Article Text

Abstract
Background: Early events in the progression of 90% of sporadic colorectal cancers depend on constitutive activation of Wnt signalling. Recent data also indicate a close association between the Hedgehog (Hh) and Wnt pathways in colonic epithelial cell differentiation.
Aims: To investigate whether expression of Gli1, a transactivator of Hh signalling, can suppress Wnt signalling and inhibit proliferation of human colorectal cancer cells.
Methods: Gli1 and nuclear β-catenin expression were examined in a series of 40 human colorectal cancers by immunohistochemistry. We quantified Gli1 and nuclear β-catenin staining as markers of Hh and Wnt pathway activation, respectively. Two human colon cancer cell lines, SW480 and HCT116, with mutations in APC and β-catenin, respectively, were used. The effects of Gli1 overexpression on Wnt transcriptional activity, β-catenin subcellular distribution, and proliferation in these cells were analysed.
Results: Nuclear accumulation of β-catenin and the Gli1 staining level were inversely associated in the 40 human colorectal cancers. Wnt transcriptional activity was reduced in Gli1 transfected cells. These effects were observed even in Gli1 transfected cells cotransfected with mutated β-catenin. Furthermore, nuclear accumulation of β-catenin was diminished compared with that in empty vector transfected cells, and downregulated transcription of c-Myc was observed in Gli1 transfected cells. Proliferation of Gli1 transfected cells was also significantly suppressed compared with that in empty vector transfected cells.
Conclusions: Our data suggest that Gli1 plays an inhibitory role in the development of colorectal cancer involving Wnt signalling, even in cases with the stabilising mutation of β-catenin.
- APC, adenomatous polyposis coli
- TCF, T cell factor
- LEF, lymphoid enhancer factor
- Hh, hedgehog
- Shh, Sonic hedgehog
- Ihh, Indian hedgehog
- Ptch, patched
- Smo, smoothened
- PBS, phosphate buffered saline
- UICC, International Union Against Cancer
- HA, haemagglutinin
- MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- RT-PCR, reverse transcription-polymerase chain reaction
- GSK3β, glycogen synthase kinase 3β
- Hedgehog signalling
- Wnt signalling
- Gli1
- β-catenin
- colorectal cancer
Statistics from Altmetric.com
- APC, adenomatous polyposis coli
- TCF, T cell factor
- LEF, lymphoid enhancer factor
- Hh, hedgehog
- Shh, Sonic hedgehog
- Ihh, Indian hedgehog
- Ptch, patched
- Smo, smoothened
- PBS, phosphate buffered saline
- UICC, International Union Against Cancer
- HA, haemagglutinin
- MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- RT-PCR, reverse transcription-polymerase chain reaction
- GSK3β, glycogen synthase kinase 3β
The Wnt signalling pathway is evolutionarily conserved and controls many events during embryonic development. In the absence of Wnt signalling, β-catenin contained within a multiprotein complex of axin, adenomatous polyposis coli (APC), and β-catenin is phosphorylated by glycogen synthase kinase 3β (GSK3β) and is subsequently degraded by ubiquitin dependent proteolysis.1,2 Following binding of Wnt to its receptor, frizzled, GSK3β is inactivated, and non-phosphorylated β-catenin is released from the complex.1,2 Non-phosphorylated β-catenin accumulates within nuclei and forms complexes with the transcription factors T cell factor (TCF) and lymphoid enhancer factor (LEF), resulting in transactivation of c-Myc and cyclin D1.3–5 Most sporadic colorectal cancers involve constitutive activation of the Wnt signalling pathway through mutational loss of APC function or, less commonly, through stabilising mutations of β-catenin or mutations of axin2.1,6,7 Loss of APC is believed to be one of the early initiating events in colorectal tumorigenesis.8
The Hedgehog (Hh) signalling pathway is also crucial for growth, patterning, and morphogenesis of many organs.9 Signalling in mammalian cells is mediated by Hh ligands Sonic, Indian, and Desert Hh (Shh, Ihh, and Dhh); morphogen gradients determine the level of signalling in target cells.9,10 In the absence of ligand, the transmembrane receptor patched (Ptch) inhibits the activity of another transmembrane protein, smoothened (Smo), resulting in inactivation of Hh signalling.9,10 Binding of ligand to Ptch abrogates the inhibitory effect of Ptch, and Smo is derepressed, thereby activating transcription factor Gli, the vertebrate homologue of Drosophila Cubitus interruptus (Ci).9,10 Three vertebrate Gli genes (Gli1, Gli2, and Gli3) have been identified.9 Gli2 and Gli3 have distinct context dependent repressor and activator functions, and Gli3 is cleaved into a repressor form in the absence of Hh signalling in vertebrates.11–13 In contrast, Gli1, originally identified as an amplified gene in a malignant glioma,14 is a strong positive activator of downstream target genes and is itself a transcriptional target of Hh signalling.15 Recent studies have shown an association between Hh pathway activation and initiation of human tumours.10 Activating mutations of Smo or suppressing mutations of Ptch activate the Hh pathway in a constitutive and ligand independent manner. These mutations have been identified in small subsets of tumours of the brain, skin, and muscle.16,17 Recent studies have shown cell autonomous ligand dependent activation of the Hh signalling pathway in small cell lung cancer and carcinomas of the oesophagus, stomach, biliary tract, and pancreas.18–20
A close association between Hh signalling and Wnt signalling in embryonic organ patterning has been reported. Interestingly, recent studies have shown that Ihh is a negative regulator of Wnt signalling in colonic epithelial cell differentiation.21 However, the biological association between Hh signalling and Wnt signalling in human colorectal cancers has not been completely elucidated. In the present study, we analysed whether Gli1, the final effector molecule of Hh signalling, affects Wnt signalling and proliferation in human colorectal cancer.
MATERIALS AND METHODS
Clinical samples
Forty patients with primary colorectal carcinoma underwent resection at the Department of Surgery and Oncology, Kyushu University (Fukuoka, Japan). All patients provided informed consent before surgical treatment and were enrolled in the present study. Primary colorectal carcinoma surgical specimens with adjacent normal colonic mucosa were fixed in 10% formalin and embedded in paraffin. Sections were cut serially into 4 μm sections. All tumours were staged according to the TNM classification system of the International Union Against Cancer (UICC).
Immunohistochemistry
Single colour DAB immunoperoxidase staining was performed as described previously.22 Primary antibodies used were anti-Gli1 (1:250; N-16, sc-6153; Santa Cruz Biotechnology, Inc., Santa Cruz, California, USA) and anti-β-catenin (1:250; Transduction Laboratories, Lexington, Kentucky, USA). Slides were incubated with each primary antibody at 4°C overnight, washed in phosphate buffered saline (PBS) three times for five minutes each, and incubated with secondary antibodies (rabbit antigoat immunoglobulin (IgG) for Gli1 and goat antimouse IgG for β-catenin; Nichirei Corp., Ltd, Tokyo, Japan) at room temperature for one hour. Immunoreactivity was visualised by the development of brown pigment via a standard 3,3′-diaminobenzidine protocol. Sections were then counterstained lightly with haematoxylin. As a negative control, the primary antibody was preabsorbed with an anti-Gli1 blocking peptide (sc-6153P; Santa Cruz Biotechnology, Inc.) prior to incubation of the sections.
Evaluation of immunostaining
Immunohistochemical labelling of Gli1 was scored as described below. Normal colonic epithelium adjacent to each tumour tissue was used as an internal positive control (see fig 2A-1, B-1, C-1). Tumours showing no detectable staining were given a score of 0 (see fig 2A-2), tumours showing significantly reduced staining compared with that in normal colorectal mucosa were given a score of 1 (see fig 2B-2), and tumours showing Gli1 staining similar to that in normal colorectal mucosa were given a score of 2 (see fig 2C-2).
Nuclear β-catenin staining was calculated as follows: more than 100 carcinoma cells were counted in six fields, and the ratio of carcinoma cells showing strong nuclear β-catenin staining to total cells was calculated and presented as a percentage (see fig 2A-3, B-3, C-3).
Vectors
pcDNA3.1/His-Wnt1, pcDNA3.1/His-LEF1, pcDNA3.1/His-β-catenin, and pcDNA3.1/His-β-cateninΔ45 were kindly provided by Dr Hiromu Suzuki (Sapporo Medical University, Sapporo, Japan). pTOPFLASH and pFOPFLASH were kindly provided by Dr Akihide Ryo (Yokohama City University School of Medicine, Yokohama, Japan). pCS2-HA-Gli1 was kindly provided by Dr Anthony E Oro (Stanford University School of Medicine, Stanford, California, USA), and pcDNA3.1/His-Gli1, 8×3′Gli-BS-Luc, and 8× mutant3′ Gli-BS-Luc were kindly provided by Dr Hiroshi Sasaki (Riken Centre for Developmental Biology, Kobe, Japan).
Cell culture
Human colon carcinoma cell lines SW480 and HCT116, with mutations in APC and β-catenin, respectively,3,6 were obtained from American Type Culture Collection (Manassas, Virginia, USA) and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (Sigma-Aldrich, St Louis, Missouri, USA) and penicillin (100 U/ml) at 37°C in 5% CO2.
Transient transfection and luciferase assay
Cells were plated onto six well plates 24 hours before transfection and transfected with Superfect Transfection Reagent (Qiagen, Valencia, California, USA) according to the manufacturer’s instructions. Each well was cotransfected with 10 ng pRL-SV40 (Promega Corp., Madison, Wisconsin, USA), 1 μg reporter plasmid, and pcDNA3.1/His-Gli1 with or without empty vector to a total of 2 μg. For cotransfection of pcDNA3.1/His-Gli1 with pcDNA3.1/His-Wnt1, β-catenin, or LEF1, cells were transfected with 1 μg of each vector (total plasmid DNA was adjusted to 2 μg) plus the reporter plasmids. Luciferase assays were performed 24 hours after transfection with a Dual-Luciferase Reporter Assay System (Promega Corp.) according to the manufacturer’s instructions. Luciferase activities were normalised to that of Renilla luciferase. Each experiment was carried out in triplicate. The activity of empty vector was set at 1.
Western blot analysis
Cells were transfected with expression vectors by electroporation with the use of a Nucleofector device and Cell Line Nucleofector Kit T or V (Amaxa Biosystems, Cologne, Germany) according to the manufacturer’s instructions. In brief, 2.5×106 cells were subjected to centrifugation, after which the pellet was resuspended in 100 μl Nucleofector solution together with 4 μg of plasmid DNAs, transferred into Amaxa certified cuvettes, and electroporated with the Nucleofector device. Cells were harvested 16 hours after electroporation, and whole cell lysates or cytoplasmic and nuclear extracts were isolated with M-PER Mammalian Protein Extraction Reagent (Pierce Biotechnology Inc., Rockford, Illinois, USA) or NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology Inc.) according to the manufacturer’s instructions. Lysates were run on 7.5% or 10% sodium dodecyl sulphate-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, Massachusetts, USA). Anti-β-catenin (1:1000; Transduction Laboratories), anti-α-tubulin (1:1000; Sigma-Aldrich), anti-haemagglutinin (HA) (1:200; F-7, sc-7392; Santa Cruz Biotechnology Inc.), or anti-RCC1 (1:1000; kindly provided by Dr Takeharu Nishimoto, Kyushu University, Fukuoka, Japan) antibodies were used as primary antibodies. After washing, membranes were incubated with the appropriate horseradish peroxidase conjugated secondary antibody (Amersham Biosciences, Piscataway, New Jersey, USA). Blots were developed with ECL Plus Western Blotting Detection System (Amersham Biosciences).
Fluorescence immunocytochemistry
SW480 cells were transfected by Superfect Transfection Reagent (Qiagen), as described previously. At 24 hours after transfection, cells were fixed in 4% paraformaldehyde followed by permeabilisation with 0.2% Triton X-100. Cells were then incubated with primary followed by secondary antibodies. Sections were then counterstained with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich). After mounting in Vectorshield Mounting Medium (Vector Laboratories, Burlingame, California, USA), samples were examined by fluorescence microscopy (TE300; Nikon, Tokyo, Japan). Antibodies and dilutions used are as follows: anti-β-catenin (1:200; H-102, sc-7199; Santa Cruz Biotechnology Inc.); anti-HA (1:200; F-7, sc-7392; Santa Cruz Biotechnology Inc.); AlexaFluor 488 goat antirabbit IgG (1:400; Molecular Probes, Eugene, Oregon, USA), and AlexaFluor 568 goat antimouse IgG (1:400; Molecular Probes).
Proliferation assay
Cells were transfected by electroporation as described above. At 16 hours after transfection, cells were suspended by treatment with trypsin, washed once with PBS, and resuspended in RPMI 1640 medium supplemented with 10% fetal bovine serum. Cells (4×103 per well) were plated onto 96 well plates, and cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay23 at several time points.
Colony formation assay
Cells (2×105) were transfected with 2 μg of pcDNA3.1/His-Gli1 or empty pcDNA3.1 vector as a control with Superfect, as described above. Cells were plated onto 100 mm tissue culture dishes 24 hours after transfection and selected with G418 (600 μg/ml; Invitrogen, Carlsbad, California, USA). After two weeks of selection, colonies were fixed with ice cold methanol and stained with 0.5% crystal violet.
Reverse transcription-polymerase chain reaction (RT-PCR) and quantification of mRNA
HCT116 cells transfected with pcDNA3.1/His-Gli1 or empty vector were selected with G418 for two weeks, as described above. Total RNA was then extracted by the single step guanidinium thiocyanate-phenol-chloroform method.24 Primers used were 5′-CCA GGC ACC AGG GCG TGA TG -3′ and 5′-CGG CCA GCC AGG TCC AGA CG -3′ for β-actin,25 5′-AGA GAA GCT GGC CTC CTA CC -3′ and 5′-AGC TTT TGC TCC TCT GCT TG -3′ for c-Myc,26 and 5′-TCT GCC CCC ATT GCC CAC TTG-3′ and 5′-TAC ATA GCC CCC AGC CCA TAC CTC-3′ for Gli1.19 Amplification was performed for 28 cycles at 55°C (β-actin) or 58°C (c-Myc and Gli1) annealing temperature with a 72°C extension temperature. PCR products were separated on ethidium bromide stained 1.5% agarose gels. Expected RT-PCR product sizes were 436 bp for β-actin, 801 bp for c-Myc, and 460 bp for Gli1. Quantification of mRNA was performed with an RNA Counter kit (TrimGen Corp., Sparks, Maryland, USA) according to the manufacturer’s instructions. In brief, 0.2 μg of RNA for c-Myc and 0.1 μg of RNA for β-actin were extracted as described above and subjected to an isometric primer extension reaction (TrimGen Corp.) followed by signal amplification and detection at an absorbance at 405 nm.
Statistical analysis
All calculations were carried out with StatView 5.0J software (Abacus Concepts, Berkeley, California, USA). Associations between Gli1 staining and clinicopathological features listed in tables 1 and 2 were analysed by χ2 test and Kruskal-Wallis test, respectively. A p value less than 0.05 was considered significant.
Clinicopathological features and Gli1 immunostaining of 40 colorectal carcinoma specimens
Nuclear β-catenin staining and Gli1 immunostaining of 40 colorectal carcinoma specimens
RESULTS
Reverse association between Gli1 staining and nuclear localisation of β-catenin in human colorectal cancers
We analysed the association between Gli1 staining and nuclear localisation of β-catenin in 40 primary colorectal cancers by immunohistochemistry, as described in materials and methods. In normal colonic epithelium, Gli1 staining was strong and restricted to the surface epithelium (fig 1A, fig 2A-1, B-1, C-1). Gli1 staining was efficiently blocked by the blocking peptide (fig 1B). Specificity of the anti-Gli1 antibody was confirmed by western blot analysis of tissue lysates of human normal colonic mucosa; the expected sized band without background was detected (fig 1C). Seventeen (42.5%), 13 (32.5%), and 10 (25%) tumours were given scores of 0 (fig 2A-2), 1 (fig 2B-2), and 2 (fig 2C-2), respectively. Statistical analyses of the relation between Gli1 staining and clinicohistopathological data showed no association of Gli1 staining with sex, localisation, or UICC stage (table 1). Reduced Gli1 staining was more frequent in poorly differentiated carcinomas although this association was not statistically significant (table 1).

Expression of Gli1 in normal human colonic mucosa. (A) Immunostaining of Gli1 of normal human colon was restricted to the top of the crypts and the surface epithelium. (B) Gli1 staining was efficiently blocked by the blocking peptide. Bars, 10 μm. (C) Specificity of the anti-Gli1 antibody was confirmed by western blot analysis of tissue lysates of normal human colonic mucosa.
Nuclear β-catenin staining varied from 0% to 94.5%, with a mean of 15.33%. For example, nuclear β-catenin staining was 22.7% in a tumour with a score of 0 (fig 2A-3), 1.8% in a tumour with a score of 1 (fig 2B-3), and 0% in a tumour with a score of 2 (fig 2C-3). Mean nuclear β-catenin staining was 25.7%, 13.0%, and 0.71% for tumours with scores of 0, 1, and 2, respectively (table 2). Mean nuclear β-catenin staining was 1.5% in 11 tumours with a score of 1 except for two tumours showing extremely high nuclear β-catenin staining (84.8% and 67.5%). There was a significant reverse association between Gli1 staining and nuclear β-catenin staining (p = 0.0143, table 2). In addition, nine of 11 (81.8%) tumours showing more than 10% nuclear β-catenin staining were scored as 0.
Gli1 overexpression inhibits β-catenin/TCF reporter activity in colon cancer cells with mutations in either APC or β-catenin
Results suggested a reverse association between Gli1 expression and nuclear β-catenin accumulation. Therefore, the molecular mechanism underlying this reverse association was analysed in Gli1 transfected SW480 and HCT116 cells possessing mutations in APC and CTNNB1, respectively.3,6 We used 8×3′Gli-BS-Luc, carrying eight copies of a Gli binding site derived from the HNF3β floor plate enhancer.27 Gli1 transfected cells showed a concentration dependent activation of the Hh signalling reporter (fig 3A, B). Gli1 transfection failed to activate 8× mutant3′Gli-BS-Luc, carrying eight copies of a mutated Gli binding site (fig 3A, B). We examined if Gli1 overexpression affected Wnt signalling with the use of the β-catenin/TCF luciferase reporter pTOPFLASH.3 Gli1 overexpression suppressed pTOPFLASH luciferase activity in a concentration dependent manner in both cell lines (fig 3C, D). In contrast, Gli1 overexpression had no significant effect on pFOPFLASH luciferase activity in which the β-catenin/TCF responsive elements are mutated and inactive (fig 3C, D).

Immunostaining of Gli1 and β-catenin. (A-1, B-1, C-1) Gli1 staining of the surface colonic epithelium served as an internal positive control. (A-2) Tumours showing no detectable Gli1 staining were scored as 0. (B-2) Tumours showing significantly reduced Gli1 staining compared with that of normal colorectal mucosa were scored as 1. (C-2) Tumours showing Gli1 staining similar to that in normal colorectal mucosa were scored as 2. Gli1 staining was mainly cytoplasmic but nuclear staining was also found in several cancer cells (arrows). (A-3, B-3, C-3) Nuclear β-catenin staining in tumours A (arrows), B, and C was 22.7%, 1.8%, and 0%, respectively. Bars, 10 μm.

Inhibition of β-catenin/ T cell factor 4 (TCF4) responsive luciferase reporter activity by Gli1 overexpression. SW480 (A, C) and HCT116 (B, D) cells were cotransfected with increasing concentrations of pcDNA3.1/His-Gli1 plus the hedgehog signalling reporter (A, B) or the Wnt signalling reporter (C, D) and pRL-SV40 Renilla as an internal control. Luciferase activity, normalised to Renilla luciferase activity, was expressed relative to that of control vector transfected cells, set at 1.0. Values represent the mean (SD) of triplicate determinations. Results shown are representative of three independent experiments.
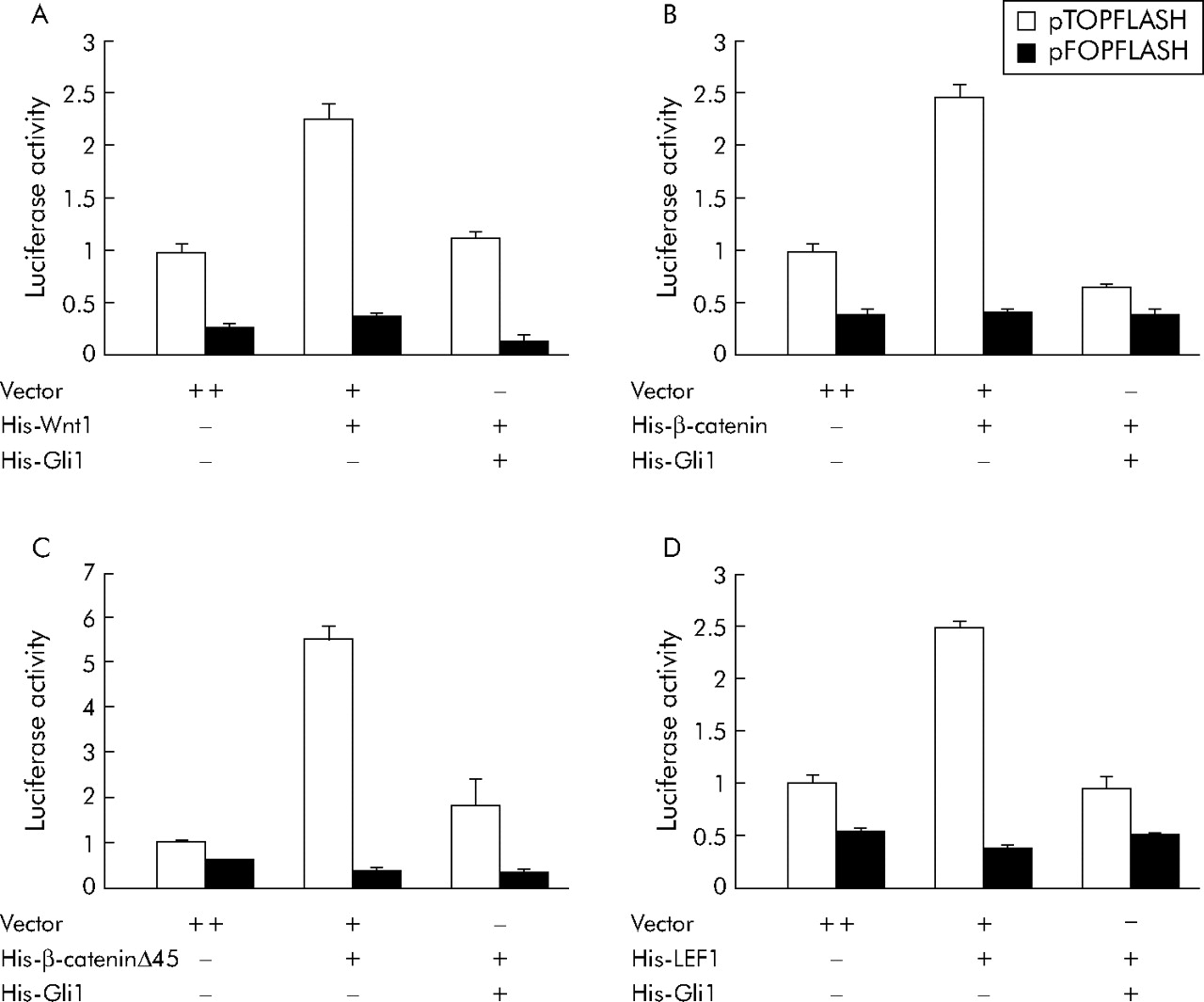
To identify target molecules of the Wnt signalling pathway that are inhibited by Gli1 overexpression, we cotransfected Gli1 with Wnt1 ligand, wild-type or mutant β-catenin (β-cateninΔ45), or LEF1 expression construct. Transfection of Wnt1 (fig 4A), wild-type β-catenin (fig 4B), or LEF1 (fig 4D) led to an approximate 2.5-fold increase in pTOPFLASH luciferase activity, and transfection of mutant β-catenin (fig 4C) led to a 5.5-fold increase in pTOPFLASH luciferase activity, similar to previously reported increases.28 Cotransfection of Gli1 with these expression constructs suppressed pTOPFLASH luciferase activity to near baseline levels (fig 4). It is notable that Gli1 overexpression inhibited even upregulated Wnt signalling driven by β-cateninΔ45, which stabilises β-catenin.6

Upregulation of the Wnt signalling pathway by Wnt1, β-catenin, and lymphoid enhancer factor 1 (LEF1), and suppression by Gli1. HCT116 cells were cotransfected with pcDNA3.1/His-Gli1 and Wnt1 (A), wild-type β-catenin (B), mutant β-catenin (C), or LEF1 (D), as indicated in the materials and methods. Values represent the mean (SD) of triplicate determinations. Results shown are representative of three independent experiments.
Gli1 overexpression reduces nuclear β-catenin levels
To determine whether Gli1 overexpression affects intracellular β-catenin levels, whole cell extracts were isolated from Gli1 transfected SW480 and HCT116 cells. Overexpression of Gli1 was confirmed by western blot analysis with a monoclonal antibody against HA (fig 5A). No detectable difference in total β-catenin levels was found between Gli1 transfected cells and control vector transfected cells in either cell line (fig 5A).

Reduced nuclear β-catenin accumulation by Gli1 overexpression. (A) SW480 and HCT116 cells were transfected with haemagglutinin (HA)-Gli1 or empty vector, followed by western blot analysis with anti-HA, anti-β-catenin, or anti-α-tubulin antibody. Results shown are representative of three independent experiments. (B) β-Catenin protein levels in nuclear and cytoplasmic fractions of SW480 and HCT116 cells were determined by western blot analysis. α-Tubulin and RCC1 were used as cytoplasmic and nuclear loading controls, respectively. Results shown are representative of three independent experiments. (C) SW480 cells were transfected with Gli1 expression construct and stained with anti-β-catenin and anti-HA antibodies to detect endogenous β-catenin (green) and exogenous Gli1 (red) expression (×600). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Bars, 10 μm.
Because nuclear translocation of β-catenin is required for transcription of target genes,1 we examined cytoplasmic and nuclear β-catenin levels by western blot analysis. Gli1 overexpression decreased nuclear β-catenin whereas cytoplasmic β-catenin was not significantly affected (fig 5B). Successful isolation of nuclear proteins and cytoplasmic proteins was confirmed by analysing the localisation of RCC1 and α-tubulin, which exist only in nuclei and cytoplasm, respectively (fig 5B).
Next, endogenous β-catenin and HA-Gli1 were detected by immunofluorescence in Gli1 transfected SW480 cells. SW480 cell overexpressing Gli1 showed a significant reduction in nuclear β-catenin staining but cells not expressing Gli1 showed abundant nuclear β-catenin staining (fig 5C).
Gli1 overexpression inhibits proliferation of colon cancer cells
Previous studies have shown that the Wnt signalling pathway is important for regulating the proliferation of colon cancer cells.4,5,29 Therefore, we examined whether induction of Gli1 overexpression affects the proliferation of SW480 and HCT116 cells by MTT and colony formation assay. Transient transfection of Gli1 in SW480 cells suppressed proliferation to 33% of that of empty vector transfected cells at 72 hours in MTT assays (fig 6A). Overexpression of Gli1 in HCT116 cells reduced proliferation to 62% of that of empty vector transfected cells (fig 6A). Similar results were obtained in colony formation assays of SW480 and HCT116 cells stably expressing Gli1 (fig 6B). We transfected Gli1 in both cell lines and counted the number of colonies after two weeks of G418 selection. The number of colonies of Gli1 transfected SW480 and HCT116 cells decreased to 54% and 52%, respectively, of that of empty vector transfected cells (fig 6B). Gli1 transfected HCT116 cells showed reduced pTOPFLASH luciferase activity and increased Gli reporter activity compared with that of vector transfected control cells, indicating stable downregulation of Wnt signalling (fig 6C).

Inhibition of colon cancer cell proliferation by Gli1 overexpression. (A) Cells were transfected with empty vector or pcDNA3.1/His-Gli1, as described in the materials and methods. Cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay at the times noted (A570 nm; six independent wells) and expressed as a percentage of controls (SD). Results shown are representative of three independent experiments. (B) For colony formation assays, SW480 and HCT116 cells were transfected with pcDNA3.1/His-Gli1 or empty vector and selected with G418. The bar graphs show average colony numbers for triplicate experiments, and error bars represent SD. Results shown are representative of three independent experiments. (C) After selection with G418 for two weeks, HCT116 cells were transfected with pTOPFLASH or 8×3′Gli-BS-Luc reporter construct, followed by luciferase assay. Results shown are representative of three independent experiments, and values represent the mean (SD) of triplicate determinations.
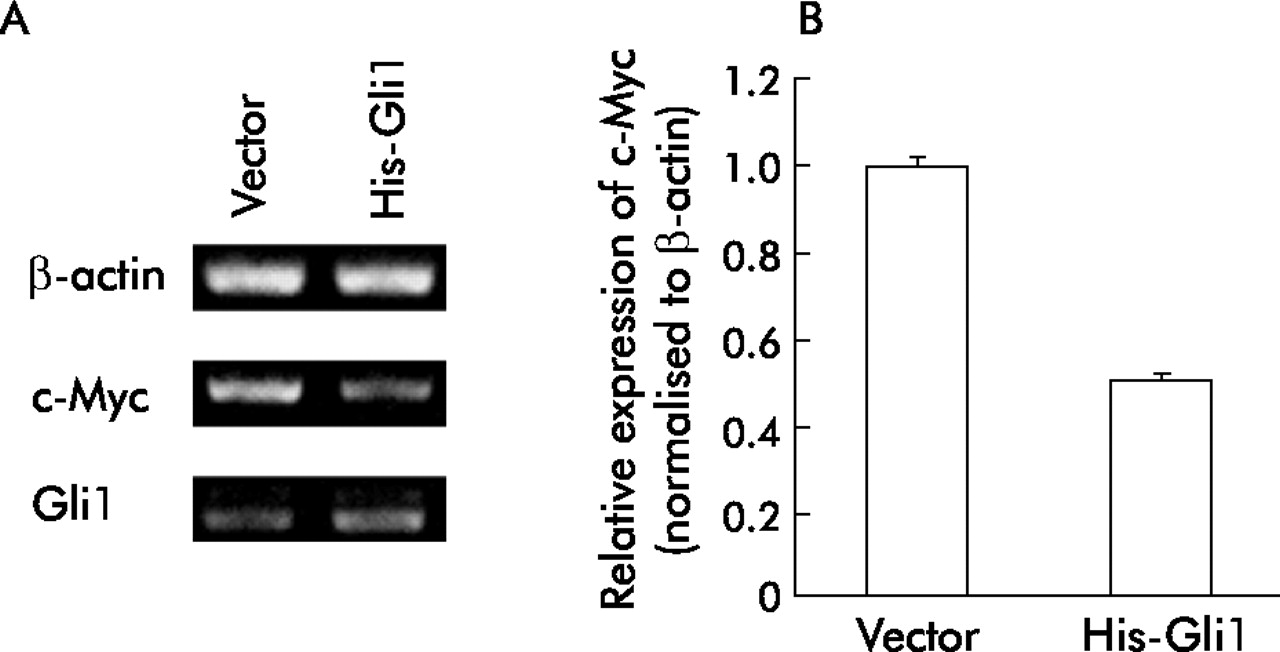
Gli1 overexpression reduces c-Myc mRNA levels
To investigate the molecular mechanisms of reduced proliferation in cells stably overexpressing Gli1, we examined whether Gli1 overexpression reduced expression of c-Myc, a canonical target of Wnt signalling4 and an oncogenic factor in colorectal cancer.29 Expression of c-Myc mRNA was reduced in Gli1 overexpressing HCT116 cells compared with that in vector transfected cells (fig 7A, B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inhibition of c-Myc mRNA level by Gli1 overexpression. (A) Total RNA was isolated from HCT116 cells transfected with pcDNA3.1/His-Gli1 or empty vector and selected with G418 for two weeks and assessed by semiquantitative reverse transcription-polymerase chain reaction. β-Actin served as an internal control. (B) c-Myc mRNA was quantified with an RNA Counter kit as described previously. Values were normalised to those of β-actin, and relative expression in Gli1 transfected cells was compared with that in empty vector transfected cells. Values represent the mean (SD) of three independent experiments.
DISCUSSION
In the present study, we showed a reverse association between Gli1 immunostaining and nuclear β-catenin accumulation in colorectal carcinoma tissues. We also showed that Gli1 overexpression reduced the proliferative ability of human colon cancer cells. Interestingly, Wnt signalling activity in these Gli1 transfected cells was significantly reduced compared with that in empty vector transfected cells.
A close association between the Hh and Wnt pathways in embryonic organ patterning and postnatal cell differentiation has been reported. For example, during Drosophila wing development, the canonical Wingless signalling pathway is involved in repression of Hh targets along the dorsal-ventral boundary.30 There is also evidence that Wnt genes are contextually regulated by Gli proteins.31 Interestingly, a recent study showed that Ihh is a negative regulator of Wnt signalling and is required for differentiation of colonic epithelial cells.21 In that study, it was shown that Ihh expression is lost in dysplastic epithelial cells in adenomatous polyps where β-catenin shows abnormal localisation.21 These observations suggest that Hh signalling is an antagonist of Wnt signalling, even in colorectal cancers. However, the role of Hh signalling in colorectal cancers remains to be elucidated. In the present study, we showed a reverse association between the Hh and Wnt pathways in a relatively large number of colorectal carcinomas. We used Gli1 as a marker of Hh signalling activity because Gli1 is the final transactivator of Hh signalling.9,11,15
Our present data also indicate that Gli1 overexpression negatively regulates Wnt signalling, at least in part by suppressing nuclear accumulation of β-catenin. Approximately 90% of sporadic colorectal cancers show stabilisation of β-catenin in response to mutation of APC, AXIN2, or CTNNB1 (encoding β-catenin).1,6,7 It is noteworthy that Gli1 overexpression suppressed Wnt signalling not only in SW480 cells possessing mutations in APC but also in HCT116 cells possessing mutations in β-catenin. This effect of Gli1 overexpression was confirmed in HCT116 cells cotransfected with Gli1 and mutant β-catenin. Our present data indicate that Gli1 overexpression inhibits the entire canonical Wnt signalling pathway in colon cancer cells. Using fluorescence microscopy, we found that nuclear accumulation of β-catenin was suppressed in SW480 cells overexpressing Gli1. However, we have no direct evidence regarding the molecular mechanism of Gli1 induced inhibition of nuclear β-catenin accumulation. Moreover, a recent study showed that nuclear β-catenin and TCF4 are both diminished by overexpression of Ihh.21 Negative regulation of Wnt signalling by Gli1 may also involve mechanisms other than suppressing nuclear accumulation of β-catenin.
The Wnt signalling pathway plays a critical role in regulating intestinal epithelial cell proliferation; a TCF4 knockout mouse model showed a critical role of the β-catenin/TCF complex in regulating intestinal epithelial cell proliferation.32 Previous studies have shown that suppression of β-catenin/TCF signalling inhibits proliferation of colon cancer cells.5,29 These results suggest that suppression of Wnt signalling inhibits proliferation of colon cancer cells. Gli1 overexpression may be a useful new therapeutic strategy against colorectal cancer involving Wnt signalling activation because Gli1 overexpression suppresses Wnt signalling not only in SW480 cells with APC mutation but also in HCT116 cells with β-catenin mutation. In the present study, we showed for the first time that Gli1 overexpression inhibits the proliferation of SW480 and HCT116 cells. Further examination of the detailed mechanism of Gli1 induced suppression of Wnt signalling and colon cancer cell proliferation is necessary. However, our data showed that Gli1 overexpression downregulates transcription of c-Myc, an oncogenic target of Wnt signalling.
In conclusion, Gli1 overexpression inhibits Wnt signalling and colorectal cancer cell proliferation, even in cells possessing the stabilising mutation of β-catenin.
Acknowledgments
This study was supported by a General Scientific Research Grant (16591326) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank Dr Hiromu Suzuki (Sapporo Medical University, Sapporo, Japan), Dr Hiroshi Sasaki (Riken Centre for Developmental Biology, Kobe, Japan), Dr Anthony E Oro (Stanford University School of Medicine, Stanford, California, USA), and Dr Akihide Ryo (Yokohama City University School of Medicine, Yokohama, Japan) for plasmids; Dr Takeharu Nishimoto (Kyushu University, Fukuoka, Japan) for reagents; Dr Masatoshi Nomura (Kyushu University, Fukuoka, Japan) for help with experiments; and Kaori Nomiyama, Nobuhiro Torata, and Miyuki Manabe for skilful technical assistance.
REFERENCES
Footnotes
-
↵* T Akiyoshi and M Nakamura contributed equally to this work.
-
Published online first 18 November 2005
-
Conflict of interest: None declared.