Article Text

Abstract
Background and aim Hepatic stellate cells (HSCs) are key participants in liver fibrosis development. 1,25(OH)2D3, the active form of vitamin D, has antiproliferative properties and antifibrotic potential, as well as a role in extracellular matrix and matrix metalloproteinase (MMP) regulation in renal and lung fibrosis. Little is known about the role of 1,25(OH)2D3 in liver and its involvement in liver fibrosis. Therefore, we investigated the antiproliferative and antifibrotic effects of 1,25(OH)2D3 in primary cultured HSCs and in a rat model of liver fibrosis induced by thioacetamide (TAA).
Methods Primary HSCs were isolated from rats' livers and treated with 1,25(OH)2D3. Proliferation was examined by bromodeoxyuridine. Vitamin D receptor (VDR) expression and several fibrotic markers were detected by western blot analysis and real-time PCR. Collagen Iα1 and MMP-9 promoter activity were measured by luciferase assay. MMP-9 enzymatic activity was investigated by zymography. VDR silencing was performed by sh-RNA. An in vivo study was performed on TAA-induced liver fibrosis model in rats treated with or without 1,25(OH)2D3. The fibrotic score and collagen deposition were determined by Masson and by Sirius red staining.
Results While VDR was highly expressed in quiescent HSCs, its expression decreased up to 40% during activation. Addition of 1,25(OH)2D3 to activated HSCs stimulated VDR expression. 1,25(OH)2D3 suppressed HSC proliferation and cyclin D1 expression by ∼50% and tissue inhibitor of metalloproteinase 1 (TIMP-1) by 60% and led to a 40% downregulation of collagen Iα1 expression. Moreover, 1,25(OH)2D3 increased MMP-9 activity by 30%. Silencing VDR by sh-RNA demonstrated that suppression of cyclin D1 and collagen Iα1 protein expression was VDR dependent. Treatment with 1,25(OH)2D3 significantly reduced extracellular matrix deposition and lowered the fibrotic score in TAA-induced liver fibrosis.
Conclusion 1,25(OH)2D3 has antiproliferative and antifibrotic effects on liver fibrosis in in vitro and in vivo models and may be considered as having potential therapeutic value.
- Liver fibrosis
- VDR
- PDGF
- TGF-β1
- TAA
- hepatic fibrosis
- hepatic stellate cell
- matrix metalloproteinase
- vitamin D receptor gene
- vitamins
Statistics from Altmetric.com
- Liver fibrosis
- VDR
- PDGF
- TGF-β1
- TAA
- hepatic fibrosis
- hepatic stellate cell
- matrix metalloproteinase
- vitamin D receptor gene
- vitamins
Significance of this study
What is already known about this subject?
The involvement of vitamin D in liver fibrosis has not been investigated in depth
Vitamin D receptor is expressed in liver nonparenchymal cells, such as hepatic stellate cells, which play a critical role in the development of liver fibrosis
Vitamin D plays a significant role in cell proliferation, differentiation and apoptosis in many normal and malignant cells
Antifibrotic effects of vitamin D were found in kidney, lung and mesenchymal multipotent cells
What are the new findings?
Vitamin D inhibited cyclin D1 expression and decreased proliferation of hepatic stellate cells (the latter being an important process in liver fibrosis)
Vitamin D demonstrated an antifibrotic effect by inhibition of collagen Iα1 promoter activity, mRNA and protein expression
Vitamin D increased MMP-9 enzymatic activity, which degraded extracellular matrix proteins, but had no detectable effect on MMP-9 promoter activity
Vitamin D inhibited TIMP-1 mRNA expression levels
Vitamin D receptor silencing impaired the effect of vitamin D on cyclin D1 and collagen Iα1 expression
Vitamin D ameliorated liver fibrosis in an in vivo model
How might it impact on clinical practice in the foreseeable future?
There are currently only limited effective treatments for liver fibrosis. Vitamin D may be considered as having potential therapeutic value for that pathology.
Introduction
The development of liver fibrosis is characterised by an accumulation of extracellular matrix (ECM) with subsequent destruction of the normal liver architecture, leading to liver cell dysfunction. Hepatic stellate cells (HSCs) play a critical role in the development of liver fibrosis, since they are responsible for excessive deposition of ECM proteins, predominantly type I collagen. Two main processes lead to liver fibrosis. First, HSCs become activated, resulting in increased cellular proliferation and biotransformation from a quiescent vitamin A-storing cell to an activated myofibroblast-like cell. Second, there is an increase in ECM protein synthesis and deposition, predominantly type I collagen. Multiple signalling pathways are implicated in HSC activation and proliferation. The most potent mitogenic factor for HSCs is platelet-derived growth factor (PDGF). The transforming growth factor (TGF)-β/Smad signalling pathway is the main stimulating factor for profibrotic ECM protein synthesis.1–5
Vitamin D is a prohormone that requires sequential enzymatic modification of 25- and 1α-hydroxylation in the liver and kidney, respectively, leading to its maximal biological activity as 1,25(OH)2D3. The effects of vitamin D are mediated through the vitamin D receptor (VDR) which is a ligand-dependent transcription factor belonging to the superfamily of nuclear hormone receptors. VDR binds to its ligand 1,25(OH)2D3, dimerises with the retinoid X receptor and attaches to specific genomic sequences (termed vitamin D response elements). Vitamin D has traditionally been associated with systemic calcium homeostasis and bone mineralisation. However, it is now recognised that VDRs are expressed in many tissues that are not associated with mineral or bone metabolism. The key enzyme, 25-hydroxyvitamin D 1α-hydroxylase (Cyp27B1), is responsible for the production of 1,25(OH)2D3 in many cell types.6 7 It has been suggested that the local extrarenal production of 1,25(OH)2D3 combined with VDR presence enables a local paracrine/autocrine action in various tissues. Indeed, it has been shown that 1,25(OH)2D3 regulates hundreds of different genes and plays a significant role in regulating cell proliferation, differentiation and apoptosis in many normal and cancer cells and also has immunomodulatory anti-inflammatory effects.8–13 Recent studies have indicated that 1,25(OH)2D3 also has an inhibitory effects on renal and lung fibrosis.14–16 Treatment with vitamin D led to the inhibition of lung fibroblast proliferation and showed an antifibrotic effect in murine lung and obstructed kidney models.15–17
There are currently few effective treatments for liver fibrosis. The main targets in exploring therapeutic possibilities are aimed at inhibiting the proliferation and biotransformation of HSCs or by inhibiting the transduction of the profibrotic TGF-β1/Smad3/2 signalling cascade. In the current study, we showed that 1,25(OH)2D3 has significant antiproliferative and antifibrotic effects on primary HSCs in vitro. We also demonstrated that 1,25(OH)2D3 has an antifibrotic effect in thioacetamide (TAA)-induced liver fibrosis using an in vivo model.
Materials and methods
Animals
Male retired breeder Wistar rats (300–400 g) were maintained in the animal facility of the Tel-Aviv Sourasky Medical Center on a standard rat chow diet with a 12 h light/dark cycle. The use of animals was in accordance with the NIH Policy on the care and use of laboratory animals and was approved by the Animal Use and Care Committee.
Isolation and culture of primary rat HSCs
HSCs were isolated by sequential pronase–collagenase perfusion followed by Nycodenz (Sigma-Aldrich, Inc., St. Louis, Missouri, USA) density gradient centrifugation, as described previously.5
Reagents
1,25(OH)2D3 (a generous gift from Dr Zeev Mazor, Teva Pharmaceutical Industries Ltd., Israel) was prepared in pure ethanol. A stock solution of 1 μg/ml PDGF-BB (Peprotech, Inc., Rocky Hill, New Jersey, USA) was prepared in water. TGF-β (R&D Systems, Inc., Minneapolis, Minnesota, USA) was dissolved in 4 mM HCl containing 1 mg/ml bovine serum albumin (BSA) at a concentration of 1 μg/ml. All stock reagents were aliquot and stored at −20°C until use. The final concentration of 2.5×10−6 M 1,25(OH)2D3 contained 0.01% ethanol as did the control sample.
Proliferation assay
Proliferation of HSCs was examined by the bromodeoxyuridine method (Exalpha Biological, Inc., Watertown, Massachusetts, USA). Primary HSCs were cultured for 14 days then trypsinised and plated at a density of 20 000 cells/well on 96-well plates in Eagle's minimal essential medium (DMEM) + 10% fetal calf serum (FCS). The cells were incubated for 24 h, after which they were starved in DMEM + 0.5% FCS overnight. The cells were treated with the various stimuli in medium containing 0.5% FCS. HSCs were exposed to either 30 ng/ml PDGF, 2.5×10−6 M 1,25(OH)2D3 or combination of the two. After 24 h, the cells were tested for proliferation following the manufacturer's instructions.
Western blot
HSCs were plated on 100 mm plates at a density of 2×106 cells/plate. After 24 h, the medium was changed to starvation medium (DMEM+0.5% FCS) overnight. On the following day, the cells were incubated for 24 or 48 h with different treatments according to the experiments. Total proteins were extracted by incubating the cells for 30 min on ice in radioimmunoprecipitation assay (RIPA) buffer containing a 1:100 dilution of a protease inhibitor cocktail (Sigma-Aldrich). After 20 min of centrifugation at 14 000 rpm at 4°C, extracts were normalised to total protein content, determined by the BCA Reagent (Sigma-Aldrich). Equal amounts of total protein were separated in 4–12% BT gels (Gibco-BRL Life Technologies, Grand Island, New York, USA), blotted onto Hybond C extramembranes, blocked overnight in 5% milk and incubated with antibodies against VDR, cyclin D1, α-smooth muscle actin (SMA), β-actin, glyceraldehyde 3-phosphate dehydrogenase - house keeping gene (GAPDH) (Santa Cruz Biotechnology, Santa Cruz, California, USA) and collagen Iα1 (Affinity Bioreagents, Golden, Colorado, USA) and then incubated with horseradish peroxidase-conjugated secondary antibody. Signals were later detected by chemiluminescent. Protein expression was normalised to either β-actin or GAPDH.
Transient transfection of MMP-9 promoter or collagen Iα1 promoter
HSCs were plated on six-well plates at a density of 250 000 cells/well. The cells were transfected using the Fugene 6 reagent (Roche Diagnostics, Mannheim, Germany), according to the manufacturer's instructions. The plasmids included the pCM/MMP-9/Luc plasmid containing the −670/+54 of the matrix metalloproteinase-9 (MMP-9) promoter cloned upstream of the luciferase gene (Luc) within the pCM vector (a kind gift of Dr Boyd, MD Anderson, Houston, Texas, USA) or the collagen Iα1 promoter plasmid, ColCAT3.6 (a kind gift of Dr David Row, Connecticut University, Farmington, Connecticut, USA), that contains 3520 bp of rat procollagen Iα1 promoter, followed by 115 bp of the first exon cloned upstream to the Luc within the pUC 12 vector. Cells were seeded on six-well plates 24 h before transfection, and then transfected with 2 μg of the MMP-9 promoter-luciferase plasmid or the collagen Iα1 promoter-luciferase plasmid together with 0.3 μg β-galactosidase expression plasmid (pCMVβ, Clontech, Mountain View, California, USA) as an internal control to normalise for transfection efficiency. Six hours after transfection, the cells were treated with 10 ng/ml TGF-β, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two for 24 h. The cells were harvested, and luciferase activity, which is indicative of promoter activity, was measured using a luminometer (Berthold Lumat LB 9507). β-Galactosidase activity was measured after addition of the substrate ortho-Nitrophenyl-β-galactoside (ONPG), and colour intensity was assessed using an ELISA plate reader, ELX 808.
Real-time PCR
Cells seeded on 100 mm plates were incubated with starvation medium overnight and then incubated for 24 h with different treatments according to the experiments. Total RNA was extracted by EZ-RNA kit (Biological Industries Ltd., Beit-Haemek, Israel) according to the manufacturer's instructions. One microgram of total RNA was reverse transcribed into cDNA using iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Inc.) and analysed using quantitative real-time PCR to determine the expression of collagen Iα1 and tissue inhibitor of metalloproteinase 1 (TIMP-1). Real-time PCR was carried out in a 15 μl reaction volume using ABsolute Blue SYBR Green ROX (Thermo Scientific, Epson, Surrey, UK) and the following primers: collagen Iα1 (5′-ACG TCC TGG TGA AGT TGG TC-3′; 5′-CAG GGA AGC CTC TTT CTC CT-3′), TIMP-1 (5′-CTT TGC ATC TCT GGC CTC) and β-actin (5′-GCT CTC TTC CAG CCT TCC TT-3′; 5′-CTT CTG CAT CCT GTC AGC AA-3′). To avoid amplification of genomic DNA, the primers were placed at the junction of two exons. Semiquantitative real-time PCR was done using β-actin as an internal control to normalise for gene expression.
Zymograms
HSCs were seeded on 24-well plates (80 000 cells/well). After 24 h of incubation with starvation media, cells were treated with 1,25(OH)2D3 in the presence or absence of TGF-β. The media were collected from cultured cells 24 h after treatment. The volume of loaded media was normalised to the cell number in the wells. Proteins were separated using zymography gels (Invitrogen; Gibco-BRL Life Technologies, Carlsbad, California, USA). Gels were washed twice, incubated for 30 min with renaturing buffer (Invitrogen) and then incubated in developing buffer (Invitrogen) at 37°C overnight with gentle agitation and then stained with Brilliant Blue R (Sigma-Aldrich). Destaining was performed with 30% methanol and 10% acetic acid.
Silencing of VDR by sh-RNA plasmid
To silence the expression of VDR in primary HSCs, we used sh-RNA plasmid containing the sequence rat VDR, 5′-CCTGTCCCTTCAATGGAGATT-3′, or a scrambled plasmid as a negative control containing the sequence 5′-GGAATCTCATTCGATGCATAC-3′ (SA Bioscience, Frederick, Maryland, USA). HSCs at 80% confluence were transfected with 2 μg of sh-RNA plasmids using the Fugene 6 reagent (Roche Diagnostics) according to the manufacturer's instructions. HSCs were treated with 30 ng/ml PDGF, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two for 24 h to examine whether cyclin D1 expression was affected by the silencing of VDR. HSCs were treated with 10 ng/ml TGF-β, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two for 24 h to check whether type I collagen expression was affected by the silencing of VDR. Total proteins were extracted by incubating the cells for 30 min on ice with lysis solution (Tropix, Bedford, Massachusetts, USA) containing a 1:100 dilution of protease inhibitor cocktail (Sigma-Aldrich) and 2 mM DL-Dithiothreitol (DTT). The detection of proteins was performed as described in the western blot section.
In vivo experiment
Male Wistar rats (n=20) weighing 250–300 g were obtained from Harlan Laboratories, Jerusalem, Israel. The experiment was approved by the local ethics committee. The animals were kept in an air-conditioned room at 21°C, received humane care and were given a standard rat diet and water ad libitum under standard environmental conditions. They were divided into controls (group 1, n=3), TAA treatment (group 2, n=5), 1,25(OH)2D3 treatment (group 3, n=6) and combined TAA and 1,25(OH)2D3 treatment (group 4, n=6).
Liver cirrhosis was induced by administration of TAA (Sigma Chemical Co., St. Louis, Missouri, USA) which was dissolved in saline (100 mg/ml) at a dose of 20 mg/100 g body weight. 1,25(OH)2D3 was diluted in saline (5 μg/ml) at a dose of 0.5 μg/100 g body weight. Both treatments were administered via intraperitoneal injections twice weekly for up to 10 weeks.
Fibrotic score
Histological assessment of the livers was performed on formalin-fixed, paraffin-embedded tissue sections after staining with H&E or Mason trichome. Evaluation of fibrosis was based on the Ludwig and Batts staining system18 using the following parameters: portal fibrosis (stage 1) characterised by mild fibrous expansion of portal tracts; periportal fibrosis (stage 2) characterised by fine strands of connective tissue in zone 1 with only rare portal–portal septa; septal fibrosis (stage 3) characterised by connective tissue bridges that link portal tracts with other portal tracts and central veins, but without regenerative nodules; and cirrhosis (stage 4) characterised by bridging fibrosis and nodular regeneration.
Quantification of collagen content
Collagen content quantification was performed on formalin-fixed, paraffin-embedded tissue sections after staining with Sirius red. Sirius red-positive areas were measured using the ImageJ software program at ×100 magnification.
Statistical analysis
The results are presented as fold induction compared to control values, considered as being 100%, and are presented as mean±SE from three separate experiments. Statistical significance was assessed by the Microsoft Excel software using an unpaired two-tailed Student t test, with p<0.05 considered significant.
Results
Expression of VDR in primary HSCs
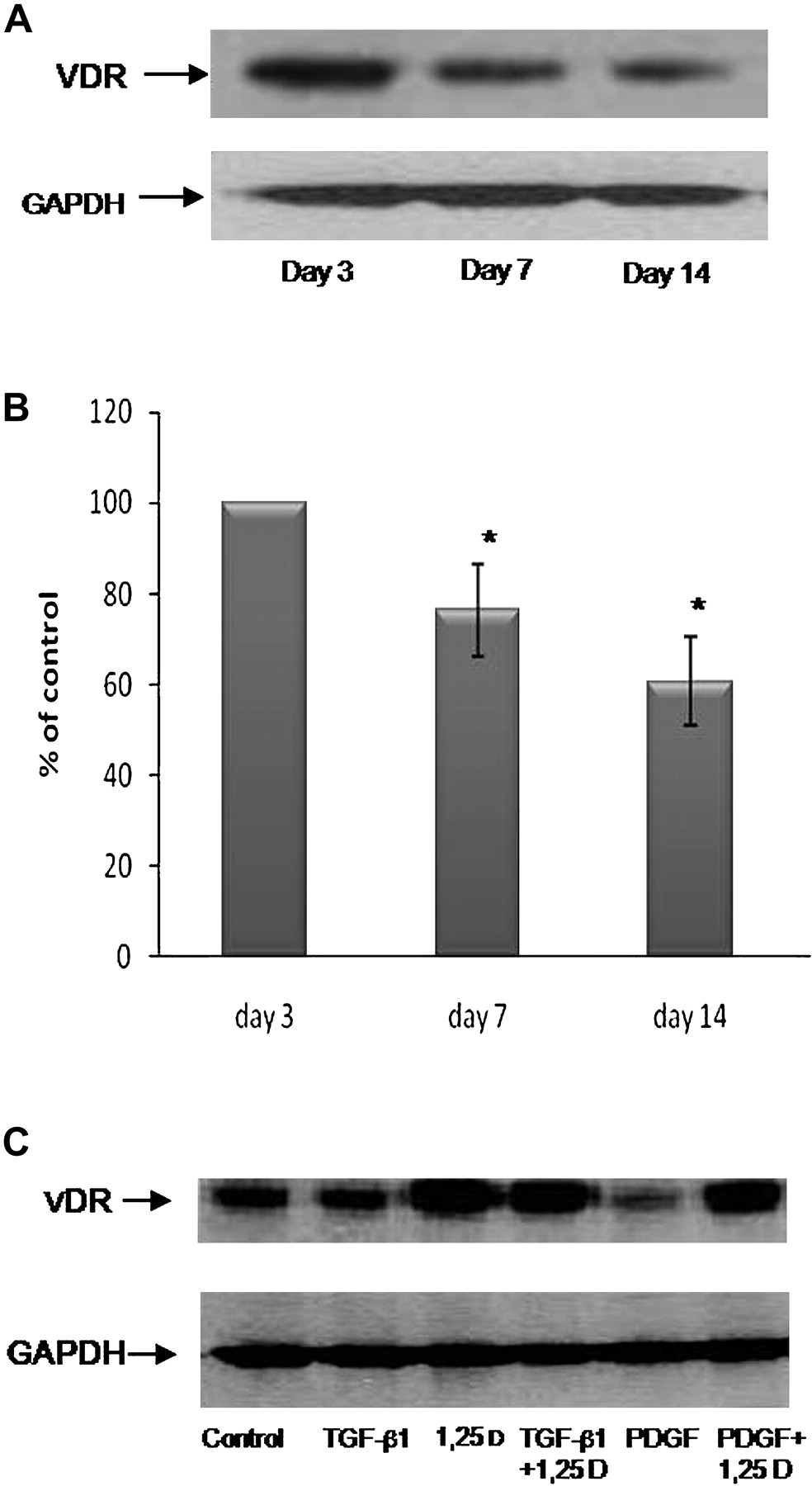
Our first goal was to determine VDR protein expression levels during HSC activation. Since primary HSCs undergo spontaneous, time-dependent activation in culture, we compared VDR expression levels on quiescent HSCs (day 3) on activated HSCs (day 7) and on fully activated HSCs (day 14). VDR expression was very high on day 3, moderate on day 7 and decreased (by 40%) on day 14 when HSCs are fully activated and differentiated to myofibroblasts (figure 1A,B). We used fully activated HSCs (ie, on day 14 after isolation) for the remaining experiments. Treatment with 2.5×10−6 M 1,25(OH)2D3 for 24 h stimulated VDR expression levels on day 14. PDGF treatment decreased VDR expression, but not to a level of significance (figure 1C).

Expression of VDR in culture-activated primary rat HSCs. Western blot analysis showing the changes in VDR expression on days 3, 7 and 14 in culture (A). Histogram showing the average±SE of densitometry results from five independent experiments. *p<0.05 vs control (B). Primary cultured HSCs were incubated for 24 h with 10 ng/ml TGF-β, 2.5×10−6 M 1,25(OH)2D3, a combination of TGF-β with 1,25(OH)2D3, 30 ng/ml PDGF or a combination of PDGF and 1,25(OH)2D3 (C).
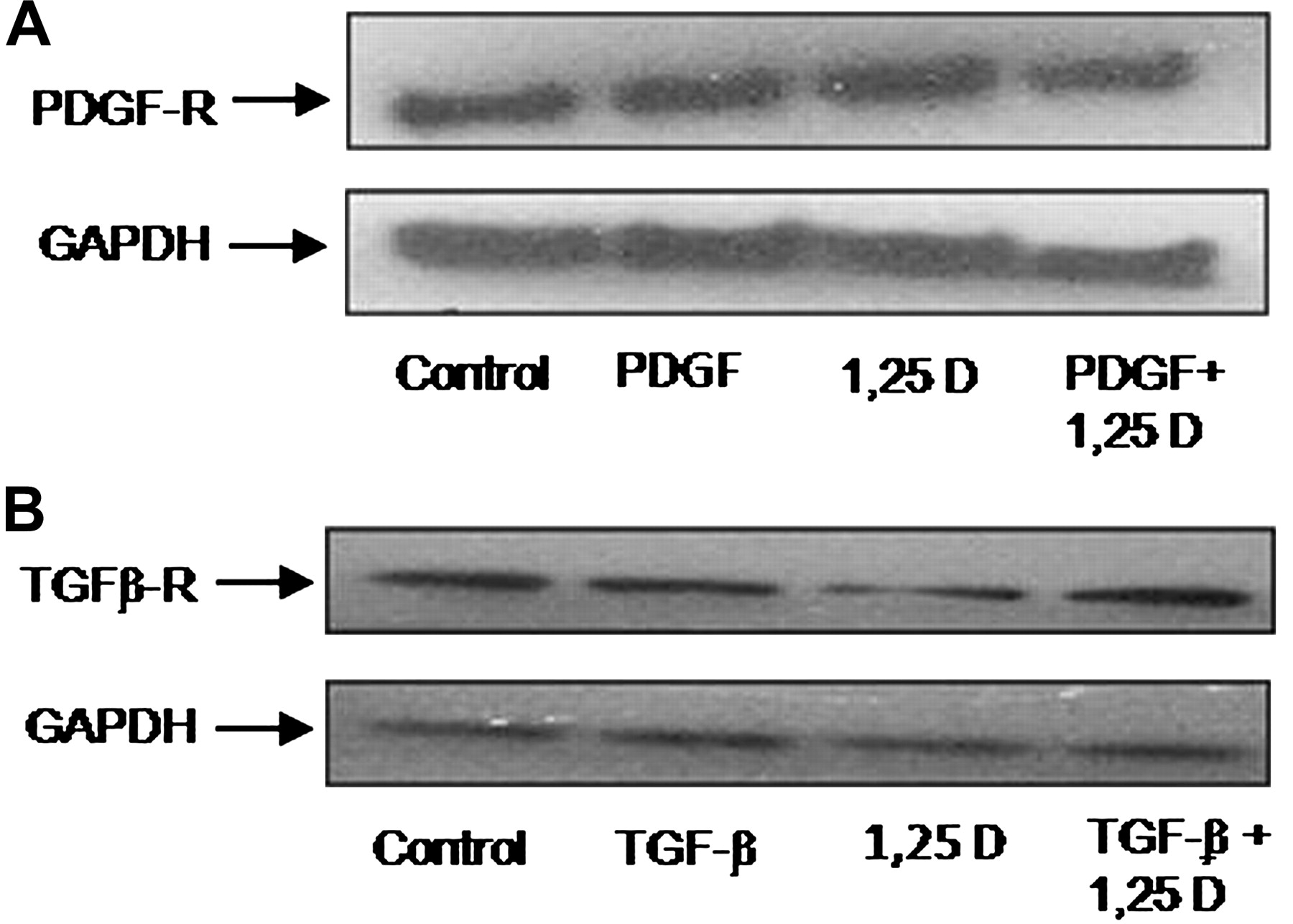
The effect of 1,25(OH)2D3 was also examined on PDGF receptor (PDGF-R) and TGF-β receptor (TGF-β-R) expressions. HSCs were treated by either PDGF or TGF-β, 1,25(OH)2D3 or a combination of the two for 24 h. PDGF-R and TGF-β-R protein expression levels were measured using western blot analysis. As expected, the results revealed that PDGF-R (figure 2A) and TGF-β-R (figure 2B) were expressed in HSC myofibroblasts, but no changes in the protein expression levels were observed following treatment with 1,25(OH)2D3.

1,25(OH)2D3 did not change expression of PDGF-R or TGF-β-R. Representative western blot showing PDGF-R (A) and TGF-β-R (B) in rat primary HSCs cultured for 14 days, treated with PDGF/TGF-β in the presence or absence of 2.5×10−6 M 1,25(OH)2D3.
Inhibition of HSC proliferation by 1,25(OH)2D3
We first assessed the cytotoxic effect of 1,25(OH)2D3 and found that treatment of 1,25(OH)2D3 at 2.5×10−5 M was 80% lethal, while lower concentrations (2.5×10−6 M, 2.5×10−7 M and 2.5×10−8 M) were only 10% lethal, that is, the same as untreated HSCs (data not shown). Cell proliferation was measured following stimulation with PDGF, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two for 24 h (figure 3). PDGF increased HSC proliferation up to ∼150%, while the combined treatment of PDGF and 2.5×10−6 M 1,25(OH)2D3 decreased HSC proliferation to the control level (∼50% suppression). Interestingly, treatment with 1,25(OH)2D3 alone decreased cell proliferation by ∼40% compared to control levels, suggesting that 1,25(OH)2D3 treatment suppresses proliferation of both PDGF-stimulated and unstimulated HSCs. 1,25(OH)2D3 at a concentration of 2.5×10−7 M showed a slight, nonsignificant effect on proliferation (data not shown).

1,25(OH)2D3 inhibited proliferation of rat primary HSCs in the presence or absence of PDGF. HSCs were plated in 96-well plates. After 24 h, the medium was changed to starvation medium (DMEM with 0.5% FCS) overnight. The next day, 1,25(OH)2D3 was added at a dose of 2.5×10−6 M, with or without 30 ng/ml PDGF. After incubation for 24 h, cell proliferation was assessed using the bromodeoxyuridine assay, and the plates were read using an ELISA reader at 450 nm. Data are expressed as mean±SE. *p<0.05 vs control, **p<0.05 vs PDGF.
Downregulation of cyclin D1 expression by 1,25(OH)2D3
Since the proliferation assay showed that 1,25(OH)2D3 treatment led to inhibition of PDGF-stimulated HSC proliferation, we measured protein expression levels of cyclin D1 in primary HSCs following exposure to PDGF, 1,25(OH)2D3 or a combination of the two after 24 h. PDGF treatment increased the expression of cyclin D1 level up to 150% compared to the control treatment, while the addition of 1,25(OH)2D3 to PDGF-treated cells led to inhibition of cyclin D1 expression by 50% (figure 4). These results correlate with those obtained in the cell proliferation assay displayed in figure 3.

1,25(OH)2D3 decreased cyclin D1 expression in primary rat HSCs. HSCs were incubated for 24 h with 30 ng/ml PDGF, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two. Western blot analysis showed an increase of cyclin D1 expression in the presence of PDGF (A). Histogram showing average±SE of densitometry results from three independent experiments (B). *p<0.01 vs control, **p<0.01 vs PDGF.
Effect of 1,25(OH)2D3 on collagen Iα1 promoter activity, mRNA and protein expression levels
To investigate whether 1,25(OH)2D3 has an antifibrotic effect on HSCs, type I collagen expression was examined at three levels: collagen Iα1 promoter activity, mRNA and protein expression. TGF-β treatment increased promoter activity by ∼40%, while the addition of 1,25(OH)2D3 to TGF-β-treated HSCs suppressed promoter activity from 140% (TGF-β alone) to the control level (figure 5A). Similar results were obtained at the mRNA level using quantitative real-time PCR. TGF-β-stimulated HSCs increased mRNA expression by ∼40%, while the addition of 1,25(OH)2D3 to TGF-β decreased mRNA expression by ∼70%. Treatment with 1,25(OH)2D3 alone also suppressed collagen Iα1 mRNA expression by 30% (figure 5B), and the same effect was noted with type I collagen protein expression level (figure 5C). The effect of 1,25(OH)2D3 on collagen I protein production was also examined after 48 h, and the results were similar to those seen after 24 h (data not shown).

Suppression of collagen Iα1 promoter activity, mRNA and type I collagen protein expression by 1,25(OH)2D3. Primary rat HSCs were transfected with the collagen Iα1 promoter-luciferase plasmid together with a β-galactosidase expression vector after 14 days in culture. Following transfection, the cells were treated overnight with either 10 ng/ml TGF-β, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two reagents. Luciferase values normalised for β-galactosidase are expressed as a percentage of the luciferase activity. Experiments were performed nine times, each in duplicate. Data are expressed as mean±SE. *p<0.01 vs control, **p<0.01 vs TGF-β (A). Total RNA was isolated from HSCs treated overnight with either 10 ng/ml TGF-β, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two and analysed by quantitative real-time PCR using primers specific to collagen Iα1. The results were normalised to β-actin mRNA expression levels. Data are expressed as mean±SE. *p<0.05 vs TGF-β (B). Western blot analysis of primary cultured HSCs that were incubated for 24 h with 10 ng/ml TGF-β, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two (C).
1,25(OH)2D3 upregulates MMP-9 enzyme activity
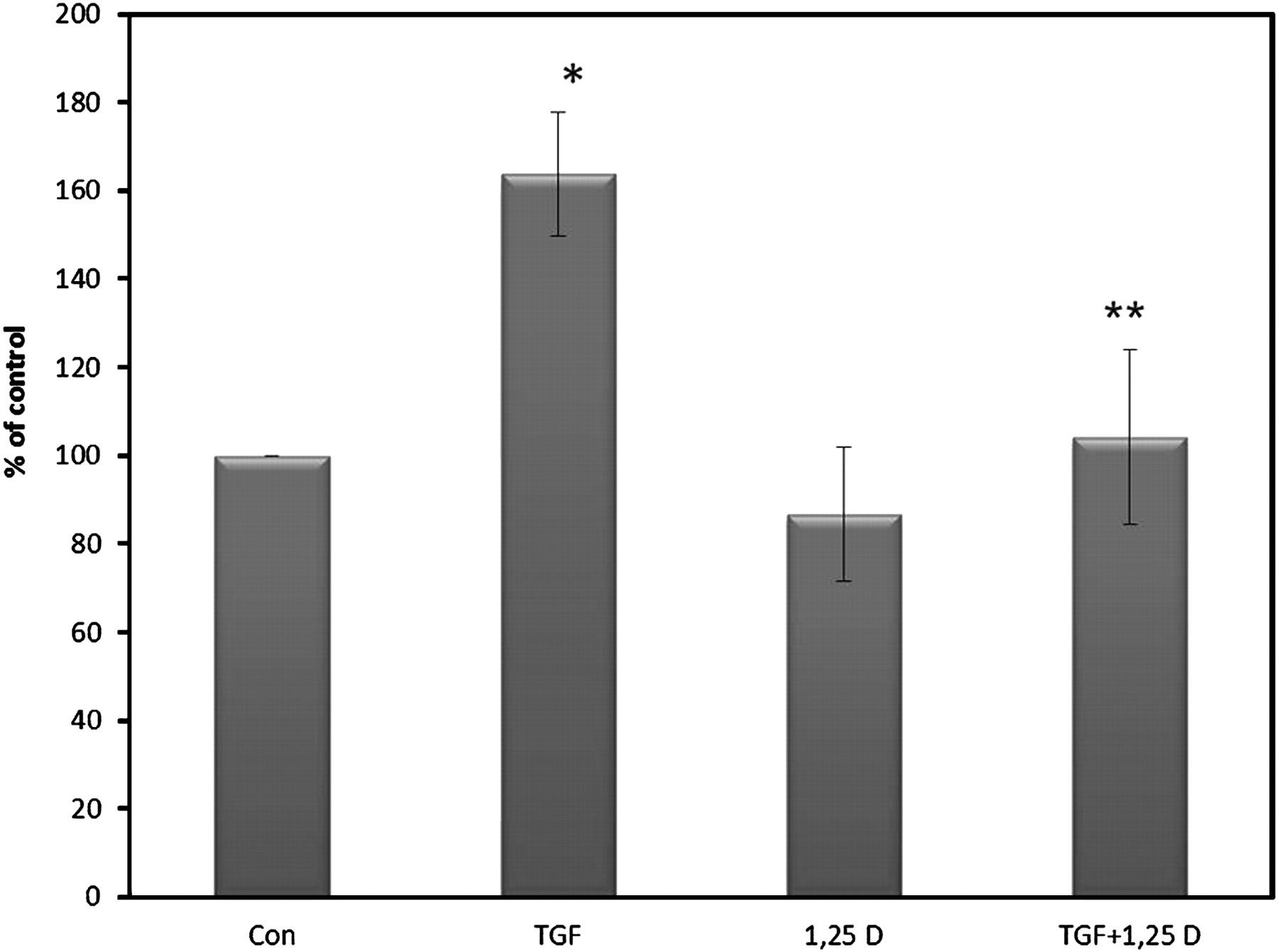
Cotransfection experiments were performed to further determine whether 1,25(OH)2D3 affects MMP-9 at the transcriptional level. TGF-β treatment decreased promoter activity by 50%. The addition of 1,25(OH)2D3 to TGF-β1-stimulated HSCs failed to increase MMP-9 promoter activity above the TGF-β1 suppression levels. Furthermore, 1,25(OH)2D3 alone was unable to increase MMP-9 promoter activity above control levels (figure 6A). In contrast, zymography analysis showed that 1,25(OH)2D3 did affect MMP-9 enzyme activity which was decreased by ∼70% in the presence of TGF-β1 (figure 6B). The addition of 1,25(OH)2D3 increased MMP-9 enzymatic by 30% in the presence of TGF-β1. MMP-2 enzyme activity also showed a similar trend, but to a lesser extent (figure 6B).

1,25(OH)2D3 did not affect MMP-9 promoter activity but increased MMP-9 and MMP-2 enzyme activity. After 14 days in culture, primary rat HSCs were transfected with the MMP-9 promoter-luciferase plasmid together with a β-galactosidase expression vector used as an internal control. After transfection, cells were treated overnight with either 10 ng/ml TGF-β, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two. Luciferase values normalised to β-galactosidase are expressed as a percentage of the luciferase activity. Experiments were performed six to nine times, each in duplicate. Data are expressed as mean±SE. *p<0.001 vs control (A). A representative zymogram result showing enzyme activity of MMP-9 and MMP-2 after treatment with either 10 ng/ml TGF-β, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two (B).
Lack of 1,25(OH)2D3 effect on α-SMA expression level
Following the inhibitory effect of 1,25(OH)2D3 on type I collagen expression and decreased MMP-9 enzyme activity, we investigated its effect on α-SMA levels. HSCs were treated with either TGF-β, 1,25(OH)2D3 or a combination of the two for 24 h. α-SMA protein expression levels were measured by western blot analysis: α-SMA expression levels did not change following 1,25(OH)2D3 treatment (figure 7).

1,25(OH)2D3 did not change expression of α-SMA. Representative western blot showing α-SMA protein expression. Primary rat HSCs cultured for 14 days, treated with TGF-β in the presence or absence of 2.5×10−6 M 1,25(OH)2D3.
Effect of 1,25(OH)2D3 on TIMP-1 mRNA expression level
HSCs were treated with either TGF-β, 1,25(OH)2D3 or a combination of the two for 24 h. TGF-β increased TIMP-1 mRNA expression by ∼60% (figure 8), while the addition of 1,25(OH)2D3 to TGF-β1-stimulated HSCs suppressed TIMP-1 expression to control levels.

Suppression of TIMP-1 mRNA expression by 1,25(OH)2D3. Total RNA was isolated from primary rat HSCs treated overnight with either 10 ng/ml TGF-β, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two and then analysed by quantitative real-time PCR using primers specific to TIMP-1. The results were normalised to β-actin mRNA expression levels. Data are expressed as mean±SE. *p<0.05 vs control, **p<0.05 vs TGF-β.
1,25(OH)2D3 operating via VDR
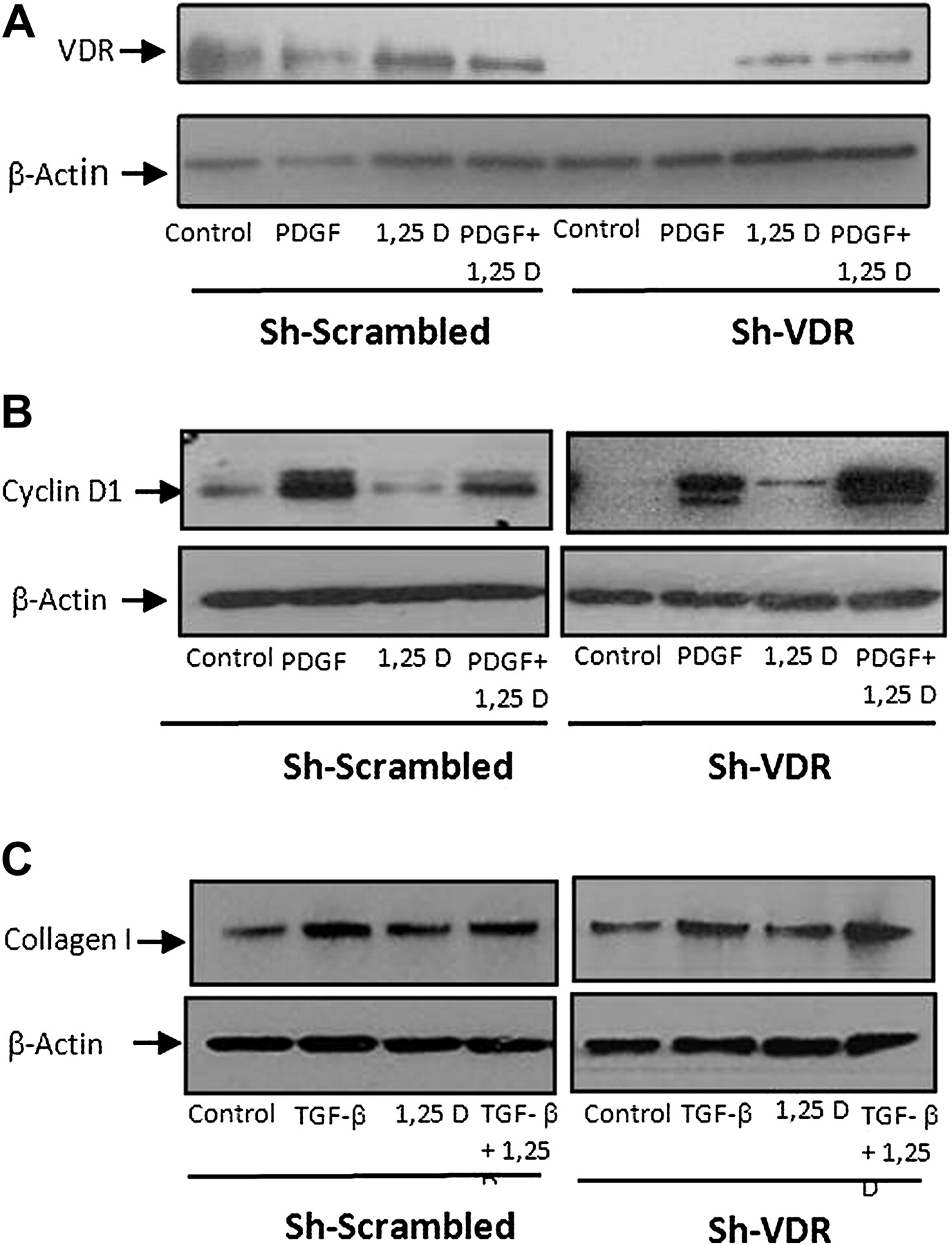
We investigated whether 1,25(OH)2D3 operates via the VDR. To effectively silence the expression of VDR, we transfected primary HSCs with an sh-RNA plasmid directed against the VDR and used a scrambled plasmid for negative control. The sh-VDR successfully suppressed VDR expression (figure 9A). Treatment with 1,25(OH)2D3 barely increased the VDR expression after an sh-VDR plasmid transfection; however, HSCs transfected with the scrambled control plasmid sustained the ability to increase VDR expression following 1,25(OH)2D3 treatment.
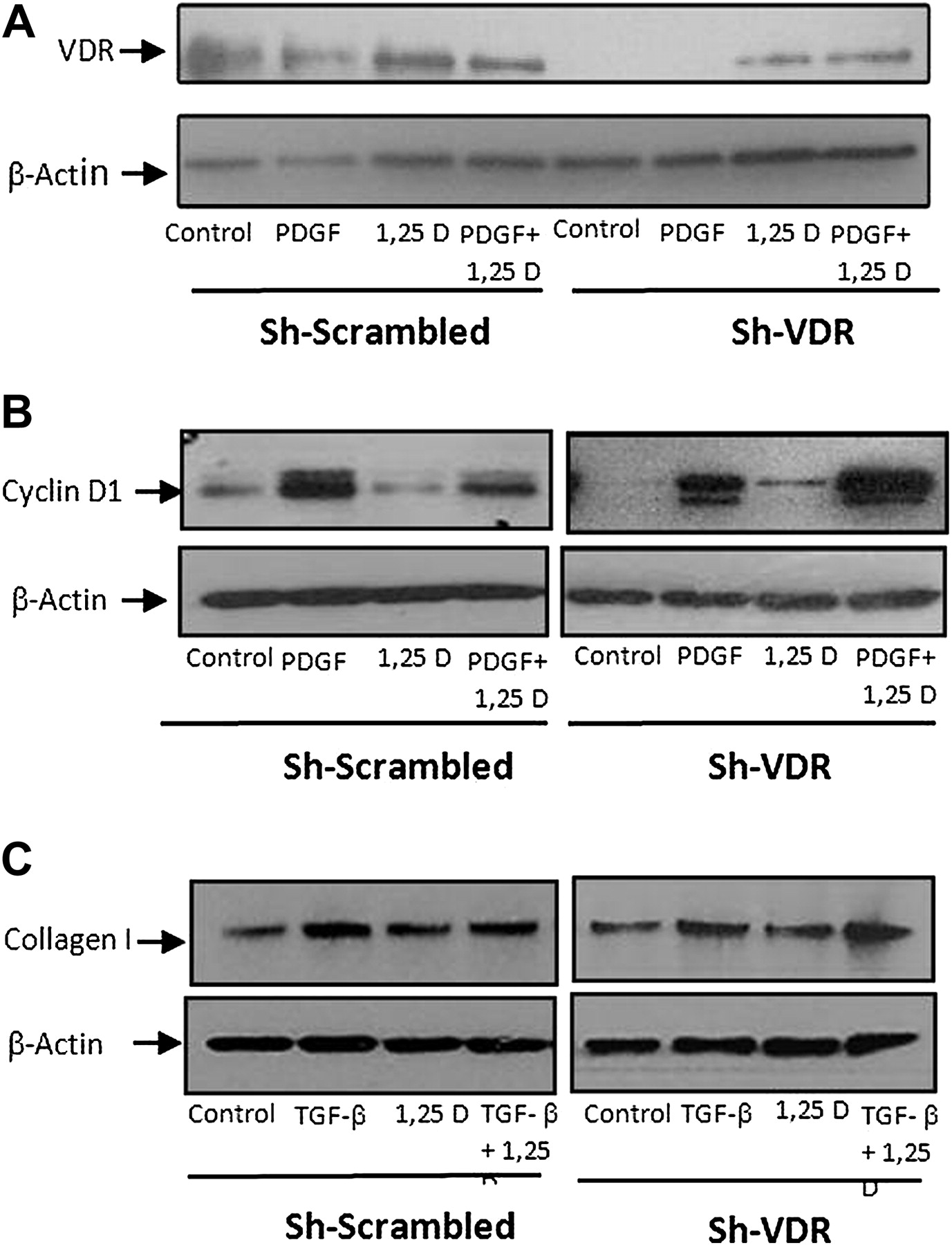
Suppression of cyclin D1 and type I collagen by 1,25(OH)2D3 through VDR. Primary rat HSCs were transfected with sh-VDR for VDR silencing or with sh-scramble as a negative control. Representative western blot showing VDR expression following treatment with 30 ng/ml PDGF, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two (A). Representative western blot showing cyclin D1 expression following treatment with 30 ng/ml PDGF, in the presence or absence of 2.5×10−6 M 1,25(OH)2D3 (B). Representative western blot showing type I collagen protein expression following incubation for 24 h with 10 ng/ml TGF-β, 2.5×10−6 M 1,25(OH)2D3 or a combination of the two (C).
Treatment with PDGF or TGF-β increased cyclin D1 or type I collagen expression in HSCs, respectively, while the addition of 1,25(OH)2D3 suppressed their expression levels. In contrast, while VDR expression was silenced in HSCs transfected with the sh-VDR plasmid, there was no decrease in cyclin D1 or type I collagen expression. HSCs transfected with scrambled plasmid showed similar results as those obtained from untransfected cells (figure 9B,C), indicating that the effect of 1,25(OH)2D3 is mediated by VDR.
Amelioration of TAA-induced liver fibrosis in rats by 1,25(OH)2D3
We examined the effect of 1,25(OH)2D3 in a liver fibrosis model following TAA induction.
The fibrotic score was determined by H&E and Masson trichrome staining (table 1). Histological examination of untreated control rats and those treated with 1,25(OH)2D3 alone revealed normal liver morphology (a fibrotic score of 0) (figure 10). In contrast, rats treated with TAA displayed a periportal fibrosis characterised by portal–portal septa surrounding the hepatic lobules (a mean fibrotic score of 3.9±0.1). Rats treated with the combination of TAA and 1,25(OH)2D3 demonstrated only mild fibrosis (a mean fibrotic score of 2.6±0.4) (p<0.05). Quantification of collagen content following Sirius red staining yielded similar results. The amount of collagen in rats treated with TAA was 16-fold higher compared to the untreated group. However, administration of TAA combined with 1,25(OH)2D3 significantly decreased collagen levels by fourfold compared to the TAA group, strongly suggesting that the administration of 1,25(OH)2D3 ameliorates liver fibrosis (figure 11).
Evaluation of the fibrotic score following treatment of TAA, 1,25(OH)2D3 and a combination of the two

1,25(OH)2D3 prevented TAA-induced liver fibrosis and decreased ECM deposition, liver morphology demonstrated by H&E staining (×100 magnification) (A) and ECM production detected by Masson trichrome staining (×100 magnification, TAA ×200 magnification) of rat livers after 10 weeks of treatment with TAA, 1,25(OH)2D3 and combined treatment of the two (B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1,25(OH)2D3 suppressed collagen deposition in a TAA-induced fibrosis model. Collagen content assessed by Sirius red-stained sections of rat livers after 10 weeks of treatment with TAA, 1,25(OH)2D3 and combined treatment of the two (×100 magnification) (A). Quantification of fibrosis by Image J analysis of Sirius red-stained sections (B). *p<0.05 vs TGF-β.
Discussion
Our understanding of the function of vitamin D has expanded over the past three decades, and many unexpected nonclassical biological actions have been uncovered. VDR and 25-hydroxyvitamin D hydroxylase, the enzyme responsible for the production of 1,25(OH)2D3, are present in many normal and malignant cell types which are not associated with calcium or bone metabolism.6 7 It has been suggested that 1,25(OH)2D3 produced in the kidney plays a hormonal role as a major regulator of calcium homeostasis, whereas the local extrarenal production of 1,25(OH)2D3 combined with the presence of VDR enables a local paracrine/autocrine action in various tissues. Indeed, 1,25(OH)2D3 regulates hundreds of different genes and plays a significant role in the modulation of cell proliferation, differentiation and apoptosis in many normal and malignant cells.8 This diverse biological function enables vitamin D to moderate the immune system and to influence the pathophysiology of cancer and cardiovascular disease.6 8 Epidemiologic studies have associated vitamin D deficiency with an increased risk of colorectal and breast cancers8–10 and autoimmune disease, such as multiple sclerosis11 and cardiovascular events.12 13
Recent studies have demonstrated that vitamin D also has antifibrotic effects in the kidney and lung. Ramirez et al14 tested the effects of 1,25(OH)2D3 on murine lung fibroblasts treated with TGF-β1 and found that 1,25(OH)2D3 inhibited TGF-β1-induced fibroblast proliferation and α-SMA expression, and prevented the upregulation of fibronectin and collagen expression. 1,25(OH)2D3 also inhibited the transdifferentiation of TGF-β1-stimulated lung epithelial cells into myofibroblasts.
Tan et al15 found that paricalcitol (19-nor-1,25 hydroxyvitamin D2), a synthetic analogue of vitamin D, significantly reduced the fibrotic lesions in a mouse obstructed kidney model, as demonstrated by reduced expression and deposition of interstitial matrix components, including fibronectin and type I and III collagen. Zhang et al16 recently subjected VDR-null mice to unilateral kidney obstruction for 7 days. Compared with wild-type mice, the VDR-null mice developed more severe renal damage and interstitial fibrosis in the obstructed kidney as well as increased deposition of collagen and other ECM proteins. Their study emphasised the importance of a functional VDR in the suppression of the fibrotic process.
The role of vitamin D in the liver remains largely unexplored. Clinical observations have recently demonstrated that 25-OH D serum levels were significantly lower in patients with chronic hepatitis C than in controls and that low 25-OH D serum levels were associated with more severe fibrosis and lower responsiveness to interferon-based therapy in those patients.17 Other studies have shown that low 25-OH D serum levels are associated with poor liver function and more advanced stages of liver fibrosis in hepatitis C virus patients.19 Moreover, a possible role for vitamin D in liver fibrosis has gained further support from the finding that VDR is expressed in human as well as in rat liver nonparenchymal cells, such as HSCs.20 It has recently been suggested that VDR polymorphism is associated with primary biliary cirrhosis.21
Based on the finding of these studies, we examined the hypothesis that vitamin D may exhibit similar antiproliferation and antifibrotic effects in HSCs. First, we showed that VDR expression is regulated by the state of HSC activation. The highest level of VDR was observed on day 3 following HSC activation and progressively declined after 14 days in culture. The declining pattern of VDR had been observed in other nuclear hormone receptors. Hellemans et al22 showed that culture activation of HSCs resulted in a gradual reduction of retinoic acid receptors and retinoid X receptors. Zvibel et al23 found that the expression of thyroid hormone receptor-α was also downregulated following HSC activation. She et al24 demonstrated that peroxisome proliferation-activated receptor-γ was reduced in activated HSCs and that overexpression of these receptors in activated HSCs induced a quiescent phenotype. Interestingly, VDR expression in HSCs was upregulated following the addition of 1,25(OH)2D3. In contrast, two stimulatory factors of HSCs, PDGF and TGF-β1, had no effect on VDR expression.24 Similar results were found by Gascon-Barre et al20 who showed that in vivo administration of 1,25(OH)2D3 upregulates the expression of VDR gene transcription in HSCs.
Second, we showed that 1,25(OH)2D3 inhibited the proliferation of PDGF-stimulated HSCs by 50%. We confirmed that this inhibition was not due to a cytotoxic effect of 1,25(OH)2D3. Previous studies on normal and malignant cells demonstrated that 1,25(OH)2D3 inhibits cell proliferation by suppression of cell cycle progression, mainly by G0/G1 arrest. We showed that 1,25(OH)2D3 suppressed the expression of cyclin D1, a key factor in the progression of cell cycle in activated HSCs.25 26
Third, we examined the antifibrotic effect of 1,25(OH)2D3 on the expression of different profibrotic and antifibrotic markers, such as collagen Iα1, α-SMA, MMP-9 and TIMP-1. 1,25(OH)2D3 treatment led to significant inhibition of collagen Iα1 at all three regulatory levels: promoter activity, mRNA expression and protein production.
Unlike the inhibitory effect of 1,25(OH)2D3 on α-SMA expression that was found in renal and lung fibrosis,14 27 we found no such effect on α-SMA expression in HSCs. These contradictory results may be related to the different cell types or may result from the fact that we used fully activated HSCs that already expressed high levels of α-SMA. Therefore, it is possible that the addition of 1,25(OH)2D3 could not overcome the high levels of α-SMA expression in activated HSCs.
The antifibrotic effect of 1,25(OH)2D3 was also demonstrated by upregulation of MMP-9 activity, although it was not observed at the promoter level but rather on the enzyme activity level. Several studies have reported that vitamin D has a regulatory function on MMP expression. Vitamin D, however, promotes both up- and downregulation of MMP activity, depending on the specific cell type. For example, 1,25(OH)2D3 decreased MMP-9, while it increased the activity of its counterparts, TIMP-1, in prostate cancer cells.28 Interestingly, it did not suppress MMP-9 expression at the transcriptional level but reduced the stability of its mRNA.28 Examination of cardiovascular cells revealed that MMP-2 and MMP-9 were upregulated in VDR knockout mice.29 In contrast, several other studies have confirmed that vitamin D stimulates MMPs. 1,25(OH)2D3 induced MMP2 activation in human osteoblast-like cells by upregulating membrane-type matrix metalloproteinase-I (MTI)-MMP, which may contribute to bone formation and resorption.30 In addition, 1,25(OH)2D3 stimulated vascular endothelial growth factor (VEGF) and MMP-9 gene expression and promoted neovascularisation of the epiphysial growth plate.31
TIMP-1 is another profibrotic marker known to be correlated with hepatic fibrosis.32 Therefore, we investigated TIMP-1 expression following 1,25(OH)2D3 treatment and found that TGF-β stimulated TIMP-1 mRNA levels by 60%, while the addition of 1,25(OH)2D3 significantly decreased TIMP-1 mRNA to the control level. These findings suggest that 1,25(OH)2D3 has both antifibrotic and profibrolytic effects.
Our current study findings confirmed that HSCs are indeed capable of responding to 1,25(OH)2D3 through a functional VDR. Using the sh-mRNA for VDR, we were able to demonstrate that silencing of VDR expression results in the inability of 1,25(OH)2D3 to decrease cyclin D1 and type I collagen expression. This indicates that the effects of 1,25(OH)2D3 on HSCs are mediated by VDR.
Finally, our in vivo experiment revealed that 1,25(OH)2D3 administration, as a preventive treatment, significantly inhibited the development of liver fibrosis, as shown by improved liver morphology and decreased collagen deposition.
The present study and previous ones on lung cells and the kidney indicate that the biological pathways through which vitamin D treatment influences the fibrotic process in various tissues have many similarities. Vitamin D inhibits the proliferation of specific activated cells responsible for profibrotic activity in these tissues, suppresses the expression of collagen and other profibrotic ECM proteins and stimulates the expression of antifibrotic factors.
Recent studies in mesenchymal multipotent cells demonstrated that vitamin D contains an antiproliferative effect by inducing cell cycle arrest and promoting the accumulation of cells in the G0/G1 phase without inducing apoptosis.33 Moreover, vitamin D reduced the collagen expression and other key profibrotic factors and increased the expression of antifibrotic factors, such as BMP7 and MMP8, in those cells.34 Those results support the notion that vitamin D has a potential preventive capacity to inhibit fibrosis in various tissues. The precise molecular mechanism by which 1,25(OH)2D3 exerts its antifibrotic effect remains to be determined. Several studies have indicated that 1,25(OH)2D3 suppresses the profibrotic TGF-β1/Smad 2/3 signalling pathway and upregulates the antifibrotic BMP7 and Smad1/5/8 and Smad7 pathways.35 36 Aschenbrenner et al37 demonstrated coimmunoprecipitation of VDR and Smad3, suggesting that vitamin D may affect expression within the TGF-β1/Smad3 signalling pathway and alter the effect of Smad3 on fibrosis.
Based on the results we obtained from the in vitro studies, 1,25(OH)2D3 appears to have antiproliferative and antifibrotic effects on liver fibrosis via ligation to the VDR. Moreover, our in vivo experiment confirms that 1,25(OH)2D3 prevents the development of liver fibrosis and should be considered as a potential therapeutic option for hepatic fibrosis.
Acknowledgments
We would like to thank Esther Eshkol for her editorial assistance.
References
Footnotes
Shirley Abramovitch and Liora Dahan-Bachar contributed equally to the work and should be considered ‘first authors’.
Funding This study was supported by grant no. 3000-4894 from the Chief Scientist of the Ministry of Health, Israel.
Competing interests None to declare.
Provenance and peer review Not commissioned; externally peer reviewed.