


Abstract
Bcr-Abl-independent signaling pathways are known to be involved in imatinib resistance in some patients with chronic myelogenous leukemia (CML). In this study, to find new targets for imatinib-resistant CML displaying loss of Bcr-Abl kinase target dependence, we isolated imatinib-resistant variants, K562/R1, K562/R2, and K562/R3, which showed profound declines of Bcr-Abl levels and its tyrosine kinase activity, from K562 cells. Importantly, the imatinib resistance mechanism in these variants also included aberrant acetylation of nonhistone proteins such as p53, Ku70, and Hsp90 that was due to upregulation of histone deacetylases (HDACs) and down-regulation of histone acetyltransferase (HAT). In comparison with K562 cells, the imatinib-resistant variants showed up-regulation of HDAC1, -2, and -3 (class I HDACs) and class III SIRT1 and down-regulation of CBP/p300 and PCAF with HAT activity, and thereby p53 and cytoplasmic Ku70 were aberrantly acetylated. In addition, these were associated with down-regulation of Bax and up-regulation of Bcl-2. In contrast, the class II HDAC6 level was significantly decreased, and this was accompanied by an increase of Hsp90 acetylation in the imatinib-resistant variants, which was closely associated with loss of Bcr-Abl. These results indicate that alteration of the normal balance of HATs and HDACs leads to deregulated acetylation of Hsp90, p53, and Ku70 and thereby leads to imatinib resistance, suggesting the importance of the acetylation status of apoptosis-related nonhistone proteins in Bcr-Abl-independent imatinib resistance. We also revealed that imatinib-resistant K562 cells were more sensitive to suberoylanilide hydroxamic acid, an HDAC inhibitor, than K562 cells. These findings may have implications for HDAC as a molecular target in imatinib-resistant leukemia cells.
The constitutively activated Bcr-Abl tyrosine kinase has been defined as the pathogenic principle of Philadelphia-positive human leukemias (Deininger et al., 2000). Imatinib mesylate (imatinib) is a tyrosine kinase inhibitor that competitively inhibits ATP binding in the kinase domains of both the Bcr-Abl and c-Abl kinases (Druker et al., 2001). However, the acquired resistance to imatinib in patients leads to a serious clinical problem. It has been suggested that resistance to imatinib stems from Bcr-Abl gene amplification, leading to overexpression of Bcr-Abl protein or point mutations in the Bcr-Abl gene (le Coutre et al., 2000; Gorre et al., 2001; Shah et al., 2002). However, several groups suggested recently that there might be other forms of Bcr-Abl-independent imatinib resistance (Nimmanapalli et al., 2002; Dai et al., 2004; Donato et al., 2004). Specifically, Bcr-Abl-positive chronic myelogenous leukemia (CML) cells cultured in the continuous presence of imatinib display a decline in Bcr-Abl protein and/or mRNA levels and a corresponding increase in expression/activity of the Lyn and Hck kinases (Dai et al., 2004). Indeed, imatinib resistance in some patients mediated through loss of kinase target dependence and acquired Bcr-Abl-independent signaling characteristics has been reported (Donato et al., 2004).
Recently, it has been reported that changes in histone deacetylase (HDAC) expression in leukemic cells could be involved in mechanisms for abnormal cellular proliferation that operate through chromatin-independent pathways (Bradbury et al., 2005) and thereby could lead to acquired drug resistance of the cells. The known HDACs can be divided into three classes based on their homology to a prototypical HDAC found in yeast. Class I HDACs (HDAC1, -2, -3, and -8) show homology to the yeast RPD3 protein; class II HDACs (HDAC4, -5, -6, -7, -9, and -10) have a high degree of homology to Hda1 protein; and class III HDACs (SIRT1–7) are homologous to the yeast Sir2 protein (de Ruijter et al., 2003).
It has been shown that HDACs are responsible for modifying the activity of diverse types of nonhistone proteins such as Hsp90, p53, Ku70, Rel A, and STATs as well as histone proteins, each of which plays an important role in regulating cell proliferation, survival, and apoptosis (Luo et al., 2001; Glozak et al., 2005). Depletion of HDAC6 levels leads to acetylation of Hsp90 and disruption of its chaperone function, resulting in polyubiquitylation and depletion of pro-growth and prosurvival Hsp90 client proteins such as Bcr-Abl (Bali et al., 2005). Activation of the tumor suppressor p53 is known to play a direct role in the apoptotic pathway, and acetylation increases its transcriptional activity. It has been reported that HDAC1, -2, and -3 reduce the ability of p53 to activate the Bax promoter through p53 deacetylation (Juan et al., 2000).
SIRT1 also catalyzed p53 deacetylation, which resulted in negative regulation of p53-mediated transcriptional activation; thus, it prevented apoptosis induced by DNA damage and stress (Kume et al., 2006). Recently, it has been shown that SIRT1 deacetylates the DNA repair factor Ku70 that complexes with Ku80 to form the DNA-binding component of the DNA-dependent protein kinase and stimulates DNA repair in the nucleus (Muller and Salles, 1997). Ku70 is also known as an acetylation-sensitive binding partner for proapoptotic Bax in the cytoplasm, causing it to sequester the Bax away from mitochondria and thereby inhibit stress-induced apoptotic cell death (Sawada et al., 2003; Cohen et al., 2004).
In addition, the expression of SIRT1 increased in drug-resistant cancer cells and in biopsy samples from cancer patients treated with chemotherapeutic agents both at the RNA and protein levels (Chu et al., 2005). Therefore, inhibition of HDACs has emerged as a novel therapeutic strategy against cancer.
Recently, to overcome imatinib resistance, the combination of imatinib with chemotherapeutic drugs, alternative Bcr-Abl inhibitors, and inhibitors of kinases downstream of Bcr-Abl and other several approaches have been studied (Yoshida and Melo, 2004). However, it is important to understand that these approaches differ in efficiency, which is often dependent on the mechanisms of resistance. Therefore, to find new molecular mechanisms of imatinib resistance in vitro, we have generated three imatinib-resistant variants from K562 cell lines by culture in gradually increasing concentrations of imatinib. We developed imatinib-resistant variants with a new resistance mechanism, which showed the severe modulation of HDAC and HAT and subsequent altered expression of Bcr-Abl and apoptosis-related molecules compared with parental K562 cells. Therefore, to overcome imatinib resistance of these variants, we evaluated the ability of suberoylanilide hydroxamic acid (SAHA), a HDAC inhibitor, to sensitize imatinib-resistant K562 cells based upon its imatinib-resistant mechanism.
Materials and Methods
Cell Line and Generation of Resistant Sublines. Human CML K562 cells were obtained from American Type Culture Collection (Manassas, VA) and maintained in culture in RPMI 1640 medium containing 10% fetal bovine serum. The cells were passaged once a week. To establish imatinib-resistant sublines, logarithmically growing cells were exposed to increasing concentrations of imatinib, starting with a concentration of 0.05 μM and increasing gradually by 0.1 μM increments. After the cells acquired the ability to grow in the presence of a specific concentration of imatinib, the level of resistance was determined. A proportion of cells then were frozen, and the remaining cells were grown at the next highest drug level. In this way, three imatinib-resistant variants with different degrees of resistance were isolated and used for further studies. The level of resistance was defined by the imatinib concentration at which the growth rate of cells was comparable with that of untreated parental K562 cells.
Cell Proliferation Assays. Cell proliferation was measured either by counting viable cells by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich, St. Louis, MO) colorimetric dye-reduction method. Conditioned medium was removed after 5 days of imatinib or SAHA exposure, and cells in 96-well plates were incubated with 0.5 mg/ml MTT for 90 min. After centrifugation, the medium was aspirated, and cells were solubilized in 200 μl of dimethyl sulfoxide. The optical density of each sample at 550 nm was measured using a microplate reader (Molecular Devices, Sunnyvale, CA). The optical density of the media was proportional to the number of viable cells. The level of inhibition was measured as a percentage of control growth (no drug in the sample). All experiments were repeated twice in triplicate. To measure drug resistance, the MTT assay was used throughout. The drug concentration resulting in 50% inhibition of the growth (IC50) was determined. The resistance ratio was calculated by dividing the IC50 for resistant cells by the IC50 for the parental cells.
Western Blot Analysis and Immunoprecipitation. Whole-cell lysates or nuclear extracts containing an equal amount of protein were separated by 7% SDS-polyacrylamide gel electrophoresis. After electrophoretic transfer from the gel onto nitrocellulose membranes, the positive signals from the membranes were detected with the reagents in the chemiluminescent detection kit (ECL system; GE Healthcare, Piscataway, NJ) according to the manufacturer's instruction. Western blot analysis was performed with the following antibodies: anti-Bcr-Abl, p-Tyr (PY99), Bax, Bcl-2, Hsp70, Hsp90, STAT5, CBP, PCAF, GCN5, β-actin, HDAC1, -2, and -6, SIRT1, Ku70/80, and acetylated α-tubulin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and acetylated p53 (K382) (Cell Signaling Technology Inc., Danvers, MA). The expression level of β-actin was used to assess the amount of protein loaded.
For immunoprecipitation of Ku70 (or Hsp90), 100 μg of protein extracted from K562 and its imatinib-resistant K562/R3 cells was precleared by incubation with protein A/G Sepharose beads (Santa Cruz Biotechnology, Inc.). The supernatant was incubated with agarose-conjugated goat polyclonal anti-Ku70 (or Hsp90) antibody, followed by three washes in 1% Triton X-100 in phosphate-buffered saline. The immunocomplex was separated by SDS-polyacrylamide gel electrophoresis, and proteins were detected with a rabbit polyclonal anti-pan-acetyl lysine (panAc-K) antibody (Santa Cruz Biotechnology, Inc.). Coimmunoprecipitation of endogenous Ku70/Bax from K562 and K562/R3 cells was performed as described previously (Um et al., 2004).
RT-PCR Analysis and Sequence Analysis of the Abl Kinase Domain. Endogenous mRNA was isolated, using an RNeasy Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer's protocol, from K562 and its imatinib-resistant variants and was assessed via RT-PCR. The Titan One Tube RT-PCR System (MJ Research, Reno, NV) was used with the following primers. For cDNA synthesis, 1 μg of RNA was retrotranscribed, and semiquantitative PCR was performed with oligonucleotides: Bcr-Abl (forward), 5′-GACATGC CATAGGTAGCAATTTCCC-3′, and (reverse), 5′-ACATCACGCCAGTCAACAGTCTGG-3′; HDAC1 (forward), 5′-GACTCGAGGAGGCGAGCAAGATGGC-3′, and (reverse), 5′-GATGTCGACGTCCATTCAGGCCAACTTGAC-3′; HDAC2 (forward), 5′-AATATGGGGAATACTTTCCT-3′, and (reverse) 5′-ACAAACGCTGTTTTATC TTT-3′; HDAC3 (forward), 5′-GACAGAACTTGGGTTGATGA-3′, and (reverse) 5′-AGGTCTTCTGGAATCTGGT-3′; HDAC4 (forward), 5′-CAAGAACAAGGAGAAGGGCAAAG-3′, and (reverse), 5′-GGACTCTGGTCAAGGGAACTG-3′; HDAC6 (forward), 5′-TGATGCTGACTACCTAGCTG-3′, and (reverse), 5′-CCACAGCTGTCTCTCTGAGTTA-3′; SIRT1 (forward), 5′-CTTGCCTCATCTGCATTTT-3′, and (reverse), 5′-ATTAGGCCAGCATTTTCTCA-3′; p53 (forward), 5′-TGCATTTCTTTTTCTGGATT-3′, and (reverse), 5′-ACGTAAGTACGGCACAAAGT-3′; p300 (forward), 5′-ACCAGAAGAACTACGACAG-3′, and (reverse), 5′-GCACTCTGTACATTCAACAA-3′; CBP (forward), 5′-CTGGGAAATAATCCAATGAA-3′, and (reverse), 5′-GTTTATGTAAACGCGACCTC-3′; Bax (forward), 5′-TGGCAGCTGACATGTTTTCTGAC-3′, and (reverse), 5′-TCACCCAACCACCCTGGTCTT-3′; p21 (forward), 5′-GACAACTCACTCGTCAAATC-3′, and (reverse), 5′-ACAGCACTGTTAGAATGAGC-3′; and β-actin (forward), 5′-ACCAACTGGGACGACATGGAG-3′, and (reverse), 5′-GTGAGGATCTTCATGAGGTAGTC-3′.
To analyze the sequence of Abl kinase domain, cDNA was synthesized from total RNA of K562 and its imatinib-resistant variants using SuperScrip II Reverse Transcriptase (Invitrogen, Carlsbad, CA) and amplified with a PTC-100 Peltier Thermal Cycler (MJ Research) using PCR primers, including two 3′ Abl-specific primers (5′-GCCAGGCTCTCGGGTGCAGTCC-3′ and 5′-GACATGCCATAGGTAGCAATTTCCC-3′) and two 5′ Abl-specific primers(5′-GCGCAACAAGCCCACTGTCTATGG-3′ and 5′-ACATCACGCCAGTCAACAGTCTGG-3′). Amplified fragments (579 and 969 base pairs) were directly sequenced with two 3′ Abl-specific primers (5′-GCCAGGCTCTCGGGTGCAGTCC-3′ and 5′-CAAGTTCCCCATCAAATG-3′) and two different 5′ Abl-specific primers (5′-GCGCAACAAGCCCACTGTCTATGG-3′ and 5′-ATGGAGGTGGAAGAGTTC-3′), respectively.
Apoptosis Assessment by Annexin V Staining. After K562 and its imatinib-resistant variants (1 × 105 cells/ml) were treated with or without various concentrations of SAHA or sirtinol for 48 h, the cells were centrifuged and resuspended in 500 μl of the staining solution (containing annexin V fluorescein and propidium iodide in a HEPES buffer (Annexin V-FITC Apoptosis Detection Kit; BD PharMingen, San Diego, CA). After incubation at room temperature for 15 min, cells were analyzed by flow cytometry. Annexin V binds to cells that express phosphatidylserine on the outer layer of the cell membrane, and propidium iodide stains the cellular DNA of cells that have a compromised cell membrane. These stainings allow for discrimination of live cells (unstained with either fluorochrome) from apoptotic cells (stained only with annexin V) and necrotic cells (stained with both annexin V and propidium iodide).
Results
Involvement of Aberrant Modulation of Apoptosis-Related Proteins in Bcr-Abl-Independent Imatinib Resistance. To understand the mechanisms of acquired imatinib resistance, we selected three variants having different degrees of resistance to imatinib derived from parental K562 cells by culturing in the presence of gradually increasing concentrations of imatinib. The imatinib resistance was evaluated with MTT assays after 5 days of incubation with the drug. IC50 values of K562/R1 (8.4 μM), K562/R2 (10.0 μM), and K562/R3 (12.6 μM) sublines, the imatinib-resistant variants, showed approximately 42-, 50- and 63-fold resistance to imatinib compared with that of imatinib-sensitive K562 (0.2 μM) cells, respectively (Fig. 1A). Then, we compared the protein and mRNA levels and autophosphorylation activity of Bcr-Abl tyrosine kinase between K562 and its imatinib-resistant variants (Fig. 1B). Compared with K562 cells, the Bcr-Abl level was profoundly decreased, and phospho-Bcr-Abl (pBcr-Abl) was barely detected in the imatinib-resistant K562/R1, -R2, and -R3 variants. We also determined whether loss of Bcr-Abl protein in the imatinib-resistant variants was due to the decreased transcription of the fusion gene using an RT-PCR assay. The level of Bcr-Abl mRNA was significantly reduced in the imatinib-resistant variants relative to K562 cells. However, mutation was not observed in the Abl kinase domain (data not shown). These findings suggested that loss of target molecules may be a mechanism of imatinib resistance and that these imatinib-resistant cells may be dependent on other signals for survival.
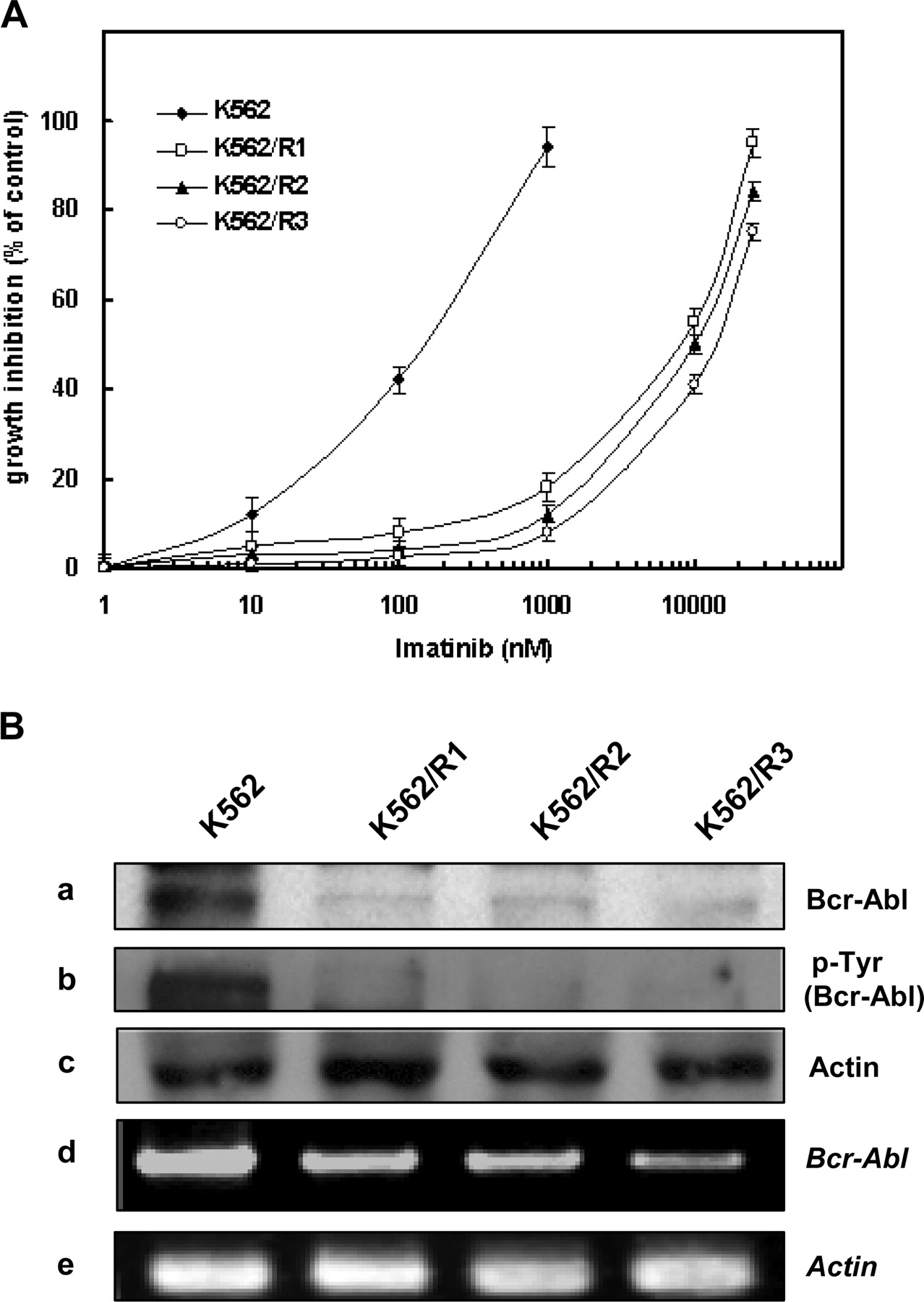
Altered drug sensitivity and loss of Bcr-Abl in imatinib-resistant K562 cells. A, K562 and its imatinib-resistant K562/R1, -R2, and -R3 cells were treated with various doses of imatinib, and growth inhibition of these cells by imatinib treatment was determined by an MTT assay. Data points on the curve represent means percent growth inhibition in at least two experiments in triplicate. B, whole-cell lysates obtained from these cells were subjected to Western blot analysis to monitor levels of Bcr-Abl (a), phosphorylated Bcr-Abl (b), and the level of β-actin (Actin) (c) was used as a loading control. In addition, total RNA was extracted from K562 and its imatinib-resistant variants and subjected to RT-PCR analysis to monitor Bcr-Abl mRNA levels (d) and actin mRNA was amplified to normalize data (e).
Therefore, we compared the expression of other determinants of apoptosis in the imatinib-sensitive and -resistant K562 cells (Fig. 2A). Compared with K562 cells, the levels of antiapoptotic Bcl-2 and proapoptotic Bax were remarkably increased and conversely decreased in the K562/R1, -R2, and -R3 cells, respectively. It has been reported that Bcr-Abl expression results in up-regulation of the Hsp70 and induction of the levels and activity of STAT5 in human leukemia cells (Guo et al., 2005). In agreement with these findings, our result showed that the antiapoptotic Hsp70 and STAT5 were significantly down-regulated in the imatinib-resistant variants displaying loss of Bcr-Abl, whereas the level of Hsp90 in these variants was slightly increased compared with that of K562 cells. On the other hands, it has been reported that Ku70 binds and sequesters the proapoptotic protein Bax in cytoplasm, and inhibition of HDAC increases acetylation of Ku70, releasing and activating Bax, which is a critical target for proapoptotic regulation (Cohen et al., 2004). Our result showed that the cytoplasmic Ku70/80, especially the cytoplasmic level of Ku70 in the K562/R1, -R2, and -R3 cells, was markedly increased compared with that in K562 cells, suggesting that up-regulation of cytoplasmic Ku70 could contribute to the down-regulation of Bax. In addition, the levels of these proteins were transcriptionally regulated as shown in Fig. 2B. These findings support the possibility that loss of Bcr-Abl tyrosine kinase may be compensated for by changes in levels of apoptosis-modulating molecules for cell survival.

Alteration of apoptosis-related proteins and the mRNA levels in Bcr/Abl-independent imatinib-resistant K562 cells. A, whole-cell lysates obtained from K562 and its imatinib-resistant variants, K562/R1, -R2, and -R3 cells were subjected to Western blot analysis to monitor levels of Bcl-2, Bax, Hsp70, Hsp90, and STAT5. C, cytoplasmic extracts isolated from these cells. The level of cytoplasmic Ku70/80 was assayed by Western blot analysis. Blots were subsequently reprobed with actin for normalization. B, total RNA was extracted from K562 and its imatinib-resistant variants were subjected to RT-PCR analysis to Bcl-2, Bax, Hsp70, Hsp90, STAT5, and Ku70/80 mRNA levels. Actin mRNA was amplified to normalize data.
Relation between Deacetylated Cytoplasmic Ku70 and Bax Sequestration in Imatinib-Resistant K562 Cells. It has been known that acetylation of the C-terminal linker of Ku70 adjacent to the Bax interaction domain by CBP with HAT activity inhibits the ability of Ku70 to suppress Bax-mediated apoptosis (Cohen et al., 2004). Therefore, we determined the Ku70-Bax interaction and the acetylation status of Ku70 in imatinib-resistant cells with immunoprecipitation. The amount of Ku70 bound to Bax was much greater in K562/R3 cells than in K562 cells despite the low level of Bax in K562/R3 cells (Fig. 3A), suggesting that the binding affinity of Ku70 to Bax may be stronger in K562/R3 than in K562 cells. The amount of acetylated Ku70 was significantly reduced in K562/R3 cells compared with that in K562 cells (Fig. 3B). These results suggest that the decreased acetylation of cytoplasmic Ku70 may contribute to high affinity between Ku70 and Bax in the imatinib-resistant cells, and, consequently, even low amounts of Bax can be captured by deacetylated cytoplasmic Ku70, leading to suppression of Bax-mediated apoptosis.
Imbalance between HAT and HDAC Expressions in Imatinib-Resistant K562 Cells. Because imatinib-resistant K562/R3 cells showed the reduction of cytoplasmic Ku70 acetylation, we determined levels of HATs and HDACs in imatinib-resistant variants. For HATs (Fig. 4A, left), the level of CBP was remarkably decreased in K562/R1, -R2, and -R3 cells compared with that in K562 cells, and the levels of GCN5 and PCAF were slightly reduced in these imatinib-resistant variants, suggesting that decreased acetylase activities might be associated with the development of imatinib-resistance. For HDACs (Fig. 4A, right), the levels of class I HDAC1, -2, and -3 and class III SIRT1 were significantly increased in K562/R1, -R2, and -R3 cells compared with those in K562 cells. In contrast, the level of class II HDAC6 was decreased in the imatinib-resistant variants compared with that in K562 cells. Therefore, our results suggest that upregulation of HDAC1, -2, and -3 and SIRT1 and down-regulation of HDAC6 and HATs such as CBP, GCN5, and PCAF could be associated with imatinib-resistance of K562/R1, -R2, and -R3 cells.

The increase of cytoplasmic Ku70 combined with Bax in imatinib-resistant K562 cells. A, cell lysates isolated from K562 and K562/R3 cells were immunoprecipitated with anti-Bax antibody or control mouse IgG antibody and blotted with anti-Ku70 antibody. B, immunocomplexes were precipitated from K562 and K562/R3 cell extracts with a polyclonal antibody against an anti-panAc-K antibody and blotted with an anti-Ku70 antibody for determination of acetylated Ku70 (Ac-Ku70) level. IP, immunoprecipitation.

Imbalance between HAT and HDAC expressions and altered susceptibility to sirtinol in imatinib-resistant K562 cells. A, nuclear or cytoplasmic extracts obtained from K562 and its imatinib-resistant variants were subjected to Western blot analysis to monitor levels of CBP, GCN5, and PCAF with HAT activity (left) and the levels of HDAC1, -2, and -6 and SIRT1 with HDAC activity (right). B, K562/R3 and K562 cells were treated with or without the indicated concentration of sirtinol, a SIRT1 inhibitor, for 48 h. After this, the percentage of apoptotic cells was determined by annexin V staining and flow cytometry. FITC, fluorescein isothiocyanate.
It has been reported that SIRT1 deacetylates Ku70, causing it to sequester the Bax away from mitochondria (Cohen et al., 2004), and also catalyze p53 deacetylation, which results in down-regulation of its target genes including Bax and p21 (Kume et al., 2006). Therefore, we compared susceptibility to apoptosis induced by sirtinol, a specific SIRT1 inhibitor, between K562 cells and K562/R3 cells with up-regulated SIRT1. Our data showed that K562/R3 cells were more susceptible to sirtinol relative to the K562 cells (Fig. 4B), suggesting that SIRT1 plays an important role in mediating the Bcr-Abl-independent form of imatinib resistance.
To determine whether the levels of HDACs were regulated transcriptionally, RT-PCR was used (Fig. 5A). Consistent with the results shown in Fig. 4A, the mRNA levels of HDAC1, -2, -3, and -4 and SIRT1, the class I and III HDACs, were increased, whereas the level of HDAC6 mRNA was decreased in the imatinib-resistant variants compared with those in K562 cells (Fig. 5A). Because previously it had been reported that targeted inhibition of HDAC6 by its small interfering RNA or LAQ824 leads to acetylation of Hsp90 and disruption of its chaperone function, resulting in polyubiquitylation and depletion of Bcr-Abl, the Hsp90 client protein in K562 cells (Bali et al., 2005), we determined the acetylation status of Hsp90 in K562/R3 cells with a low level of HDCA6 by immunoprecipitating panAc-K and probing the immunocomplex for Hsp90 (Fig. 5B). Hsp90 was highly acetylated in K562/R3 cells compared with K562 cells. Concomitantly, acetylation of α-tubulin, which is a well known substrate of HDAC6, was also increased in K562/R3 cells compared with K562 cells, indicating the possibility that the decrease of HDAC6 in the imatinib-resistant cells led to increased acetylation of Hsp90, and this would disrupt the chaperone association of Bcr-Abl to Hsp90, leading to the proteasomal degradation and thereby down-regulation of Bcr-Abl expression. These findings indicate that alteration of HDACs as well as an imbalance between HATs and HDACs in the imatinib-resistant cells leads to deregulated protein acetylation, which may contribute to activation of the Bcr-Abl-independent cell survival signaling pathway in the imatinib-resistant cells.
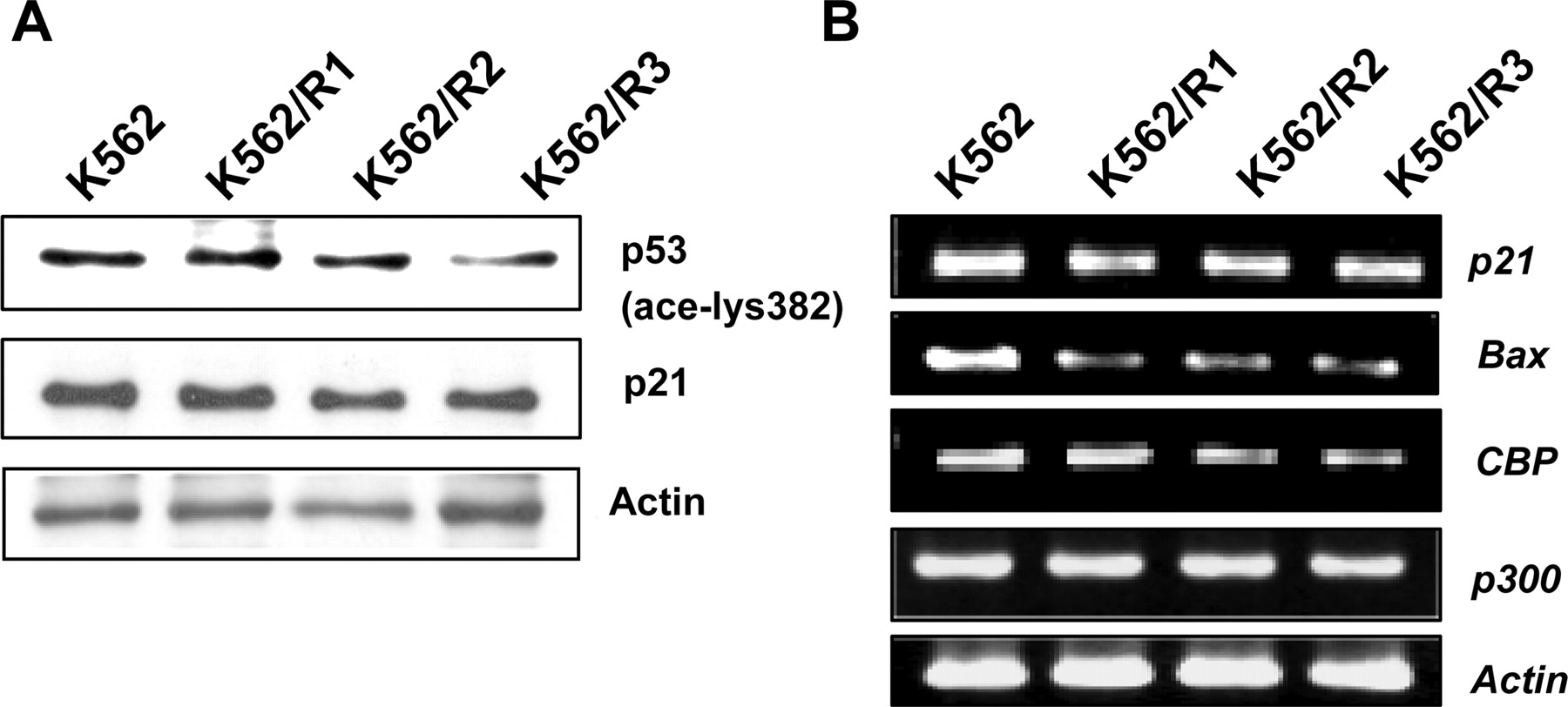
Association of Down-Regulated HAT and Up-Regulated Class I HDACs with Decreased Acetylation of p53. Because activation of the tumor suppressor p53 is known to play a direct role in the apoptotic pathway and acetylation of p53 increases its transcriptional activity (Bandyopadhyay et al., 2004), we determined the status of p53 acetylation and its downstream molecules in imatinib-resistant variants (Fig. 6A). Interestingly, the acetylation of p53 was decreased, and this decrease was followed by down-regulation of expression of p21 and Bax at both protein and mRNA levels in K562/R1, -R2, and -R3 cells compared with K562 cells, and we also found alterations of CBP and p300 mRNAs in the imatinib-resistant variants (Fig. 6B). These results suggest that down-regulation of p21 and Bax may be due to disruption of p53 acetylation by a decrease of CBP/p300 and an increase of class I HDACs as shown in Fig. 5A, because acetylation of p53 could be related to CBP/p300mediated HAT activities and SIRT1 and class I HDACs activities, which are known to acetylate and deacetylate p53, respectively (Juan et al., 2000; Zhao et al., 2006), and these changes may compensate for the loss of Bcr-Abl during acquisition of the imatinib-resistant phenotype.

Involvement of aberrant regulation of HDACs in imatinib-resistant K562 cells. A, total RNA was extracted from K562 and its imatinib-resistant variants subjected to RT-PCR analysis to class I, II, and III HDACs mRNA levels. Actin mRNA was amplified to normalize data. B, immunocomplexes were precipitated from K562 and K562/R3 cell extracts with anti-panAc-K antibody and blotted with an anti-Hsp90 antibody for determination of acetylated Hsp90 (Ac-Hsp90) level and the level of IgG was used as a loading control (top). The state of tubulin acetylation was detected with the antiacetylated α-tubulin (Actubulin) antibody, and the level of tubulin was used as a loading control (bottom).

Reduction of p53 acetylation and decreased levels of p21 and Bax in imatinib-resistant variants. A, cellular levels of acetylated p53 and p21 in K562 cells and its imatinib-resistant variants were measured by Western blotting. Blots were subsequently reprobed with anti-actin for normalization. B, total RNA was extracted from K562 and its imatinib-resistant variants were subjected to RT-PCR analysis to p21, Bax, CBP, and p300 mRNA levels. Actin mRNA was amplified to normalize data.
HDAC Inhibitor Restores Imatinib-Induced Molecular Changes. Because imatinib-resistant cells with upregulated class I and III HDACs and down-regulated HATs showed hypoacetylation of nonhistone proteins such as Ku70 and p53, we determined whether the HDAC inhibitor is capable of restoring the molecular changes obtained during imatinib resistance. After treatment with SAHA, one of the HDAC inhibitors, the acetylation of cytoplasmic Ku70 of K562/R3 cells was significantly increased compared with that of K562 cells (Fig. 7A). Next, we tested the effects of SAHA on the expression of the apoptosis-modulating molecules, which were changed in imatinib-resistant variants (Fig. 7B). After treatment with SAHA, the level of pBcr-Abl was significantly reduced in K562 cells, but this inhibitory effect of SAHA was not detectable in Bcr-Abl- and HDAC6-depleted K562/R3 cells. In contrast, the levels of up-regulated Bcl-2 and cytoplasmic Ku70 in K562/R3 cells were significantly suppressed by SAHA treatment. In addition, the level of Bax was increased in both K562 cells and the Bax-depleted K562/R3 cells by SAHA treatment. These results suggest that Bcr-Abl-negative imatinib-resistant cells might be sensitive to SAHA through inhibition of up-regulated class I HDACs and class III SIRT1 and subsequent modulation of apoptosis-related molecules.

Effect of SAHA on the Ku70 acetylation and apoptosis-related proteins of K562 and K562/R3 cells. A, immunocomplexes were precipitated from K562 and K562/R3 cell extracts with anti-panAc-K antibody and blotted with an anti-Ku70 antibody. B, the cells were treated with various concentrations of SAHA for 24 h, and the changed levels of Bcl-2, cytoplasmic Ku70, and Bax were assayed by Western blot analysis and electrophoretic mobility shift assays, respectively.
Comparison of Sensitivity to SAHA-Induced Cell Death between K562 and Imatinib-Resistant Variants. Recently, it has been found that HDAC inhibitors can trigger both mitochondria-mediated apoptosis and caspase-independent autophagic cell death, indicating the potential benefit of HDAC inhibitors in treating cancers with apoptotic defects (Marks and Jiang, 2005). Our result showed that imatinibresistant K562/R1, -R2, and -R3 variants were more sensitive to SAHA-induced cytotoxicity than K562 cells in a resistance degree-dependent manner, showing the highest sensitivity in K562/R3 cells, which were 7.3-fold more sensitive to SAHA than K562 cells when growth inhibition was evaluated by the IC50 value 96 h after SAHA treatment using an MTT assay (Fig. 8A, left). Furthermore, K562/R3 cells were more sensitive to SAHA-induced apoptosis than K562 cells when annexin V-fluorescein isothiocyanate staining was used to detect early apoptosis 48 h after SAHA treatment (Fig. 8A, right). Consistently, the degradation of poly(ADP-ribose) polymerase-1, a 113-kDa enzyme that functions as a sensor of DNA damage, was observed at lower doses and earlier in K562/R3 cells than in K562 cells after treatment with SAHA (Fig. 8B). These results demonstrated that imatinib-resistant K562 cells with loss of Bcr-Abl and overexpressed class I and III HDACs were more susceptible to HDAC inhibitor than imatinib-sensitive parental cells.

The effect of SAHA-induced cytotoxicity and apoptosis on the K562 and its imatinib-resistant variants. A, K562 and its imatinib-resistant K562/R1, -R2, and -R3 cells were treated with or without the indicated concentration of SAHA for 96 h. After this, a growth inhibition assay was performed by the MTT method. Data are calculated as the mean ± S.D. of triplicate samples (left). K562/R3 and K562 cells were treated with or without the indicated concentration of SAHA for 48 h. After this, the percentage of apoptotic cells was determined by annexin V staining and flow cytometry. Values represent the mean ± SD of three separate experiments (right). B, K562/R3 and K562 cells were treated with various concentrations of SAHA for 12 or 24 h, and the changed level of poly(ADP-ribose) polymerase-1 (PARP-1) was assayed by Western blot analysis.
Discussion
Although it has been known that in most cases resistance against imatinib results from kinase domain mutations and/or overexpression of the Bcr-Abl gene (Azam and Daley, 2006), there are clearly some cases of resistance that appear to occur through mechanisms independent of Bcr-Abl. Indeed, it has been reported that imatinib resistance in some patients with CML is associated with activation of the HDAC signaling pathway mediated through loss of kinase target dependence (Donato et al., 2004). Therefore, further understanding of Bcr-Abl-independent imatinib resistance may be required to develop new therapeutic modalities for imatinib-resistant chronic myelogenous leukemia.
In the present study, we demonstrated that imatinib-resistant K562 variants displayed a dramatic reduction in levels of Bcr-Abl, the imatinib-target protein, without mutations in the Abl kinase domain and concurrent up-regulation of antiapoptotic molecules such as Bcl-2 and cytoplasmic Ku70 and down-regulation of Bax and p21, suggesting a new mechanism for the Bcr-Abl-independent form of imatinib resistance. In addition, we demonstrated for the first time that an alteration in the normal balance of HATs and HDACs leads to deregulated acetylation of nonhistone proteins such as Hsp90, p53, and Ku70. These molecular changes might be involved in the compensation for Bcr-Abl loss after exposure to imatinib by activation of the Bcr-Abl-independent cell survival signaling pathway.
Although it is not known how Bcr-Abl gene expression was suppressed, a loss of Bcr-Abl in the imatinib-resistant variants seemed to be associated with not only transcriptional inhibition but also the reduced stability of Bcr-Abl that was due to decreased activity of HDAC6. It has been shown that targeted inhibition of HDAC6 in K562 cells induced acetylation of Hsp90, and this disrupted the chaperone association of the Hsp90 client protein Bcr-Abl to Hsp90, leading to the proteasomal degradation and depletion of Bcr-Abl (Bail et al., 2005). Therefore, our study suggests that the decreased level of Bcr-Abl in K562/R3 cells might result in part from a decreased level of HDAC6. Nimmanapalli et al. (2002) isolated Bcr-Abl-negative imatinib-resistant K562 cells that showed marked declines in mRNA and protein levels and activity of Bcr-Abl that are associated with reduced AKT kinase and STAT5 activity, and Dai et al. (2004) reported a pronounced reduction in expression of Bcr-Abl accompanied by an increase in activated Lyn in imatinib-resistant chronic myelogenous leukemia cells (K562 and LAMA84). In addition, our imatinib-resistant variants showed several changes in levels of apoptosis-modulating molecules such as an increase in antiapoptotic Bcl-2 and Ku70 levels and a decrease in proapoptotic Bax and reduction of antiapoptotic Hsp70 and STAT5 levels showing characteristics similar to those of the Bcr-Abl-negative imatinib-resistant cells of Nimmanapalli et al. Although we isolated three imatinib-resistant variants dose dependently, the down-regulation of Bcr-Abl and Hsp70 levels seemed to be almost saturated at the lowest concentration of imatinib used for isolation of K562/R1 cells. However, STAT5 and GCN5 showed dose-dependent declines. Therefore, the molecular changes during acquisition of imatinib resistance seem to be very complex and may be dependent on cell type and drug treatment conditions.
A controlled balance between HATs and HDACs is known to be essential for acetylation of nonhistone proteins as well as histones, which seems to be essential for normal cell growth. The dual roles of Ku70, the regulatory subunit of DNA-protein kinase, in DNA repair and the regulation of apoptosis place it in a key position to coordinate these two processes in response to cellular damage. Our data showed that the cytoplasmic levels of Ku70/80 in K562/R1, -R2, and -R3 cells (Fig. 2A) as well as total Ku70/80 levels in the imatinib-resistant variants (data not shown) were increased compared with those of K562 cells. Moreover, up-regulation of cytoplasmic Ku70 might be contributing to the down-regulation of Bax. Recently, it has been found that sequestration of Bax by cytoplasmic Ku70 is increased or decreased by deacetylation or acetylation at lysines 539 and 542 of Ku, which is catalyzed by SIRT1 or CBP and PCAF, respectively (Cohen et al., 2004). The current data showed that up-regulation of SIRT1 and concurrent down-regulation of CBP and PCAF in imatinib-resistant variants were associated with the deacetylation of cytoplasmic Ku70, which sequesters the Bax away from mitochondria (Cohen et al., 2004), and of p53, which results in down-regulation of its target genes including Bax and p21 (Kume et al., 2006). These molecular changes in K562/R3 cells led to increased susceptibility to sirtinol-induced apoptosis relative to the K562 cells. In addition, it has been reported that expression of SIRT1 increased in drug-resistant cancer cells and could be a potential marker of the drug resistance phenotype in cancer (Chu et al., 2005). Therefore, the increased SIRT1 seemed to be associated with compensation for Bcr-Abl loss during the obtaining of imatinib resistance.
Alterations in the expression of HATs and HDACs occur in many cancers (Chambers et al., 2003; Yoshida et al., 2004), and, in addition, perturbation of this balance can lead to transcriptional dysregulation of genes involved in the regulation of apoptosis (Romanski et al., 2004). Our results suggest that alterations in the normal balance of HATs and HDACs such as up-regulation of class I and III HDACs (HDAC1, -2, and -3 and SIRT1) and down-regulation of class II HDAC6 and HATs involving CBP and PCAF could be also involved in the compensation for Bcr-Abl loss during obtaining of imatinib resistance.
It has been reported that overexpression of class I HDACs (HDAC1, -2, and -3) repressed Bax promoter activity of p53 (Juan et al., 2000). In addition, deacetylation of p53 by SIRT1 and HDAC1 decreases the ability of p53 to transcriptionally activate the cell cycle inhibitor p21 (Wang et al., 2005), and, conversely, the p300/CBP-mediated acetylation of p53 significantly potentiates p53-mediated transactivation and growth inhibition (Zhao et al., 2006). Therefore, our results showed that up-regulation of class I and III HDACs and down-regulation of p300/CBP in the imatinib-resistant cells seemed to be associated with the decrease in the acetylation of p53 and the levels of its target genes including Bax and p21.
Recently, it has been demonstrated that the expression of Bcl-2 could be regulated by activities of HDACs and HATs (Duan et al., 2005; Kikuchi et al., 2005). Because overexpression of HDAC2 or GCN5 deficiency has been known to result in an increase in the expression of Bcl-2, both up-regulation of HDAC2 and down-regulation of GCN5 in the imatinib-resistant cells might be associated with up-regulation of Bcl-2.
We suggest that the imbalance of HDACs and HATs leads to deregulated acetylation of p53, Ku70, and Hsp90, which was closely associated with inactivation of p53 and down-regulation of Bax as well as Bcr-Abl loss and subsequently up-regulation of Bcl-2, and these molecular changes might be involved in the compensation for Bcr-Abl loss during the obtaining of imatinib resistance. This finding suggests the possibility that deregulation of HDAC could be a potential target to circumvent Bcr-Abl-independent imatinib resistance in CML cells.
Recently, HDAC inhibitors have been shown to be potent apoptosis inducers in a variety of cancer cells and currently are being tested for their efficacy in a variety of clinical trials (Chinnaiyan et al., 2005). The activity of HDAC inhibitors has been linked to their ability to induce gene expression through acetylation of nonhistone proteins as well as histone proteins. SAHA, a prototype of a synthetic hydroxamic acid-based hybrid polar HDAC inhibitor, inhibits the activity of HDACs (Suzuki et al., 2005) and is synergistic in its anticancer activity with radiation, kinase inhibitors, cytotoxic agents, and differentiating agents (Liu et al., 2006). It has been reported that coadministration of imatinib with SAHA results in a dramatic increase in mitochondrial damage and apoptosis in Bcr-Abl-positive imatinib-resistant human leukemia cells, accompanied by down-regulation of the Bcr-Abl protein (Yu et al., 2003). In contrast, in our Bcr-Abl-negative imatinib-resistant cells, which showed up-regulation of class I and III HDACs, treatment with SAHA was accompanied by down-regulation of Bcl-2 and Ku70 and up-regulation of Bax. These responses to SAHA treatment might be responsible in part for increased susceptibility to SAHA in K562/R3 cells compared with K562 cells.
In conclusion, our results suggest that imatinib resistance can be obtained by loss of Bcr-Abl, a target of imatinib, and subsequent up-regulation of class I and III HDACs and down-regulation of HATs. These findings support the possibility that HDAC inhibitors might be exploited for treatment of patients with imanitib-resistant CML.
Footnotes
-
This work was supported by the Medical Research Center program of the Ministry of Science and Technology/Korea Science and Engineering Foundation (R13-2005-009) (S.H.K.).
-
S.H.K. and C.D.K. share equal authorship.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.124461.
-
ABBREVIATIONS: CML, chronic myelogenous leukemia; HDAC, histone deacetylase; Hsp, heat shock protein; STAT, signal transducer and activator of transcription; HAT, histone acetyltransferase; SAHA, suberoylanilide hydroxamic acid; MTT, 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide; CBP, cAMP-response element-binding protein binding protein; panAc-K, pan-acetyl lysine; RT, reverse transcriptase; PCR, polymerase chain reaction; LAQ824, (2E)-N-hydroxy-3-[4-[[(2-hydroxyethyl)[2-(1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2-propenamide.
- Received April 30, 2007.
- Accepted June 13, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}