recently described.32 We examined vessel density of xenograft tissue samples by staining with antimouse CD31 and counting positive cells. TRAIL and SF strongly reduced blood vessel density, suggesting that both agents have antiangiogenic activity. Furthermore, application of both agents together had an additive effect on prevention of blood vessel formation (Supplementary fig 5).
Together, these data demonstrate that SF alone or combined with TRAIL potentially reduces growth of tumours enriched in TICs by repression of NF-κB activity, tumour angiogenesis, proliferation and induction of apoptosis without exhibiting toxicity to normal tissue.
SF reduces viability of tumour cells from patients harbouring CD44+/CD133+ TICs
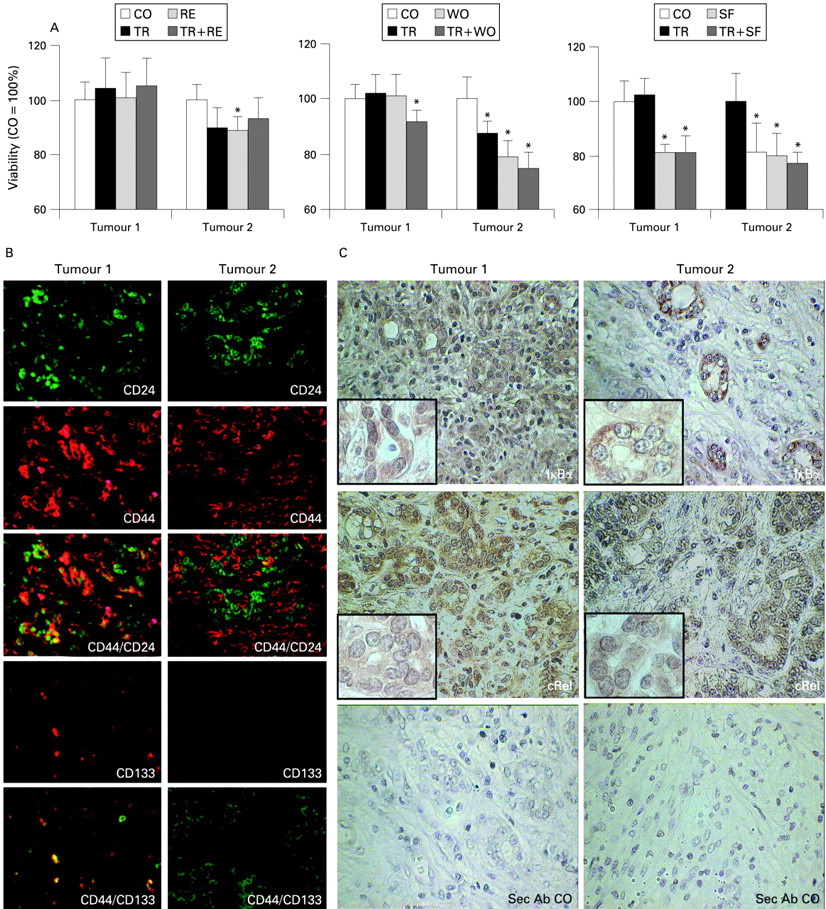
To see whether chemopreventive agents may be able to overcome resistance of tumours freshly resected from patients with pancreatic cancer, we isolated primary tumour cells, treated them with the chemopreventive agents RE, WO or SF, and TRAIL alone, or the chemopreventive agents and TRAIL together. As measured 48 h later by MTT assay (fig 7A), cells from tumour 2 were sensitive towards chemopreventive agents and TRAIL, while cells from tumour 1 were only sensitive towards SF. The high grade of resistance of tumour 1 corresponded to a moderate to poorly differentiated ductal adenocarcinoma and the presence of rare populations of CD44+/CD133+ (7.5% of total cellular mass) and CD44+/CD24+ (25% of total cellular mass) cells (fig 7B). These TIC marker-expressing populations were also present in carcinoma-free tissue from two other patients with chronic pancreatitis. Patient and tumour characteristics are summarised in table 3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Tumour cells from patients with pancreatic cancer were isolated immediately after resection from tissue and cultivated in 96-well plates at 5×105/ml. As indicated, cells were left untreated (CO) or were treated with resveratrol (RE; 50 μM), wogonin (WO; 50 μM) or sulforaphane (SF; 10 μM). After 24 h, recombinant tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) was added to untreated (TR, 5 ng/ml), RE-treated (TR+RE), WO-treated (TR+WO) or SF-treated (TR+SF) cells and 48 h later mitochondrial activity/viability was measured by MTT assay. Due to limitations in the amount of patient material, experiments were performed only once but in octuplicate, and the SD is shown. An asterisk marks a significant difference from the corresponding control, p<0.05 (t test). (B) Paraffin-embedded pancreatic tumour tissue sections were subjected to double immunofluorescence staining. CD24 expression was detected by mouse monoclonal antibodies (mAbs) and goat antimouse Alexa 488-conjugated immunoglobulin G (IgG; green). CD44 expression was analysed using mouse mAb and visualised by goat antimouse Alexa 594–IgG (red). CD44+/CD24+ cells were detected by rabbit anti-CD44 mAb with goat anti-rabbit Alexa 594–IgG (red) and CD24 mouse mAb with goat antimouse Alexa 488–IgG (green). CD133 expression was detected by rabbit polyclonal antibody (Ab) and visualised by Alexa 594-conjugated goat antirabbit IgG (red). For double staining, mouse mAb CD44 conjugated to goat antimouse Alexa 488–IgG (green) was used. (C) IκBα and cRel were detected in tumour samples by regular immunohistochemistry with rabbit polyclonal Abs, and 3,3′ diaminobenzidine (DAB) was used as a chromogen. Tissue samples were analysed under ×400 magnification and insets are shown under ×1000 magnification. Sec Ab CO, secondary antibody control.
Since our in vitro data demonstrate enhanced NF-κB activity as a reason for resistance, we examined expression and localisation of IκBα and cRel proteins in untreated tissue sections of tumours 1 and 2. IκBα and cRel expression was detectable and no obvious difference was observed between tumour 1 expressing TIC markers and tumour 2 without markers (fig 7C). These data confirm our hypothesis that enhanced binding of cRel and not necessarily expression or translocation of NF-κB subunits is the underlying reason for enhanced NF-κB activity in TIC-like tumour cells.
DISCUSSION
Our report provides evidence that treatment resistance of TICs is due to increased binding of transactivation-competent NF-κB complexes, which prevent induction of apoptosis. We demonstrate that chemopreventive agents such as WO, RE and SF are able to re-sensitise TICs by interfering with NF-κB activity to confer reduced vitality and clonogenicity, induction of apoptosis and caspase activity. This effect is strongest for SF and our study is the first to demonstrate that SF can selectively potentiate apoptotic effects in TICs without exhibiting pronounced cytotoxic side effects.
Definition of TIC populations in pancreatic carcinoma
We found >90% of a CD44+/CD24− population in highly resistant established pancreatic cancer cell lines MIA-PaCa2, AsPC-1 and Capan-1 but only 17.5% in sensitive BxPc-3 cells. This CD44+/CD24− population corresponds to a similar expression pattern suggested for TICs detected in aggressive breast cancer.2 A percentage of >90% TICs within established tumour cell lines is extremely high but mimics the aggressive phenotype of pancreatic cancer. Another research group identified the same percentage of 90% CD133+ TICs in the established human liver cancer cell line Hep3B.33 A strong indication for the TIC-like nature of the CD44+/CD24− population in our study is that TIChigh pancreatic cancer cell lines AsPC-1 and Capan-1 were originally isolated from low differentiated patient tumour cells derived from ascites or metastases. These cells, together with MIA-PaCa2, but not BxPc-3 have a high ability to form colonies and spheroids, are ALDH positive and highly treatment resistant. In contrast, the TIClow BxPc-3 pancreatic cancer cell line harbouring only 17.5% of CD44+/CD24− cells was isolated from a biopsy of a patient tumour of a well-differentiated pancreatic carcinoma and this pathological classification may be a hint as to why they contain a low amount of TICs. Accordingly, we found in BxPx-3 cells a low degree of resistance, low colony formation, no ALDH activity and no sphere formation. However, gemcitabine treatment of BxPc-3 cells led to enrichment of the CD44+/CD24−-expressing cells combined with enhanced treatment resistance. This finding supports the recent idea that cytotoxic cancer treatment may eliminate the majority of normal tumour cells, but not the highly resistant TICs, which survive cytotoxic treatment and are enriched in this way.34 The TIC characteristics are further underlined by the documented capacity of all our pancreatic cell lines used to grow in immunodeficient mice.24–26 Notably, BxPc-3 cells were found to grow very slowly,27 and this was confirmed in our experiments, where 103 BxPc-3 cells needed 28 days to form xenograft tumours in nude mice while the same amount of MIA-PaCa2 cells formed tumours in 17 days. A hint at the multipotency of TIChigh pancreatic cancer cell lines is the reported finding that xenografted Capan-1 and AsPC-1 cells differentiate into phenotypically diverse populations which show morphological, biological and biochemical characteristics similar to the tumour of origin.28 29 These data are supported by our in vitro spheroid-forming assays: only part of the whole cell population of Capan-1, AsPC-1 and MIA-PaCa2 acquired the ability to grow detached in spheroids; other cells from the same culture did not and continued to grow adherent. These results strongly hint at the differentiation of TICs to daughter cells with different properties. However, additional markers seem to be expressed by pancreatic TICs. ABCG2 is such a candidate marker since this high capacity drug transporter mediates efflux of fluorescent dyes such as rhodamine 123 or Hoechst 33342 which often correlates with a multidrug-resistant phenotype.4 35 In the same way, the pancreatic cell lines Panc1, Panc89, Colo357, PancTu1 and A818-6 expressed the ABCG2 protein on the cell surface.4 Additionally, PancTu1 and A818-6 cells isolated from patient tumours with a poor differentiation grade were positive for CD133. These data are confirmed by Hermann and colleagues,6 who found a small population of CD133+ cells on the established human pancreatic cancer cell lines L3.6pl and MIA-PaCa2 and were able to link CD133 expression to invasiveness. We did not examine ABCG2 expression and could not detect expression of CD133 in our pancreatic cancer cell lines. This might be due to differences in antibodies available. However, the CD133 antibody used in our studies is functional and gave positive staining in paraffin sections of three of four examined tumour/pancreatitis tissue samples from patients. This higher expression of CD133 in tissue could be best explained by clonal selection during conditions of prolonged cell culture and by the missing stem cell niche in vitro. We assume that CD133 expression might have been lost under normoxic in vitro conditions, since it has been reported that CD133 expression as well as invasiveness can be induced by hypoxia.36 In view of the fact that a microenvironment of low oxygen is a pronounced feature of pancreatic cancer,37 hypoxia might be the reason for expression of CD133 in patient tumours but not in established cells lines. In addition to expression of CD133, we found CD44+/CD24+ and CD133+/CD44+ double-positive cells in three of four examined tissues. The CD44+/CD24+ phenotype differs from the CD44+/CD24− phenotype which we identified to characterise a TIC population in vitro. We assume that the in vivo CD44+/CD24+ population comprises TICs as well as non-TICs, and cell culture conditions may have led to loss of CD24 expression in the TIC population. Interestingly, primary fresh cells isolated from marker-positive tissues were totally resistant towards ex vivo treatment with TRAIL but could be sensitised by SF. Two very recent studies in pancreatic cancer have utilised a marker combination of CD44+/CD24+/ESA+5 and of CD133+/CXCR4+6 in patient-derived tumours after serial transplantation on mice. FACS analysis showed that these two populations overlap but are not identical.6 We did not examine expression of ESA in patient tumours, due to a lack of correlation with a TIC phenotype in our in vitro experiments and the correlation between ESA and CD24 expression. Interestingly, CD44+ cells have been shown to represent a highly tumourigenic TIC-like population in breast, prostate, and head and neck cancer.2 38 39 In this context, a variant of glycoprotein CD44 has been known since 1991 to be expressed only in metastasising cell lines and whose overexpression was sufficient to establish full metastatic behaviour of a non-metastasising pancreatic cancer cell line.40 The same CD44 variant is expressed on B and T lymphocytes and is required for the lymphatic spread of tumour cells.41 This finding supports the recent idea that metastasising tumour cells, which most probably represent TICs, may mimic lymphocyte behaviour.
Binding of distinct NF-κB subunits in TIChigh and TIClow cells
From basic studies with highly resistant tumour cells, we know that two types of events are necessary to induce cell death: (1) inhibition of NF-κB-dependent survival signals and (2) activation of the cellular stress response.42 In several studies we have demonstrated that chemotherapy or radiation induces the cellular stress response, which, however, alone may not be effective to induce apoptosis in a cellular environment of increased NF-κB activity. Elevated NF-κB DNA-binding activity of transactivation-competent dimers may be involved in the pronounced resistance in TICs. By performing EMSA, we identified binding of distinct NF-κB complexes in TIChigh and TIClow pancreatic carcinoma cell lines. In supershift experiments we documented that complex 3, most probably composed of transactivation-potent cRel homodimers, binds only in TIChigh cells. In contrast, complex 1, which consists of transactivation-inactive p50 proteins, was selectively found in TIClow cells. Complex 2, which represents RelA-containing heterodimers, is detected in both TIChigh and TIClow cells, with a higher abundance in TIClow cells. Taken together, these data suggest that in TIClow cells competition between transactivation-competent and -incompetent NF-κB dimers exists, which most probably leads to a lower level of NF-κB-dependent gene transcription. In contrast, in TIChigh cells, this competition is rather low, leading to enhanced NF-κB target gene expression, such as that found in the present study for XIAP, cIAP and FLIP (see fig 3; and data not shown).
SF inhibits DNA binding of transactivating NF-κB in TIChigh cells
Many plant products have been proven to inhibit NF-κB together with inducing apoptosis of tumour cells. NF-κB is a target of SF, a natural isothiocianate found in high concentration in broccoli. We found impaired DNA binding of transactivation-potent NF-κB subunits in SF-treated cell lines and these data were confirmed by SF-mediated downregulation of expression of antiapoptotic NF-κB target genes. Since we could enhance SF-induced apoptosis by siRNA-mediated downregulation of cRel or, vice versa, were able to block SF-induced apoptosis completely by additional activation of NF-κB via siRNA-mediated inhibition of IκBα, NF-κB appears to be the key step by which SF abrogates resistance of TICs. Quite similar results were demonstrated in murine macrophages,43 where SF selectively reduced DNA binding of NF-κB without interfering with nuclear translocation of NF-κB. Moreover, a recent report indirectly confirms our finding that SF is able to sensitise TICs. In that study, SF is described to induce death in chemotherapy- and TRAIL-resistant Hep3B hepatoma cells.44 This is the same cell line which has just been described to harbour 90% of CD133+ TICs.33 The suitability of SF for eradication of TICs is underscored by our experiments in which we could also sensitise TIChigh pancreatic carcinoma cells to TRAIL-induced apoptosis by pretreament with RE or WO, although the effects were less pronounced than those we observed with SF. Most importantly, another recent publication suggests that the plant-derived compound parthenolide from traditional Mexican medicine is useful to kill leukaemia and AML (acute myeloid leukaemia) stem cells while sparing normal haematopoietic cells. Similarly to our results, parthenolide eradicated TICs via inhibition of NF-κB without obvious side effects on normal cells.45
Involvement of PKC in SF-mediated apoptosis
Trauzold et al46 described a TRAIL receptor-mediated activation of PKC in the pancreatic adenocarcinoma cell line PancTuI, which contributes to resistance through inhibition of the mitochondrial apoptosis pathway and activation of NF-κB. From this point of view, it was interesting to evaluate whether SF overcomes resistance of TIChigh cells upon activation of PKC. As expected, SF-mediated apoptosis was reduced in the presence of the PKC activator PMA, but not, however, completely blocked. These data indicate that SF-mediated apoptosis involves a block of PKC, which may result in the observed downregulation of NF-κB signalling and antiapoptotic target genes.
In vivo applicability of TRAIL and SF
We used TRAIL in our study for targeting of TICs since clinical evaluation in a phase I study confirmed that TRAIL is well tolerated by patients with advanced solid malignancies such as colorectal, lung, prostate, ovarian or renal cancer.47 These results are reflected by our data, in which TRAIL was non-toxic to fibroblasts and mesenchymal stem cells while it induced apoptosis and reduced viability in TIClow, but not in TIChigh pancreatic cancer cell lines after single treatment. Only after prolonged culture in clonogenic assays was TRAIL-induced cytotoxicity seen in TIChigh cells. Accordingly, in our studies with TIChigh cells xenografted to nude mice, repeated intratumoural application of recombinant TRAIL protein was able to inhibit tumour growth without apparent significant effects on body weight or liver function. In contrast, a recent report suggests that TRAIL promotes invasion and metastasis in an apoptosis-resistant orthotopically xenografted Colo357 pancreatic ductal adenocarcinoma whose growth has been observed during 40 days.48 In our subcutaneous MIA-PaCa2 xenograft model no micrometastasis could be detected during the observation period of 10 days. This was obvious from analysis of human EpCAM (epithelial cell adhesion molecule) and human major histocompatibility complex (MHC) class I. No positive cells were seen in the liver, spleen and lungs of untreated or treated xenografted mice as examined by immunohistochemistry and flow cytometry (data not shown). The difference between our findings and the results of Trauzold et al48 may be due to different models used and to the fact that metastasis in subcutaneous mouse xenograft models used by us is rather untypical. However, TRAIL alone did not completely eliminate pancreatic TICs after three consecutive injections in vivo. A more pronounced reduction of tumour growth was obtained with SF in combination with TRAIL. This is most probably due to the SF-mediated strong inhibition of NF-κB binding to DNA. Thus, SF itself was quite effective in eradication of TICs and increased the antitumour effects of TRAIL. Besides, our results in mice reflect data of a clinical phase I study showing that daily consumption of 100 μmol of glucosinolate, the precursor of SF, is well tolerated and has no adverse side effects—for example, on body weight or liver function.49 Most importantly, another prospective study of fruit and vegetable intake in 1338 patients with prostate cancer among 29 361 men (average follow-up 4.2 years) demonstrated that among several phytonutrients only a high intake of cruciferous vegetables, including broccoli and cauliflower, was associated with reduced risk of aggressive prostate cancer, particularly extraprostatic disease.50
In conclusion, these findings indicate the effectiveness of SF in eradication of pancreatic TICs and its ability to potentiate the antitumour effects of TRAIL by repression of NF-κB activity and downregulation of expression of antiapoptotic genes, leading to the inhibition of viability, clonogenicity and tumour growth, with inhibition of angiogenesis and induction of caspase activity followed by apoptosis. The dose of SF (4.4 mg/kg/day) used in the current animal studies is quite relevant to that used in human subjects. Therefore, the combination of SF with, for example, gemcitabine and/or TRAIL, has significant potential as an effective therapy for pancreatic cancer that can overcome chemoresistance of TICs and enhance the therapeutic effect. Further clinical studies are necessary to confirm our findings in patients with pancreatic cancer.
Acknowledgments
We thank Dr W Rittgen for statistical analysis, Dr R Saffrich for help with immunofluorescence microscopy, and Dr H-J Stark for providing human primary skin fibroblasts. This study was supported by grants from the Bundesministerium für Bildung und Forschung (IH: 01GU0611), Tumorzentrum Heidelberg/Mannheim (IH: D10027(6)350), Stiftung Chirurgie Heidelberg (IH) and Deutsche Krebshilfe (IH: 107254), and by the Deutsche Forschungsgemeinschaft (BB: SFB 518/A17).
REFERENCES
Supplementary materials
Web only appendices 58;7:949
Files in this Data Supplement:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
See Commentary, p 900
▸ Additional figures are published online only at http://gut.bmj.com/content/vol58/issue7
Competing interests: None.
Ethics approval: Patient material was obtained with the approval of the ethical committee of the University of Heidelberg.