


Abstract
The present study was undertaken to determine the underlying mechanism of silibinin-induced cell death in human breast cancer cell lines MCF7 and MDA-MB-231. Silibinin-induced cell death was attenuated by antioxidants, N-acetylcysteine (NAC) and 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid, suggesting that the effect of silibinin was dependent on generation of reactive oxygen species (ROS). Western blot analysis showed that silibinin induced downregulation of extracellular signal-regulated kinase (ERK) and Akt. When cells were transiently transfected with constitutively active (ca) mitogen-activated protein kinase (MEK), an upstream kinase of ERK and caAkt, they showed resistance to silibinin-induced cell death. Silibinin decreased the cleavage of Notch-1 mRNA and protein levels. Notch-1–overexpressed cells were resistant to the silibinin-induced cell death. Inhibition of Notch-1 signaling was dependent on ROSgeneration. Overexpression of Notch-1 prevented silibinin-induced inhibition of ERK and Akt phosphorylation. Silibinin-induced cell death was accompanied by increased cleavage of caspase-3 and was prevented by caspase-3 inhibitor in MDA-MB-231 cells but not in MCF7 cells. Silibinin induced translocation of apoptosis-inducing factor (AIF), which was blocked by NAC, and transfection of caMEK and caAkt. Silibinin-induced cell death was prevented by silencing of AIF expression using small interfering AIF RNA in MCF7 cells but not in MDA-MB-231 cells. In conclusion, silibinin induces cell death through an AIF-dependent mechanism in MCF7 cells and a caspase-3–dependent mechanism in MDA-MB-231 cells, and ROS generation and Notch-1 signaling act upstream of the ERK and Akt pathway. These data suggest that silibinin may serve as a potential agent for induction of apoptosis in human breast cancer cells.
Introduction
Breast cancer is the most common cancer in Korean women (Jung et al., 2012). Although effective treatment includes surgery, radiation, and chemotherapy, breast cancer frequently shows resistance to these therapies (Trape and Gonzalez-Angulo, 2012). Therefore, there is a great need for new agents to treat breast cancer.
Silibinin is a major bioactive component of silymarin, which is isolated from milk thistle (Silybum marianum), and has been used as a traditional medicine for hepatoprotective therapeutics in Europe and Asia (Singh and Agarwal, 2002). Many studies have shown that silibinin has anticancer activities in various cancer cells such as colon, prostate, skin, glioma, and breast cancer cells, and the effect mainly results from targeting proliferation, inflammation, apoptosis, and cancer cell metabolism (Deep et al., 2011; Jeong et al., 2011; Kim et al., 2011; Kauntz et al., 2012). However, an underlying mechanism of changes induced by silibinin has not been clearly elucidated.
Extracellular signal-regulated kinase (ERK) and Akt signaling pathways serve to coordinate the cellular response to a variety of extracellular stimuli (Steelman et al., 2011). These pathways are implicated in cancer cell proliferation, invasion, and tumorigenesis and drug resistance in many different cell types (Gan et al., 2010; Huynh et al., 2010; Roy et al., 2010; Cheng et al., 2011). Silibinin induces cancer cell death through the increase of ERK and Akt activities in human glioma (Kim et al., 2009), prostate (Singh et al., 2007), and melanoma cancer cell lines (Jiang et al., 2011). However, inhibition of ERK and Akt signaling by silibinin induces human lung (Chen et al., 2005) and renal cancer cell death (Li et al., 2008). These studies suggest that the effect of silibinin on ERK and Akt activation may depend on cell types.
Notch signaling is important for cell-cell communications, which control multiple processes of cell differentiation during embryonic and adult cell fate decision (Allenspach et al., 2002). Recent studies suggest that Notch signaling plays critical roles in numerous cancers. When Notch signaling and related transcriptional factors are upregulated in invasive human cancer, cells become resistant to apoptosis. Previous reports have shown that suppression of Notch-1 expression inhibits human breast cancer cell tumorigenesis and metastasis (Wang et al., 2011; Simmons et al., 2012). Flavonoids such as genistein and quercetin induce cancer cell death by inhibition of the Notch signaling pathway (Wang et al., 2006; Zhou et al., 2012). However, whether the Notch-1 signaling pathway is involved in silibinin-induced breast cancer cell death has not been determined.
The aim of the present study is to determine the molecular mechanisms of the silibinin-induced cell death in MCF7 and MDA-MB-231 breast cancer cell lines. Our data demonstrated that silibinin induced cell death through the reactive oxygen species (ROS)/Notch-1/ERK/Akt pathway, followed by the nuclear translocation of apoptosis-inducing factor (AIF) in MCF7 cells and caspase-3 activation in MDA-MB-231 cells.
Materials and Methods
Reagents.
Silibinin, 3-[4,5-dimethylthiazol2-yl]-2,5-diphenyltetrazolium bromide (MTT), N-acetylcysteine (NAC), 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid, and propidium iodide were purchased from Sigma-Aldrich (St. Louis, MO). An FITC Annexin V Apoptosis Detection Kit was purchased from BD Biosciences (San Jose, CA). 2′,7′-Dichlorofluorescein diacetate (DCFH-DA) and Hoechst dye 33258 were obtained from Molecular Probes (Eugene, OR). Constitutively active mitogen-activated protein kinase (MEK) and Akt-expressing pCMV plasmids were kindly provided by Dr. Suh (Pohang University, Pohang, Korea). Tween 20, Z-VAD-FMK, and Ac-DEVD-CHO were purchased from Calbiochem (San Diego, CA). Plasmid EF.hlCN1.CMV.GFP was purchased from Addgene (Cambridge, MA). All antibodies were obtained from Cell Signaling Technology Inc. (Danvers, MA). All other chemicals were of the highest commercial grade available.
Cell Culture.
MCF7 and MDA-MB-231 cells were obtained from the American Type Culture Collection (Manassas, VA) and maintained by serial passages in 75-cm2 culture flasks (Costar, Cambridge, MA). The cells were grown in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) containing 10% heat inactivated fetal bovine serum (HyClone, Logan, UT) at 37°C in a humidified 95% air/5% CO2 incubator. When the cultures reached confluence, subculture was prepared using 0.02% EDTA–0.05% trypsin solution. The cells were grown on well tissue culture plates and used 1–2 days after plating. Unless otherwise stated, cells were treated with silibinin in serum-free medium.
Measurement of Cell Viability.
Cell viability was evaluated using an MTT assay. After washing the cells, culture medium containing 0.5 mg/ml MTT was added to each well. The cells were incubated for 2 hours at 37°C, the supernatant was removed, and the formed formazan crystals in viable cells were solubilized with 0.11 ml of dimethylsulfoxide. A 0.1-ml aliquot of each sample was then transferred to 96-well plates, and the absorbance of each well was measured at 550 nm with an ELISA Reader (FLUOstar OPTIMA; BMG LABTECH, Offenburg, Germany). Data were expressed as the percentage of control measured in the absence of silibinin. Unless otherwise stated, the cells were exposed to 30 µM silibinin for 48 hours. Test reagents were added to the medium 30 minutes before exposure to silibinin.
Measurement of Apoptosis.
Cell apoptosis was estimated using an FITC Annexin V Apoptosis Detection Kit (BD Biosciences). The annexin V binding assay was performed as described in the manufacturer’s manual. The cells were treated with 30 µM silibinin for 24 hours. Cells were harvested with 0.025% trypsin and washed with ice-cold phosphate-buffered saline (PBS) and then resuspended in annexin V binding buffer. The cells were then incubated for 15 minutes with binding solution containing FITC annexin V and propidium iodide in the dark and using a FACSort Becton Dickinson Flow Cytometer (BD Bioscience), and data were analyzed with CELLQuest Software (BD Bioscience). The percentage of apoptotic cells was calculated as the quadrant statistics of the early and late apoptotic region to the entire cell population (1 × 104 cells).
Measurement of Reactive Oxygen Species.
The intracellular generation of ROS was measured using DCFH-DA. The nonfluorescent ester penetrated into the cells and was hydrolyzed to DCFH by the cellular esterases. The probe (DCFH) was rapidly oxidized to the highly fluorescent compound 2′,7′-dichlorofluorescein (DCF) in the presence of cellular peroxidase and ROS such as hydrogen peroxide or fatty acid peroxides. Cells cultured in a 24-well plate were preincubated in the culture medium with 30 µM DCFH-DA for 1 hour at 37°C. The cells were then exposed to 30 µM silibinin at various periods. Changes in DCF fluorescence was assayed using a FACSort Becton Dickinson Flow Cytometer (BD Bioscience), and data were analyzed with CELLQuest Software.
Western Blot Analysis.
Cells were harvested at various times after silibinin treatment and disrupted in lysis buffer (1% Triton X-100, 1 mM EGTA, 1 mM EDTA, 10 mM Tris-HCl, pH 7.4). Cell debris was removed by centrifugation at 10,000g for 10 minutes at 4°C. The resulting supernatants were resolved on a 10% SDS-PAGE under denatured reducing conditions and transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dried milk at room temperature for 30 minutes and incubated with different primary antibodies. The membranes were washed and incubated with horseradish peroxidase–conjugated secondary antibodies. The signal was visualized using an enhanced chemiluminescence (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK).
Transfection.
To modulate the activity of ERK and Akt, a transient transfection of constitutively active forms of MEK1, an upstream kinase of ERK, and Akt was performed. Cells were seeded in six-well plates and grown to 70% confluence. A 2-µg cDNA each was transiently transfected using Lipofectamine (Invitrogen) according to the manufacturer’s guidelines. After 4-hour incubation at 37°C, cells were maintained in normal culture media for 24 hours. To overexpress intracellular Notch-1, we transferred EF.hlCN1.CMV.GFP (Addgene) according to the manufacturer’s instructions.
Measurement of AIF Nuclear Translocation.
Cells were harvested at various times after silibinin treatment and washed twice with PBS. To measure AIF nuclear translocation, the cells were incubated with cytosol lysis buffer (10 mM HEPES, 250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 0.05% digitonin, and 1 mM phenylmethylsulfonyl fluoride) at 4°C for 30 minutes with vortexing every 10 minutes and then centrifuged at 10,000g for 10 minutes at 4°C. The pellet was incubated in the nuclear lysis buffer (350 mM NaCl, 1 mM EGTA, 1 mM EDTA, 10 mM Tris-HCl, pH 7.4, and protease inhibitors) at 4°C for 30 minutes with vortexing every 10 minutes and then centrifuged at 10,000g for 10 minutes at 4°C. Western blot analysis of AIF nuclear fractions obtained was performed as described earlier.
Immunocytochemistry.
Cells were cultured on cover glasses and treated with silibinin. Cells were fixed with 4% paraformaldehyde for 15 minutes and blocked with 1% bovine serum albumin in PBS for 1 hour, remove from blocking solution, and incubated overnight at 4°C with rabbit anti-AIF (1:500 dilution). Cells were washed and incubated with Alexa Fluor 488-conjugated antirabbit secondary antibody (Invitrogen) at room temperature for 1 hour. After washing, counterstaining was carried out with Hoechst dye 33258 for 15 minutes. Cells were viewed under a fluorescent microscope (Leica, Wetzlar, Germany).
RNA Interference.
Silencing of AIF expression was achieved by the small interfering RNA technique. We used the BLOCK-iT Pol miR RNAi Expression Vector Kits (Invitrogen) to facilitate the expression of microRNA. MicroRNA sequences for AIF were designed using online software (BLOCK-iT RNAi Designer from Invitrogen). The target sequence was 5′-ATGCAGAACTCCAAGCACGTT-3′. This single-stranded oligonucleotide generated a double-stranded oligonucleotide, which was constructed into pcDNA 6.2-GW/EmGFP-miR vector. Cells were transiently transfected with these plasmids using Lipofectamine (Invitrogen).
Reverse Transcription-Polymerase Chain Reaction Analysis.
Total RNA was isolated from MCF7 and MDA-MB-231 cells treated or untreated with silibinin. Reverse transcription was performed according to conventional protocols. cDNA was synthesized in a reaction mixture containing oligodT primer, dNTP mixture, RNase inhibitor, reverse transcriptase, reverse transcription buffer, and total cellular RNA. cDNA amplification was performed using the following primer sets: Notch-1 (5′-GCAACAGCTCCTTCCACTTC-3, 5′- GCCTCAGACACTTTGAAGCC-3′); and GAPDH (5′-TCCATGACAACTTTGGTATCG-3′, 5′-TGTAGCCAAATTCGTTGTCA-3′). The primer sequences were determined using established GenBank sequences. We performed polymerase chain reaction for 30 cycles, and the products were electrophoresed on 1.5% agarose gels, stained with ethidium bromide, and visualized (ethidium bromide) with UV illumination. The optical density of the bands was quantified using an image analysis system.
Statistical Analysis.
The data are expressed as means ± S.E.M., and the difference between two groups was evaluated using Student’s t test. Multiple-group comparison was performed using one-way analysis of variance followed by the Tukey post-hoc test. A probability level of 0.05 was used to establish significance.
Results
Silibinin Induces Inhibition of Cell Viability.
To investigate the effect of silibinin on cell viability in MCF7 and MDA-MB-231 cells, cells were exposed to various concentrations of silibinin for 24 and 48 hours, and cell viability was measured by MTT assay. Silibinin significantly decreased cell viability in both cell lines in a dose-dependent manner (Fig. 1A).
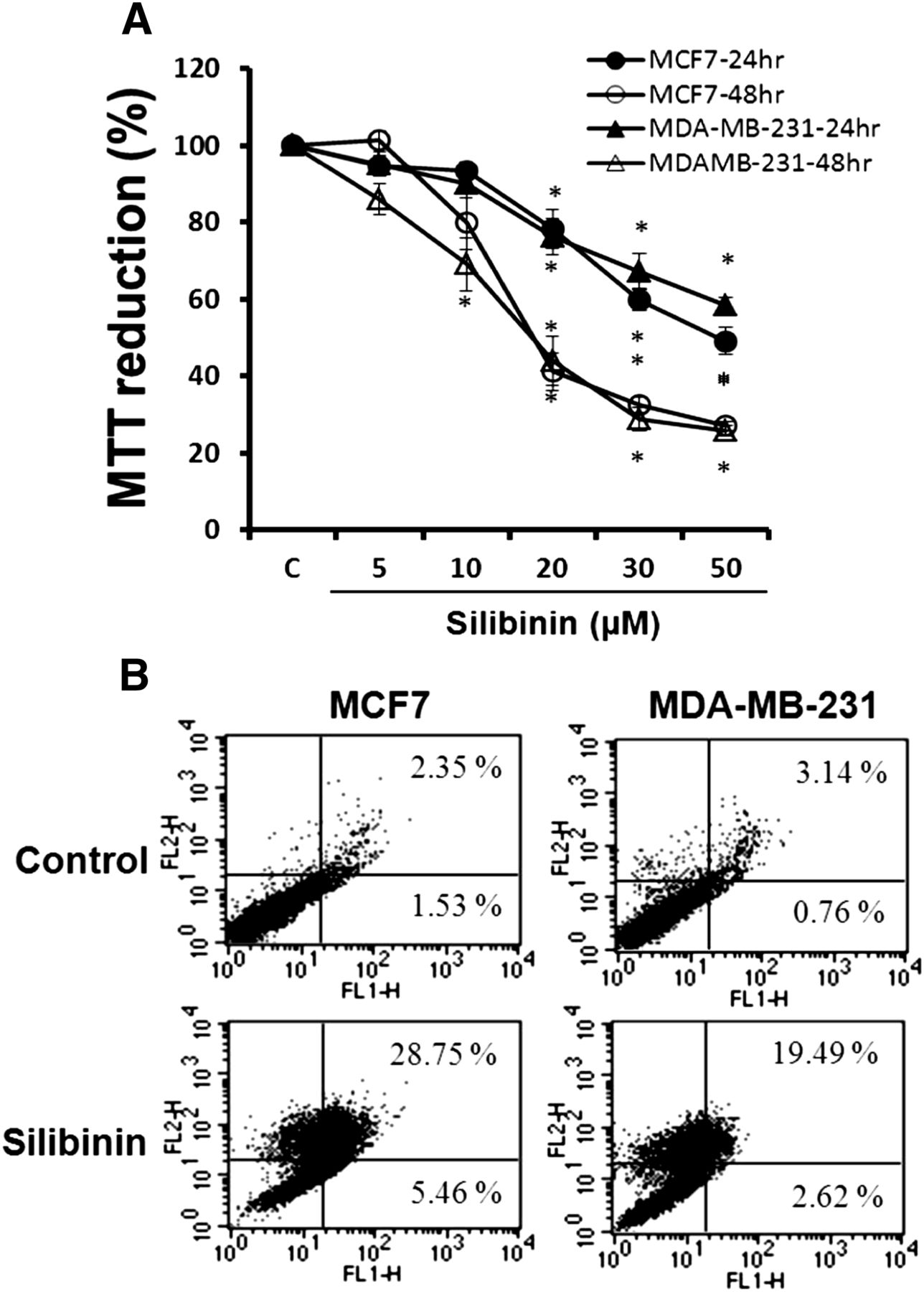
Effects of silibinin on cell viability and apoptosis. Cells were treated with various concentrations of silibinin (5–50 µM) for 24 and 48 hours. Cell viability was determined by a MTT assay (A), and cell apoptosis was estimated by FITC-conjugated annexin V binding assay (B). Data in (A) are the mean ± S.E.M. of three independent experiments performed in duplicate. *P < 0.05 compared with control without silibinin. C, control.
To evaluate whether silibinin-induced decrease of cell viability is attributed to the induction of apoptosis, cells were treated with 30 µM silibinin, and the apoptosis was evaluated using an annexin V binding assay. As shown in Fig. 1B, 34.2% of the MCF7 cells and 22.1% of the MDA-MB-231 cells were annexin V-positive. These results suggest that silibinin decreases cell viability through the induction of apoptosis.
Silibinin Induces ROS Generation.
Many studies have shown that oxidative stress has antitumor effects in cancer cells (Dewaele et al., 2010; Sesti et al., 2012). To examine the effect of silibinin on ROS generation, cells were exposed to silibinin and the changes in DCF fluorescence was measured with flow cytometry. Silibinin increased ROS generation in a time-dependent manner, and it was blocked by an antioxidant, NAC (Fig. 2, A and B). To determine whether ROS generation is involved in silibinin-induced cell death, we tested the effect of antioxidants on cell viability. Silibinin-induced cell death was significantly prevented by antioxidants NAC and 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Fig. 2C). These results suggest that silibinin-induced cell death is associated with ROS generation.

Role of ROS generation in silibinin-induced cell death. Cells were loaded with DCFH-DA for 1 hour and treated with 30 µM silibinin for 12 hours (A) in the presence or absence of 2 mM NAC and indicated various times (B). DCF fluorescence intensity was measured by flow cytometry. (C) Cells were exposed to 30 µM silibinin in the presence or absence of 2 mM NAC and 1 mM 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid for 48 hours. Cell viability was determined by MTT assay. Data in (B) and (C) are the mean ± S.E.M. of three independent experiments performed in duplicate. *P < 0.05 compared with silibinin alone. C, control.
Silibinin Induces Downregulation of Cell Survival Kinases.
ERK and Akt play critical roles in cell proliferation, survival, and differentiation. It is well known that ROS generation is closely related to ERK and Akt signaling. Therefore, we examined whether silibinin-induced cell death is associated with these kinases. Cells were exposed to 30 µM silibinin at various durations, and the activities of these kinases were estimated by measuring their phosphorylation by Western blot analysis. Silibinin-induced downregulation of phospho-ERK and phospho-Akt in a time-dependent manner (Fig. 3A).

Role of cell survival kinases in silibinin-induced cell death. Cells were exposed to 30 µM silibinin at various times. (A) Expression of phospho-ERK (p-ERK), total ERK (t-ERK), phospho-Akt (p-Akt), and total Akt (t-Akt) was evaluated using the specific antibodies. (B) Effect of modulation of ERK and Akt signaling on silibinin-induced cell death. Cells were transfected with EV and caMEK and caAkt. Cells were exposed to 30 µM silibinin for 48 hours. Expression of p-ERK, t-ERK, p-Akt, and t-Akt was estimated by Western blot analysis and quantitative densitometry data. Data are the mean ± S.E.M. of three independent experiments. *P < 0.05 compared with EV with silibinin. (C) Cell viability was estimated by MTT assay. Data are the mean ± S.E.M. of three independent experiments performed in duplicate. *P < 0.05 compared with EV with silibinin. (D) Relationship between cell survival kinases and ROS generation on silibinin-induced cell death. Cells were treated with 30 µM silibinin in the presence or absence of 2 mM NAC for 48 hours. Expression of p-ERK, t-ERK, p-Akt, and t-Akt was estimated by Western blot analysis and quantitative densitometry data. Data are the mean ± S.E.M. of three independent experiments. *P < 0.05 compared with silibinin alone. C, control.
To confirm that the decrease of phosphorylation in these kinases is involved in silibinin-induced cell death, the effect of silibinin on cell death was evaluated in cells transfected with constitutively active (ca)MEK and caAkt. The transfection efficiency was determined to be more than 70% by immunofluorescence (data not shown), and phospho-ERK and phospho-Akt expression were determined by Western blot analysis. Expression of both kinases was increased compared with cells transfected with the empty vector (EV) (Fig. 3B). The cells that were transfected with caMEK and caAkt showed increased resistance to silibinin-induced cell death (Fig. 3C).
To investigate whether the silibinin-induced downregulation of phospho-ERK and phospho-Akt was attributed to ROS generation, cells were exposed to silibinin in the presence or absence of NAC. The decrease of phospho-ERK and phospho-Akt by silibinin was prevented by NAC in both cells (Fig. 3D). These data suggest that the downregulation of phospho-ERK and phospho-Akt is dependent on ROS generation and plays a critical role in silibinin-induced cell death.
Silibinin Inhibits Notch-1 Signaling in Breast Cancer Cells.
We investigated whether silibinin-induced cell death is associated with the Notch signaling pathway. The expression of Notch-1 in silibinin-treated cells was assessed by reverse transcription-polymerase chain reaction and Western blot analysis. As shown in Fig. 4A, silibinin induced downregulation of Notch-1 mRNA expression. Likewise, it decreased Notch-1 protein synthesis time dependently in both cancer cells (Fig. 4B).

Role of the Notch-1 signaling pathway in silibinin-induced cell death. (A) Cells were incubated for 48 hours in the presence (Sili) or absence (Con) of 30 µM silibinin, and Notch-1 mRNA level was detected by reverse transcription-polymerase chain reaction in breast cancer cells. GAPDH was used as a loading control. (B) The cleavage (C.N) and total (T.N) forms of Notch-1 protein level were measured by Western blot analysis at various times. (C) Effect of Notch-1 in silibinin-induced cell death. Cells were transfected with EV and EF.hlCN1.CMV.GFP (Notch1), and cell viability was measured by MTT assay. Data are the mean ± S.E.M. of three independent experiments performed in duplicate. *P < 0.05 compared with EV with silibinin. (D) Relationship between Notch-1 signaling and ROS generation in cells with silibinin. Cells were treated with 30 µM silibinin in the presence or absence of 2 mM NAC for 48 hours. Expression of cleavage (C.N) and total (T.N) forms of Notch-1 protein level was measured by Western blot analysis and quantitative densitometry data. Data are the mean ± S.E.M. of three independent experiments. *P < 0.05 compared with silibinin alone. (E) The association between the Notch-1 signaling pathway and cell survival kinases. Cells were transfected with EV and EF.hlCN1.CMV.GFP (Notch1), and changes in phospho-ERK (p-ERK), total ERK (t-ERK), phospho-Akt (p-Akt), and total Akt (t-Akt) were evaluated by Western blot analysis and quantitative densitometry data. Data are the mean ± S.E.M. of three independent experiments. *P < 0.05 compared with EV with silibinin. C, control.
To evaluate whether the downregulation of Notch-1 is involved in silibinin-induced cell death, cell viability was measured in cells transfected with EF.hlCN1.CMV.GFP. The effective transfection was confirmed by observing the fluorescence of GFP, which was inserted into the CMV promoter, under a fluorescence microscope (data not shown). Both breast cancer cells overexpressing Notch-1 exhibited increased resistance to silibinin-induced cell death (Fig. 4C), indicating that silibinin-induced cell death is associated with downregulation of the Notch-1 signaling pathway in both MCF7 and MDA-MB-231 cells.
To determine whether silibinin inhibits Notch-1 signaling through the increase of ROS generation, cells were pretreated with NAC before exposure to silibinin, and the changes in Notch-1 expression were determined by Western blot analysis. As shown in Fig. 4D, silibinin-induced inhibition of Notch-1 signaling was prevented by the treatment with NAC.
To determine whether silibinin-induced inhibition of Notch-1 signaling is responsible for downregulation of ERK and Akt, the cells were transfected with EF.hlCN1.CMV.GFP, and the changes in phospho-ERK and phospho-Akt were determined by Western blot analysis. Phospho-ERK and phospho-Akt were increased by the EF.hlCN1.CMV.GFP transfection in both breast cancer cells, and transfected cells were recovered by the silibinin-induced downregulation of phospho-ERK and phospho-Akt (Fig. 4E). Results suggest that silibinin-induced inhibition of phospho-ERK and phospho-Akt was attributed to downregulation of Notch-1 signaling in both breast cancer cells.
Silibinin Induces Caspase-Dependent Cell Death in MDA-MB-231 Cells.
Caspases are essential in the apoptosis process. Among them, caspase-3 plays a central role in the execution phase of apoptosis. To determine whether silibinin induces apoptosis through a caspase-dependent pathway, the effect of silibinin on the activation of caspase-3 was examined. Silibinin induced the caspase-3 cleavage in a time-dependent manner in MDA-MB-231 cells. Caspase-3 was not detected in MCF7 cells (Fig. 5A).

Role of caspase-3 in silibinin-induced cell death. (A) Cells were exposed to 30 µM silibinin at various times, and the uncleaved caspase-3 forms (Cas-3) and cleaved caspase-3 forms (Cl. Cas-3) were measured by Western blot analysis using the specific antibody. GAPDH was used as a loading control. (B) Effect of caspase inhibitor on silibinin-induced cell death. Cells were exposed to 30 µM silibinin for 48 hours in the presence or absence of both 10 µM Ac-DEVD-CHO (CHO) and Z-VAD-FMK (FMK). Cell viability was estimated by MTT assay. Data are the mean ± S.E.M. of three independent experiments performed in duplicate. *P < 0.05 compared with silibinin alone. (C) Relationship between caspase-3 cleavage and upstream signaling such as Notch-1, ERK, and Akt on silibinin-induced cell death. Cells were transfected with EV, EF.hlCN1.CMV.GFP (Notch1), caMEK, and caAkt. Cells were exposed to 30 µM silibinin for 48 hours. The uncleaved caspase-3 forms (Cas-3) and cleaved caspase-3 forms (Cl. Cas-3) were estimated by Western blot analysis and quantitative densitometry data. Data are the mean ± S.E.M. of three independent experiments. *P < 0.05 compared with EV with silibinin. GAPDH was used as a loading control. C, control.
To evaluate whether caspase is involved in silibinin-induced cell death, the effect of caspase inhibitors on cell viability was examined. Cells were exposed to silibinin in the presence or absence of the caspase-3–specific inhibitor Ac-DEVD-CHO and the general caspase inhibitor Z-VAD-FMK. Silibinin-induced cell death was prevented by these inhibitors in MDA-MB-231 cells. In MCF7 cells, cell death was prevented by the general caspase inhibitor, whereas it was not affected by the caspase-3–specific inhibitor (Fig. 5B).
To determine whether the effect of silibinin on the activation of caspase-3 is dependent on downregulation of Notch-1 signaling, ERK, and Akt in MDA-MB-231 cells, cells were transfected with EF.hlCN1.CMV.GFP, caMEK, and caAkt and exposed to silibinin, and the activation of caspase-3 was determined. Silibinin-induced caspase-3 activation was prevented by the transfection of EF.hlCN1.CMV.GFP, caMEK, and caAkt (Fig. 5C). These data suggest that silibinin causes caspase-3 activation through downregulation of Notch-1, ERK, and Akt in MDA-MB-231 cells.
Silibinin Induces MCF7 Cell Death through AIF Nuclear Translocation.
To determine whether nuclear translocation of AIF is involved in silibinin-induced cell death, the effect of silibinin on AIF nuclear translocation was measured by Western blot analysis. Silibinin increased the translocation of AIF into the nucleus in a time-dependent manner in MCF7 cells, whereas only a small amount of AIF was translocated into the nucleus in MDA-MB-231 cells compared with MCF7 cells (Fig. 6A). The translocation induced by silibinin was confirmed by immunocytochemistry analysis (Fig. 6B).

Role of nuclear translocation of AIF (Nu-AIF) in silibinin-induced cell death. (A) Cells were exposed to 30 µM silibinin at various times, and the expression levels of cytosol AIF (Cyto-AIF) and Nu-AIF were determined by Western blot analysis using the specific antibody. β-Actin and histone H1 were used as a loading control. (B) Nuclear localization of AIF was estimated by immunocytochemistry. Cells were exposed to 30 µM silibinin for 48 hours in the presence or absence of 2 mM NAC or in cells transfected with caMEK and caAkt. Nuclei were counterstained with Hoechst dye 33258. Green and blue fluorescence indicated AIF and nuclei, respectively. (C) Effect of nuclear translocation of AIF in silibinin-induced cell death. Cells transfected with nonspecific small interfering RNA control (siCon) and siRNA AIF (siAIF) were treated with 30 µM silibinin for 48 hours, and cell viability was determined by MTT assay. Data are the mean ± S.E.M. of three independent experiments performed in duplicate. *P < 0.05 compared with siCon. (D) Effect of Notch signaling, ERK, and Akt pathway on silibinin-induced AIF nuclear translocation. Cells were transfected with EV, EF.hlCN1.CMV.GFP (Notch1), caMEK, and caAkt. Cells were exposed to 30 µM silibinin for 48 hours. The expression levels of Nu-AIF were estimated by Western blot analysis and quantitative densitometry data. Data are the mean ± S.E.M. of three independent experiments. *P < 0.05 compared with EV with silibinin. Histone H1 was used as a loading control. C, control.
To determine whether AIF nuclear translocation was associated with ROS generation and phosphorylation of ERK and Akt, cells were pretreated with NAC and transfected with caMEK and caAkt before exposure to silibinin. The nuclear translocation of AIF was blocked by the treatment with NAC and the transfection of caMEK and caAkt (Fig. 6B).
To evaluate the role of AIF in silibinin-induced cell death, AIF mRNA expression was downregulated by transfection with small interfering AIF RNA. Transfection with small interfering RNA effectively prevented silibinin-induced cell death in the MCF7 cells but not in MDA-MB-231 cells (Fig. 6C).
To determine whether silibinin-induced nuclear translocation of AIF was attributed to downregulation of Notch-1 signaling, phospho-ERK, and phospho-Akt in MCF7 cells, cells transfected with EF.hlCN1.CMV.GFP, caMEK, and caAkt were exposed to silibinin. Silibinin-induced nuclear translocation of AIF was prevented by EF.hlCN1.CMV.GFP, caMEK, and caAkt (Fig. 6D), suggesting that silibinin induces AIF nuclear translocation through downregulation of Notch-1, ERK, and Akt.
Discussion
Silibinin has been shown to have antitumor and anti-inflammation activity in various cancer models. In our previous study, silibinin inhibited glioma cell proliferation in vitro and tumor growth in vivo (Kim et al., 2009). Silibinin is a dietary ingredient that we can take easily from foods such as fruits, vegetables, grains, and tea. It has a wide range of pharmacological effects such as inhibition of DNA synthesis, cell proliferation, cell cycle progression, and apoptosis in various cancer cell lines, including breast cancer cells (Cheung et al., 2010). However, the molecular mechanism of silibinin-induced cell death has not been clearly defined.
ROS is a natural byproduct of the normal metabolism of oxygen and has important roles in cell signaling and homeostasis (Sena and Chandel, 2012). However, the increased ROS level by environmental stress induces DNA damage and thus enhances tumor development (Alfadda and Sallam, 2012). It is well known that flavonoids exhibit antioxidant actions (Agati et al., 2012). However, flavonoids may also behave as a pro-oxidant, which is responsible for cell death in some cancer cells (Chen et al., 2013). In the present study, silibinin induced ROS generation, and the antioxidant NAC prevented silibinin-induced cell death (Fig. 2). These results indicate that silibinin-induced cell death is associated with ROS generation. These results are consistent with those reported in MCF7 cells (Wang et al., 2010). In the present study, we did not define the mechanism by which silibinin induces ROS generation. However, in our previous study, silibinin increased ROS generation through an intracellular Ca2+-dependent mechanism in glioma cells (Kim et al., 2009). Ca2+ influx induces accumulation of Ca2+ in mitochondria (Orrenius et al., 2003), leading to an increased production of ROS by the respiratory chain (Thor et al., 1984). Silibinin may cause an increase in ROS generation through a Ca2+-dependent mechanism in breast cancer cells.
Thus, we can suppose that silibinin has been shown to induce cell death through downregulation of ERK1/2 and Akt activity in various cancer cells as well (Li et al., 2008; Kim et al., 2010; Wang et al., 2012). Therefore, ERK and Akt expression levels may be critical in silibinin-induced cell death. Indeed, the present study showed that silibinin caused inhibition of ERK and Akt phosphorylation time-dependently (Fig. 3A). Silibinin-induced cell death was blocked by the transfection with caMEK and caAkt (Fig. 3C). Similar results have been reported from hepatocellular carcinoma (Cui et al., 2009) and human fibrosarcoma (Duan et al., 2011).
ROS activates multiple signaling pathways such as ERK and Akt, leading to cell survival or cell death (Martindale and Holbrook, 2002). On the other hand, ROS can trigger downregulation of the ERK and Akt pathway (Jeong et al., 2009). In the present study, silibinin-induced inhibition of ERK and Akt was blocked by the antioxidant NAC (Fig. 3D), suggesting that silibinin induces downregulation of ERK and Akt through a ROS-dependent mechanism.
Our previous studies have shown that downregulation of ERK and Akt is involved in flavonoid-induced apoptosis in various cancer cells (Jeong et al., 2009; Cho et al., 2013). However, the underlying mechanism of such a downregulation of kinases remains unclear. Notch signaling promotes cell growth migration, invasion, and apoptosis in various cancer cells. It has been reported that cell survival kinases such as ERK and Akt are involved in the Notch signaling pathway (Miyamoto et al., 2003; Dang, 2012). Therefore, Notch signaling may be involved in the silibinin-induced downregulation of ERK and Akt. To test this possibility, we examined the role of Notch signaling in silibinin-induced cell death. In the present study, silibinin decreased Notch-1 mRNA and protein level. In addition, silibinin-induced cell death was prevented by overexpression of Notch-1 in both cell lines (Fig. 4, A–C), indicating involvement of Notch-1 signaling in silibinin-induced cell death. The cleavage of Notch by a γ-secretase releases Notch intracellular domain into the cytoplasm where it activates the ERK pathway. Activated ERK also upregulates the Notch signaling pathway, resulting in the positive feedback mechanism (Takeshita et al., 2007; Kang et al., 2011). Released Notch intracellular domain into the cytoplasm can also inhibit Akt signaling by PTEN (Cornejo et al., 2011). Therefore, the silibinin-induced downregulation of ERK and Akt may be attributed to inhibition of Notch signaling. Indeed, in the present study, the silibinin-induced inhibition of phospho-ERK and phospho-Akt was prevented by transfection with EF.hlCN1.CMV.GFP (Fig. 4E).
The correlation between ROS generation and Notch-1 signaling is very intricate and follows many steps. Notch-1 suppresses ROS generation (Kim et al., 2007), and conversely, ROS generation could regulate Notch-1 signaling (Coant et al., 2010). In the present study, silibinin increased ROS generation (Fig. 2B) and inhibited Notch-1 protein expression (Fig. 4B). Furthermore, the silibinin-induced downregulation of Notch signaling and cell death was prevented by NAC (Fig. 4D). These results suggest that silibinin-induced downregulation of ERK and Akt was attributed to the inhibition of the Notch-1 signaling pathway, which is dependent on ROS generation.
It is well known that cancer cell apoptosis is associated with the caspase family; in particular, caspase-3 plays a key role during the execution phase in various forms of apoptosis (Fadeel et al., 2008; Fiandalo and Kyprianou, 2012). It is noteworthy that MCF7 breast cancer cells do not express caspase-3 and that caspase-3 is not essential during apoptosis (Mc Gee et al., 2002). In the present study, silibinin induced MDA-MB-231 breast cancer cell death through caspase-3 cleavage, as demonstrated by Western blot analysis. However, caspase-3 expression was not detected in MCF7 cells (Fig. 5A). Selective caspase-3 inhibitor Ac-DEVD-CHO and the general caspase inhibitor Z-VAD-FMK prevented silibinin-induced MDA-MB-231 cell apoptosis. Although silibinin-induced cell death was prevented by Z-VAD-FMK in MCF7 cells, cell death was not affected by Ac-DEVD-CHO (Fig. 5B). This indicates that other caspase mechanisms are involved in silibinin-induced MCF7 cell death. These results are similar to reports that silibinin induces apoptosis through a mechanism independent of caspase-8 activation in MCF7 and T47D breast cancer cells (Tiwari et al., 2011).
AIF is released from mitochondria and translocates from the cytosol to the nucleus. In the nucleus, it induces oligonucleosomal DNA fragmentation, a hallmark of caspase-independent apoptosis (Chou et al., 2009; Vittar et al., 2010; Liu and Chang, 2011). Our data show that silibinin increased nuclear translocation of AIF in both breast cancer cells as demonstrated by the Western blot analysis and immunocytochemistry (Fig. 6, A and B). AIF knockdown prevented cell death in MCF7 cells but not in MDA-MB-231 cells (Fig. 6C). Although AIF nuclear translocation was induced by silibinin treatment in MDA-MB-231 cells, the quantity of translocation may be not sufficient to induce apoptosis, or AIF does not interact with molecules of apoptosis induction in the nucleus. Taken together, these data indicate that silibinin induces MCF7 cell apoptosis through the nuclear translocation of AIF. Although silibinin induced nuclear translocation of AIF in MDA-MB-231 cell, it did not play an important role in silibinin-induced cell death.
In conclusion, silibinin induces ROS-dependent downregulation of Notch/ERK/Akt signaling, which leads to cell death through a caspase-3–dependent mechanism in MDA-MB-231 cells and an AIF-dependent mechanism in MCF7 cells. The data suggest that silibinin induces cell death through different mechanisms between MCF7 and MDA-MB-231 cells (Fig. 7).

Proposed model of silibinin-induced cell death in human breast cancer cell lines. Silibinin increased ROS generation, which inhibited the Notch-1 signaling pathway. These signal transductions led to downregulation of phosphorylation of ERK and Akt. Downregulation of ERK and Akt induced cell death through a caspase-3–dependent mechanism in MDA-MB-231 cells and an AIF-dependent mechanism in MCF7 cells.
Authorship Contributions
Participated in research design: T. H. Kim, J. S. Woo, Y. K. Kim,
K. H. Kim.
Conducted experiments: T. H. Kim.
Performed data analysis: T. H. Kim, Y. K. Kim.
Wrote or contributed to the writing of the manuscript: T. H. Kim, J. S. Woo,
Y. K. Kim, K. H. Kim.
Footnotes
- Received July 6, 2013.
- Accepted January 24, 2014.
This study was supported by the National R&D Program for Cancer Control, Ministry for Health, Welfare and Family Affairs, Republic of Korea [Grant 0920050].
Abbreviations
- Ac-DEVD-CHO
- N-acetyl-Asp-Glu-Val-Asp-Chinese hamster ovary
- AIF
- apoptosis-inducing factor
- ca
- constitutively active
- CMV
- cytomegalovirus
- DCF
- 2′,7′-dichlorofluorescein
- DCFH-DA
- 2′,7′-dichlorofluorescein diacetate
- ERK
- extracellular signal-regulated kinase
- EV
- empty vector
- GFP
- green fluorescent protein
- FITC
- fluorescein isothiocyanate
- MEK1
- mitogen-activated protein kinase 1
- MTT
- 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- NAC
- N-acetylcysteine
- PBS
- phosphate-buffered saline
- ROS
- reactive oxygen species
- Z-VAD-FMK
- N-benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethyl ketone
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}