Article Text

Abstract
Background: The CpG island methylator phenotype (CIMP), characterised by widespread promoter methylation, is associated with microsatellite instability (MSI) and BRAF mutation in colorectal cancer. The independent effect of CIMP, MSI and BRAF mutation on prognosis remains uncertain.
Methods: Utilising 649 colon cancers (stage I–IV) in two independent cohort studies, we quantified DNA methylation in eight CIMP-specific promoters (CACNA1G, CDKN2A (p16), CRABP1, IGF2, MLH1, NEUROG1, RUNX3 and SOCS1) as well as CHFR, HIC1, IGFBP3, MGMT, MINT1, MINT31, p14, and WRN by using MethyLight technology. We examined MSI, KRAS and BRAF status. Cox proportional hazard models computed hazard ratios (HRs) for colon cancer-specific and overall mortalities, adjusting for patient characteristics and tumoral molecular features.
Results: After adjustment for other predictors of patient survival, patients with CIMP-high cancers (126 (19%) tumours with ⩾6/8 methylated CIMP-specific promoters) experienced a significantly low colon cancer-specific mortality (multivariate HR 0.44, 95% confidence interval (CI) 0.22 to 0.88), whereas the BRAF mutation was significantly associated with a high cancer-specific mortality (multivariate HR 1.97, 95% CI 1.13 to 3.42). A trend toward a low cancer-specific mortality was observed for MSI-high tumours (multivariate HR 0.70, 95% CI 0.36 to 1.37). In stratified analyses, CIMP-high tumours were associated with a significant reduction in colon cancer-specific mortality, regardless of both MSI and BRAF status. The relation between CIMP-high and lower mortality appeared to be consistent across all stages. KRAS mutation was unrelated to prognostic significance.
Conclusion: CIMP-high appears to be an independent predictor of a low colon cancer-specific mortality, while BRAF mutation is associated with a high colon cancer-specific mortality.
Statistics from Altmetric.com
Epigenetic aberrations are thought to be an important mechanism in human carcinogenesis.1 2 A number of tumour suppressor genes are silenced by promoter CpG island methylation in colon cancers.2 3 A subset of colon cancers exhibits widespread promoter methylation, referred to as the CpG island methylator phenotype (CIMP),2 4–6 which is associated with microsatellite instability (MSI).7 8 CIMP-high colon cancers have been associated with older age, cigarette smoking, proximal tumour location, female gender, poor differentiation, BRAF mutation, wild-type TP53, inactive β-catenin/WNT and stable chromosomes,8–19 and many of these associations are independent of MSI status.9 16–19
Among patients with colon cancer, MSI has generally been associated with good prognosis in most,20 though not all studies.21 On the other hand, the presence of BRAF mutations in tumours has been characteristically associated with an inferior patient survival.22 In contrast, results for CIMP have been conflicting.22–28 These inconsistent results likely reflect differences in patient cohorts, methylation markers examined, and the variable inclusion of data on other potentially confounding molecular events, such as MSI and BRAF in multivariate analysis models.
We therefore examined both genetic and epigenetic alterations among colon cancer patients participating in two large prospective cohort studies, to assess the independent effect of CIMP, MSI and BRAF mutation on patient outcome. Furthermore, to assess CpG island methylation, we utilised quantitative DNA methylation assays (MethyLight technology) on a validated expanded panel of eight markers that appear to well characterise the presence or absence of CIMP-high in colorectal cancers.8 29
METHODS
Study population
We utilised the databases of two independent prospective cohort studies; the Nurses’ Health Study (n = 121 700 women followed since 1976),30 31 and the Health Professional Follow-up Study (n = 51 500 men followed since 1986).31 On each biennial follow-up questionnaire, participants were asked whether they had a diagnosis of colon cancer during the previous 2 years. When a participant (or next of kin for decedents) reported colon cancer, we sought permission to obtain medical records. Study physicians, while blinded to exposure data, reviewed all records related to colon cancer, and recorded the date of cancer diagnosis, American Joint Committee on Cancer (AJCC) stage and tumour location. For non-responders, we searched the National Death Index to discover deaths and ascertain any diagnosis of colon cancer that was a primary cause of death or a secondary diagnosis. Approximately 96% of all incident colon cancer cases were identified through these methods. We collected paraffin-embedded tissue blocks from hospitals where patients underwent resections of primary colon cancers.31 Tissue sections from all cases in this study were reviewed by a pathologist (SO). Tumour grade was categorised as high (⩽50% glandular area) or low (>50% glandular area). We excluded rectal cancers and cases that were preoperatively treated with radiation and/or chemotherapy. Based on availability of tissue samples, we included a total of 649 colon cancer cases (283 from the men’s cohort and 366 from the women’s cohort) diagnosed up to 2002. Written informed consent was obtained from all study subjects.
Measurement of mortality
Patients were observed until death or June 2006, whichever came first. Ascertainment of deaths included reporting by the family or postal authorities. The names of persistent non-responders were searched in the National Death Index. The cause of death was assigned by physicians blinded to information on lifestyle exposures and molecular changes in colon cancer. In rare patients who died as a result of colon cancer not previously reported, we obtained medical records with permission from next of kin. More than 98% of deaths in the cohorts were identified by these methods.
Genomic DNA extraction and sequencing of KRAS and BRAF
Genomic DNA from paraffin-embedded tissue was extracted, and whole genome amplification was performed by polymerase chain reaction (PCR) using random 15-mer primers.32 PCR and sequencing targeted for KRAS codons 12 and 13, and BRAF codon 600 were performed as previously described.32 33
Real-time PCR (MethyLight) for quantitative DNA methylation analysis
Bisulfite treatment on genomic DNA and subsequent real-time PCR (MethyLight)34 were validated and performed as previously described.35 We quantified DNA methylation in eight CIMP-specific promoters (CACNA1G, CDKN2A (p16), CRABP1, IGF2, MLH1, NEUROG1, RUNX3 and SOCS1)8 29 (which were selected from screening of 195 CpG islands in the human genome),8 17 as well as HIC1, MINT1, MINT31,36 MGMT,35 IGFBP3 and WRN.25 The PCR condition was initial denaturation at 95°C for 10 min followed by 45 cycles of 95°C for 15 s and 60°C for 1 min.
CIMP-high was defined as ⩾6/8 methylated markers using the eight-marker CIMP panel, CIMP-low as 1/8 to 5/8 methylated markers, and CIMP-0 as 0/8 methylated markers, according to the previously established criteria.29
Microsatellite instability analysis
Microsatellite instability analysis (MSI) status was determined using D2S123, D5S346, D17S250, BAT25, BAT26,37 BAT40, D18S55, D18S56, D18S67 and D18S487 (ie, 10-marker panel).29 A high degree of MSI (MSI-high) was defined as the presence of instability in ⩾30% of the markers, and microsatellite stability (MSS) as no unstable marker or instability in <30% of the markers. When tumours with instability in <30% of the markers (ie, MSI-low) was compared to tumours with no unstable marker, MSI-low did show no prognostic value (data not shown). Thus, we combined MSI-low tumours into MSS tumours in further analyses.
Statistical analysis
Cox proportional hazard models were used to calculate hazard ratios (HRs) of death according to molecular features in tumour (ie, MSI, CIMP and BRAF mutation), adjusted for age, sex, year of diagnosis, tumour stage, tumour location, tumour grade, and the molecular variables. In the analyses for colon cancer-specific mortality, death as a result of colon cancer was the primary end point and deaths as a result of other causes were censored. Age and year of diagnosis were used as continuous variables, and all of the other variables were used as categorical variables. When information on tumour location (1.4% missing), tumour stage (7.4% missing), KRAS (0.3% missing) or BRAF (2.8% missing) was missing, we assigned a separate (“missing”) indicator variable and included those cases in the multivariate analysis model. We confirmed that excluding cases with a missing variable did not significantly alter results (data not shown). An interaction was assessed by including the cross-product of two variables of interest in the analysis model. The Kaplan–Meier method was used to describe the distribution of colon cancer-specific and overall survival time, and the log-rank test was performed to test the null hypothesis of no difference in survival time distributions among all subtypes. The χ2 test was used to examine an association between categorical variables. The t test assuming unequal variances was used to compare mean ages. All analyses used SAS version 9.1 and all p values were two-sided.
RESULTS
CpG island methylator phenotype and microsatellite instability
Among 649 colon cancer patients with available tissue specimens, there were 281 deaths, including 163 colon cancer-specific deaths. Among all patients, 121 (19%) demonstrated MSI-high; 126 (19%) were CIMP-high (⩾6/8 methylated CIMP-specific markers;29 (ie, CACNA1G, CDKN2A, CRABP1, IGF2, MLH1, NEUROG1, RUNX3 and SOCS1); 238 (37%) demonstrated a mutation in KRAS; and 105 (17%) demonstrated a mutation in BRAF. Tumours were distributed bimodally according to the number of methylated CIMP markers (fig 1), and BRAF mutations were common in CIMP-high tumours while KRAS mutations were common in CIMP-low tumours (1/8–5/8 methylated markers). We assessed baseline patient characteristics according to MSI and CIMP status (table 1).
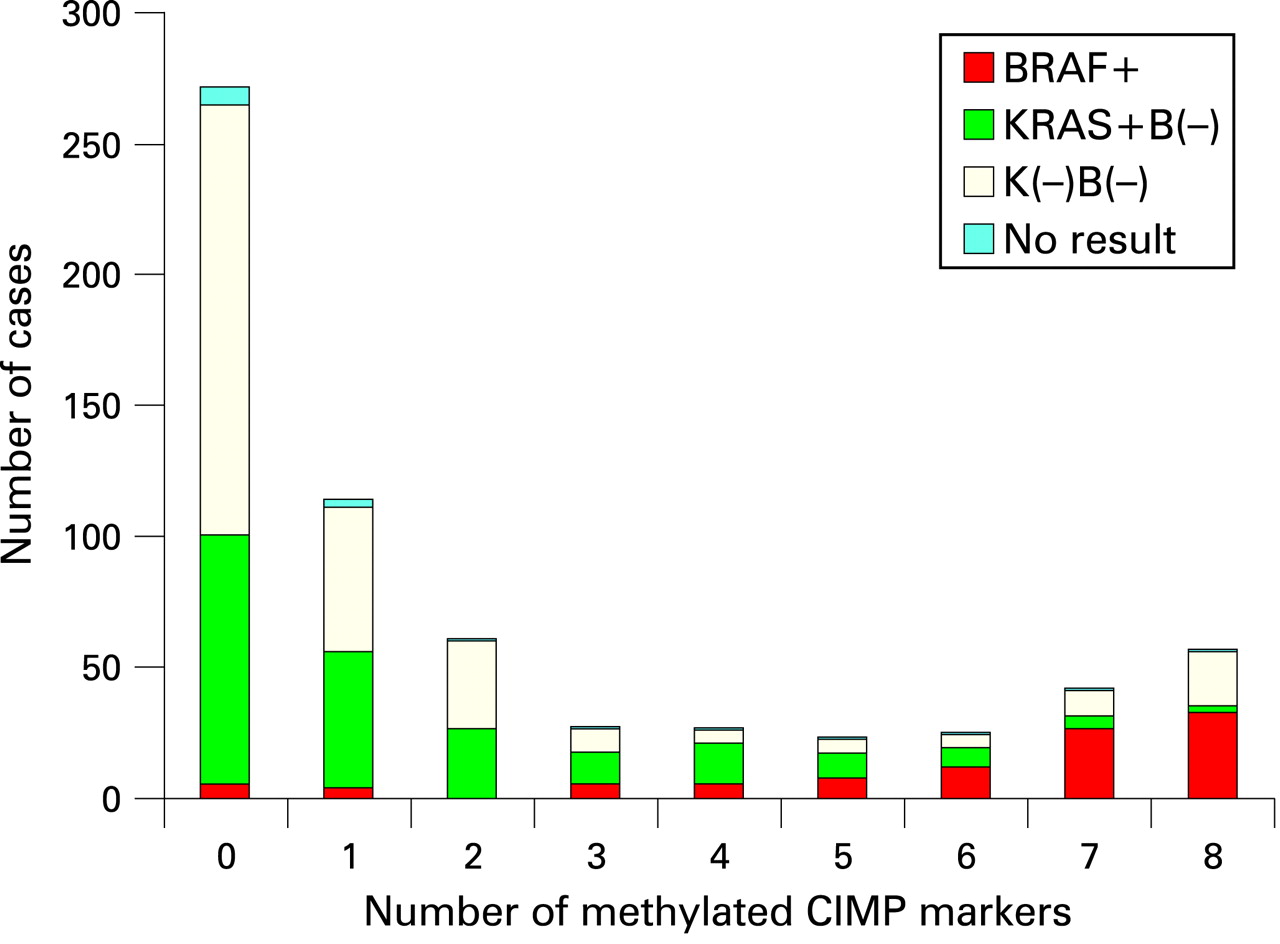
MSI-high tumours were more likely to originate in the proximal colon and possess BRAF mutations and CIMP-high status, and less likely to present with stage III or IV disease. CIMP-high tumours were also more likely to originate in the proximal colon and possess BRAF mutations.
When we jointly classified tumours by MSI and CIMP status, MSS (microsatellite stable) CIMP-high tumours had a greater prevalence of stage IV disease (36%; 13/36; p = 0.0004, compared to all other subtypes) when compared to MSI-high CIMP-high tumours (4%; 3/84), MSI-high CIMP-low/0 tumours (6%; 2/32) or MSS CIMP-low/0 tumours (14%; 65/452). In addition, BRAF mutations were found in 62% (53/86) of MSI-high CIMP-high tumours, 57% (21/37) of MSS CIMP-high tumours, none (0/32) of MSI-high CIMP-low/0 tumours, and 6% (31/476) of MSS CIMP-low/0 tumours.
Molecular features in colon cancer and patient survival
We assessed the influence of MSI, CIMP, and BRAF mutation on patient survival, independent of the clinical and other tumoural variables (table 2). Compared to patients with MSS tumours, those with MSI-high tumours experienced a significant reduction in colon cancer specific mortality in a univariate analysis (HR 0.38, 95% CI 0.22 to 0.66); however, in the multivariate model that adjusted for CIMP, KRAS, BRAF and patient characteristics, the effect of MSI-high was attenuated. This attenuation in the effect of MSI-high was principally the result of adjusting for tumour stage; when we simply adjusted for tumour stage, the HR for colon cancer-specific mortality for MSI-high tumours was 0.73 (95% CI 0.42 to 1.28).
In addition, compared to CIMP-0, CIMP-high tumours were associated with a non-significant reduction in colon cancer specific mortality in a univariate analysis (HR 0.88, 95% CI 0.57 to 1.38), which became statistically significant after adjusting for other molecular and patient characteristics (multivariate HR 0.44, 95% CI 0.22 to 0.88). The greater beneficial effect of CIMP-high status in the multivariate model was principally the result of adjusting for BRAF mutational status; when we simply adjusted for BRAF status, the HR for colon cancer-specific mortality for CIMP-high tumours was 0.45 (95% CI 0.26 to 0.79).
In both univariate and multivariate analyses, BRAF mutation was associated with a significant increase in colon cancer-specific mortality (multivariate HR 1.97, 95% CI 1.13 to 3.42). In contrast, KRAS mutation was not associated with patient outcome. Of note, the aforementioned molecular events did not significantly influence all-cause mortality.
Although statistical power was diminished for individual patient subsets, the influence of CIMP status on colon cancer-specific mortality appeared similar among patients with either early (I and II) or advanced (III and IV) pathological stages of disease (p for interaction = 0.93). Compared to CIMP-low/0 tumours, the multivariate HR for colon cancer-specific mortality in CIMP-high tumours was consistently low across all stages (I–IV) (table 3).
In contrast, any apparent effect of CIMP-high on overall mortality was limited to patients with stage III/IV disease; the multivariate HR for all-cause mortality was 1.49 (95% CI 0.66 to 3.34) for stage I/II patients and 0.58 (95% CI 0.29 to 1.15) for stage III/IV patients.
Next, we examined whether the effect of CIMP or BRAF mutation on survival differed between the cohort studies. The effect of CIMP-high did not significantly differ between the male cohort (Health Professionals Follow-up Study) and the female cohort (Nurses’ Health Study; p for interaction = 0.59). Likewise, the effect of BRAF mutation did not significantly differ between the male cohort and the female cohort (p for interaction = 0.35).
To eliminate the potential confounding effect of HNPCC, we identified 19 possible or suspected HNPCC cases; ie, MSI-high CIMP-low/0 tumours (none of which turned out to be BRAF-mutated) with any of the following: (1) positive family history of colorectal cancer in at least one first-degree relative; (2) loss of MLH1 without evidence of MLH1 methylation; (3) loss of PMS2 without evidence of MLH1 loss; (4) loss of MSH2 and/or MSH6. After we excluded these 19 cases, multivariate Cox regression analysis showed following results for colon cancer-specific mortality: HR for MSI-high, 0.68 (95% CI 0.34 to 1.35); HR for CIMP-high, 0.41 (95% CI 0.20 to 0.83); HR for BRAF mutation, 1.85 (95% CI 1.12 to 3.06). These results were similar to table 2.
We compared different CIMP panels consisting of different sets of markers. Multivariate HRs for colon cancer-specific mortality in CIMP+ vs CIMP− were as follows: HR 0.57 (95% CI 0.32 to 1.01) by a panel consisting of CACNA1G, IGF2, NEUROG1, RUNX3 and SOCS1;8 HR 0.81 (95% CI 0.55 to 1.20) by a panel consisting of CDKN2A (p16), MINT1, MINT31, MLH1 and p14;26 HR 0.74 (95% CI 0.46 to 1.20) by a panel consisting of CDKN2A, HIC1, MINT1, MINT31 and MLH1; HR 0.54 (95% CI 0.30 to 0.99) by a panel consisting of CACNA1G, CDKN2A, CRABP1, MLH1 and NEUROG1;17 HR 0.65 (95% CI 0.37 to 1.12) by a panel consisting of CACNA1G, CDKN2A, CRABP1, IGF2, MLH1, NEUROG1, RUNX3, SOCS1, IGFBP3, MINT1, MINT31, MGMT and WRN.25 These results indicate that a variation in methylation markers in CIMP panels can result in a variation in associations with patient outcome, which may, at least in part, explain the discrepancy of different studies on CIMP and patient outcome. In the current study, we utilised the validated eight-marker panel in light of our prior published work.29
Combined MSI/CIMP status and patient survival
We further stratified patients according to both MSI and CIMP status to assess the joint effect on patient outcome (table 4), because molecular classification based on MSI and CIMP status is increasingly important.38 39 Compared to patients whose tumours were both MSS and CIMP-low/0, those with CIMP-high tumours experienced a significant reduction in colon cancer-specific mortality, regardless of MSI status. A combination of MSI and CIMP determinations might differentiate ∼24% ((38+33+88)/649) of tumours (either CIMP-high or MSI-high) with good prognosis (HR estimates 0.17 to 0.40) from the other ∼76% of tumours (MSS CIMP-low/0).
Combined MSI/BRAF status and patient survival
Similarly, we stratified patients according to both MSI and BRAF status to assess joint effect on patient outcome. Compared to patients whose tumours were both MSS and BRAF-mutated, those with MSI-high/BRAF-wild-type tumours showed a significant reduction in colon cancer-specific mortality (table 4). Notably, there was no protective effect of MSI-high among BRAF-mutated tumours; compared to MSS BRAF-mutated tumours, the multivariate HR for colon cancer-specific mortality among MSI-high BRAF-mutated tumours was 1.09 (95% CI 0.48 to 2.51).
Combined CIMP/BRAF status and patient survival
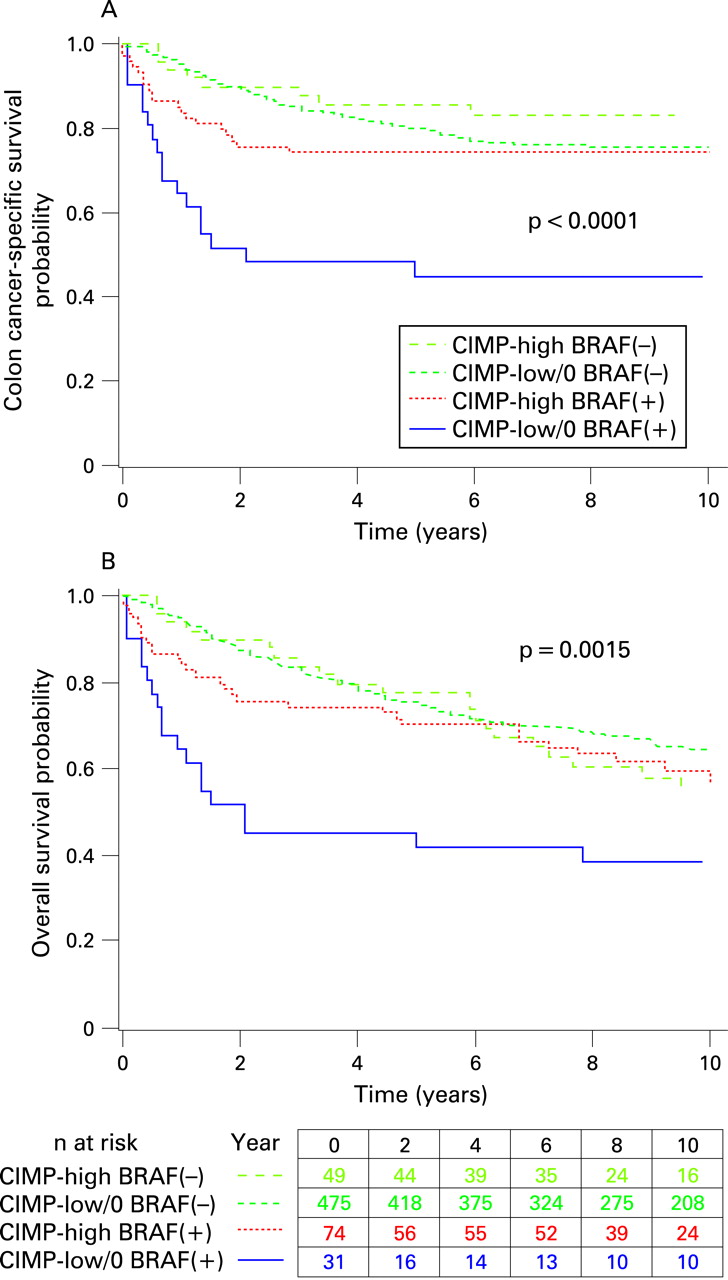
We also stratified patients according to both CIMP and BRAF status to assess the joint effect on patient outcome. Colon cancer-specific survival at 5 years was 45% for patients with CIMP-low/0 BRAF-mutated tumours, 80% for CIMP-low/0 BRAF-wild-type tumours, 74% for CIMP-high BRAF-mutated tumours, and 86% for CIMP-high BRAF-wild-type tumours (multi-group log rank p<0.0001; fig 2A). Similarly, overall survival at 5 years was lower in CIMP-low/0 BRAF-mutated tumours than the other subtypes (multi-group log rank p = 0.0015; fig 2B).
{kind=link}
{kind=link}
In a multivariate analysis, when compared to patients with CIMP-low/0 BRAF-mutated tumours, those with CIMP-high tumours demonstrated a significantly lower colon cancer-specific mortality regardless of BRAF status (table 4). Moreover, the adverse effect of BRAF mutation on patient survival was not apparent when tumours also demonstrated CIMP-high.
DISCUSSION
In this cohort of patients with colon cancer, we examined the effect of the CpG island methylator phenotype (CIMP), microsatellite instability (MSI), and BRAF mutation on patient outcome. CIMP-high status was independently associated with a low cancer-specific mortality, whereas BRAF mutation was associated with a significant increase in cancer-specific mortality. Consistent with other studies,20 we found that MSI-high tumours showed a trend towards an association with longer survival. Of note, the adverse effect of BRAF mutation appeared to be limited to tumours that were not CIMP-high. Although our observations need to be confirmed by other independent studies, the associations of CIMP and BRAF mutation with clinical outcome were consistent across the two independent prospective cohort studies in this analysis.
The relationship between CIMP, MSI, and BRAF mutations in colon cancer is complex. In our cohort, 70% of BRAF-mutated tumours exhibited CIMP-high, and 70% of CIMP-high tumours exhibited MSI-high. Among patients who do not manifest hereditary non-polyposis colorectal cancer (HNPCC), MSI-high is often the consequence of promoter methylation (and subsequent silencing) of MLH1, a DNA mismatch repair gene.8 In fact, CIMP and BRAF tests are used to exclude HNPCC among patients who exhibit MSI-high, since HNPCC seldom exhibits CIMP or BRAF mutation.8 40 41
Studying epigenetic and/or genetic alterations is increasingly important in cancer research.3 42–44 To decipher the apparently complex effect of CIMP and BRAF mutation on patient survival, we utilised a validated expanded panel of eight methylation markers for CIMP diagnosis in colorectal cancer.8 29 To determine DNA methylation status at each locus, we used a quantitative method that appears to reproducibly differentiate high-level from low-level methylation.35 Our validated criteria for CIMP-high are based on the bimodal distribution of tumours according to the number of methylated CIMP markers, and the observation that CIMP-high is associated with BRAF mutation while CIMP-low is associated with KRAS mutation.33 45 Our large sample size from the two independent cohort studies enabled us to estimate the frequencies of specific molecular features (eg, CIMP-high, etc.) and cancer death rates at the population level.
Although prognostic factors have been extensively investigated for colon cancer,20–22 46–48 previous studies of CIMP and survival in colon cancer have yielded somewhat inconsistent results.22–28 Some studies suggested an adverse effect of CIMP on survival of patients with MSS tumours.23 25 26 However, accumulating evidence has been suggested that MSI-high tumours are associated with good prognosis regardless of CIMP status,22 23 which is in agreement with our current study (table 4). BRAF mutation has been associated with worse survival in MSS tumours, but there was little prognostic value of CIMP in multivariate analysis.22 27 Our findings of good prognosis in CIMP-high tumours appear to differ from the data in the previous studies.23 25 26 These discrepant observations might have resulted from differences in patient cohorts, methylation markers, criteria for CIMP, and/or the variable inclusion of other potential confounders (such as BRAF mutation) in multivariate analysis models. In particular, we have previously observed worse prognosis associated with CIMP-high tumours in stage IV colorectal cancer in small phase I/II clinical trials.25 The two possible reasons for the discrepant results are as follows. First, a selection bias in the small clinical trials with only five CIMP-high tumours might have caused this discrepancy. Second, data in our previous study25 with only five CIMP-high tumours might simply be the result by chance in the setting of a small patient population. A p value by the log-rank test is calculated by the Mantel–Haenszel χ2 test, which can offer a far more accurate p value with a large sample size. Thus, we would emphasise the importance of a large sample size in any clinical study. Because of the use of the expanded CIMP marker panel (including the five new markers described by Weisenberger et al8) in the current study, good prognosis might be specifically associated with CIMP-high tumours defined by these new CIMP makers. Our observations of good prognosis in CIMP-high tumours appeared to be consistent across all stages (I–IV), further supporting that CIMP-high tumour is a biologically indolent subtype. In addition, we found that, after jointly examining CIMP and BRAF status, CIMP-high predicted a lower colon cancer-specific mortality (regardless of BRAF status) compared to CIMP-low/0 BRAF-mutated tumours, whereas the deleterious effect of BRAF mutation was not as evident in patients with CIMP-high tumours.
In our cohorts, data on cancer treatment are limited. Nonetheless, it is unlikely that chemotherapy use differed according to tumoural CIMP, MSI or BRAF status, especially since such data were not typically available to patients or treating physicians. It still remains a possibility that differential response to chemotherapy according to a specific molecular variable (eg, MSI) might confound our findings. Further studies are necessary to examine whether response to chemotherapy may be differentially influenced by specific molecular features in colon cancer. In addition, beyond cause of mortality, data on cancer recurrences were not available in these cohorts. Nonetheless, given the median survival for metastatic colon cancer was approximately 10–12 months during much of the time period of this study,49 colon cancer-specific mortality should be a reasonable surrogate for cancer-specific outcomes.
Despite the apparent effects of CIMP, MSI, and BRAF mutation on colon cancer-specific mortality, the influence of these tumoural events on overall mortality was markedly attenuated, which may have reflected the inclusion of earlier stage (I and II) patients in our analysis. In fact, when we limited our analysis to patients with either stage III or IV cancer, we observed similar effects of CIMP on both cancer-specific and all-cause mortality. Moreover, when we jointly classified patients according to both CIMP and BRAF status, we observed similar trends for cancer-specific and overall mortality among the entire patient cohort (table 4 and fig 2).
In conclusion, this large prospective study of colon cancer patients suggests that CIMP-high is independently associated with a low cancer-specific mortality. While BRAF mutation is associated with worse survival, CIMP-high appears to eliminate, at least in part, the adverse effect of BRAF mutation. Finally, while our data confirm the extensive body of evidence supporting a better prognosis for patients with MSI-high tumours, the good prognosis associated with MSI-high was abrogated in the presence of a BRAF mutation. Our finding that CIMP-high is an independent predictor of cancer survival may have significant clinical implications, although it needs to be confirmed by additional independent studies. Future studies to validate our observations should consider a joint examination of CIMP, MSI and BRAF mutation to decipher the role of these molecular features in biological and clinical behavior of colon cancer.
Acknowledgments
We thank the Nurses’ Health Study and Health Professionals Follow-up Study cohort participants who have generously agreed to provide us with biological specimens and information through responses to questionnaires. We thank F Speizer, W Willett, S Hankinson, G Colditz, M Stampfer, and many other staff members who implemented and have maintained the cohort studies. We deeply thank P Laird, D Weisenberger and M Campan for their assistance in the development of the MethyLight assays.
REFERENCES
Footnotes
Funding: This work was supported by The U.S. National Institute of Health grants P01 CA87969, P01 CA55075, P50 CA127003, and K07 CA122826 (to SO), the Bennett Family Fund for Targeted Therapies Research, and the Entertainment Industry Foundation National Colorectal Cancer Research Alliance. KN was supported by a fellowship grant from the Japan Society for the Promotion of Science.
Competing interests: None.
Ethics approval: This study was approved by the Human Subjects Committees at Brigham and Women’s Hospital and Harvard School of Public Health, on 30 October 2002 and 25 November 2002.
Linked Articles
- Digest