Article Text

Abstract
Background The clinical significance of tumor-specific genomic alterations in metastatic renal cell carcinoma (mRCC) is emerging, with several studies suggesting an association between PBRM1 mutations and response with immunotherapy (IO). We sought to determine genomic predictors of differential response to vascular endothelial growth factor–tyrosine kinase inhibitors (VEGF-TKIs) and IO.
Methods Consecutive patients who underwent genomic profiling were identified; patients receiving either VEGF-TKIs or IO were included. Clinical tumor-normal whole exome sequencing and tumor whole transcriptome sequencing test were performed using a Clinical Laboratory Improvement Amendments (CLIA)-certified assay (Ashion Analytics; Phoenix, Arizona, USA). Genomic findings were compared between patients with clinical benefit (CB; complete/partial response or stable disease for >6 months) and no clinical benefit (NCB) in VEGF-TKI-treated patient cohort and IO-treated patient cohort.
Results 91 patients received genomic profiling and 58 patients received VEGF-TKI and/or IO therapy. 17 received sequenced treatment involving both VEGF-TKI and IO, resulting in 32 patients in the IO cohort and 43 patients in the VEGF-TKI cohort. The most commonly used IO and VEGF-TKIs were nivolumab (66%) and sunitinib (40%). The most frequently detected alterations in the overall cohort were in VHL (64%), PBRM1 (38%), SETD2 (24%), KDM5C (17%) and TERT (12%). TERT promoter mutations were associated with NCB in the IO cohort (p=0.038); transcriptomic analysis revealed multiple differentially regulated pathways downstream of TERT. TERT promoter mutations and PBRM1 mutations were found to be mutually exclusive. While PBRM1 mutations were more prevalent in patients with CB with IO and VEGF-TKIs, no statistically significant association was found.
Conclusions Our analysis found that TERT promoter mutations may be a negative predictor of outcome with IO and are mutually exclusive with PBRM1 loss-of-function mutations.
- immunotherapy
- genetic markers
- kidney neoplasms
- tumor biomarkers
- translational medical research
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Systemic therapy for metastatic renal cell carcinoma (mRCC) is rapidly evolving. In the late 1990s and early 2000s, the use of the cytokine-based therapy with interleukin-2 and interferon-alpha was the standard of care for first-line treatment of mRCC.1 Then with the development of vascular endothelial growth factor (VEGF) and mammalian target of rapamycin (mTOR) inhibitors, the use of tyrosine kinase inhibitors (TKIs) began to supplant these cytokine-based agents due to their favorable toxicity profile and higher efficacy.1 2 Then around 2015, another paradigm shift in mRCC treatment occurred with the US Food and Drug Administration approval of immune checkpoint inhibitors or immunotherapy (IO) agents such as nivolumab or pembrolizumab.2 3 The current standard of care for first-line mRCC treatment entails using a VEGF-TKI (ie, cabozantinib), doublet IO agents (ie, nivolumab and ipilimumab), or combination of VEGF-TKI and IO agent (ie, axitinib and pembrolizumab).4–6
As a result, there are many more choices available for systemic therapy in patients with mRCC, which has greatly helped improve patient outcomes. However, due to the rapid development of these new treatments, the optimal strategy to sequence these regimens has yet to be fully established. Current expert consensus based on the best supporting evidence available recommends the use of a patient risk factor stratification model using the International mRCC Database Consortium (IMDC) criteria and other clinical factors such as relative efficacy and toxicity of the selected agent in the context of the clinical setting.7 8 However, due to the inherent subjectivity and bias introduced by using these clinical factors, there is much effort to develop objective laboratory-based biomarkers to aid in this decision-making process. A recent study done by Miao et al proposed an association between genomic alterations in PBRM1, a chromatin remodeling gene, and response to nivolumab with an OR for clinical benefit (CB) of 12.93 based on analysis of a prospective trial evaluating nivolumab monotherapy.9 Findings were consistent when this association was tested in a multi-institutional dataset comprising patients treated with either a single agent or combination immunotherapies.
While these data are compelling, one potential shortcoming is the absence of a comparator group to establish PBRM1 as predictive as opposed to prognostic biomarker. To this end, Braun et al have explored genomic predictors of IO response by analyzing the CheckMate-025 dataset.10 In this phase III clinical trial comparing nivolumab and everolimus in patients who previously received VEGF-directed therapy, there again appeared to be an association between nivolumab response and PBRM1 mutation with a hint toward predictive capability against everolimus. However, according to the most recently published National Comprehensive Cancer Network (NCCN) and European Society for Medical Oncology (ESMO) guidelines, the current standard of care for first-line treatment of mRCC revolves around choosing between VEGF inhibitors (not mTOR inhibitors) and checkpoint blockade.7 11 Despite all the advances in the field, the treating oncologist at the time of this publication still has no well-established objective biomarkers which can help guide the optimal treatment for the patient with mRCC. In order to help address this gap in knowledge, we evaluated the clinical outcomes and genomics data from a large series of patients at our institution treated with VEGF inhibitors and/or checkpoint inhibitors with the intent of identifying genomic predictors for VEGF-TKI and IO response.
Methods
Patient selection
After obtaining institutional review board approval, we retrospectively identified 91 patients with mRCC who had undergone comprehensive genomic profiling as part of routine clinical care at the City of Hope Comprehensive Cancer Center (Duarte, California, USA) between July 2018 and September 2019. Figure 1 depicts the flow diagram of patient inclusion for cohort analysis. Patients’ demographics and clinical variables including IMDC risk score criteria, treatment type, treatment response and survival outcomes were obtained by reviewing the patients’ electronic medical records and abstracted into a coded database deficient of any patient health identifiers to ensure privacy. Patients who received either an immunotherapy or a VEGF-TKI regimen for treatment of metastatic disease and had received genomic profiling of their tumor were deemed eligible. Treatment response was evaluated via RECIST 1.1 (Response Evaluation Criteria In Solid Tumors) criteria. Patients with a best response of complete response (CR) or partial response (PR) regardless of duration or patients with stable disease (SD) for a duration of at least 6 months were considered to have experienced CB. Patients with progressive disease as the best response to treatment were considered to have no clinical benefit (NCB). We only included patients whose genomic profiling was performed prior to initiation of a systemic treatment, had a follow-up duration of more than 6 months, and treatment responses were evaluable at the time of data cut-off.

Flow diagram detailing inclusion and exclusion of the participants. IO, immunotherapy; mRCC, metastatic renal cell carcinoma; TT, targeted therapy.
Genomic analysis
Genomic profiling was performed via the GEM ExTra assay in a College of American Pathologists (CAP)-accredited, Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory through Ashion Analytics (Phoenix, Arizona, USA) and encompassed comprehensive DNA (paired tumor-normal whole exome sequencing (WES)) and RNA (tumor whole transcriptome sequencing) next-generation sequencing. Tumor whole exome and transcriptome sequencing was performed on tumor samples obtained as part of routine diagnostic clinical care. Paired normal WES was performed using peripheral blood monocytes. Tumor-normal WES data were available for 58 patients in the analysis cohort, of which 49 patients also had matched tumor whole transcriptome data available. The mean target coverage for exome sequencing was 402X for tumor and 258X for normal. RNA sequencing averaged 187 million aligned reads.
Briefly, following analyte extraction with the Qiagen AllPrep kit and DNA sheering, exome libraries were generated with the KAPA HyperPrep Library Kit (Roche) and captured with a custom IDT xGen probe set targeting 19,396 genes and intronic regions containing breakpoints for 712 known translocation events as well as providing coverage for hotspot mutations within the TERT promoter. RNA libraries were prepared following ribosomal RNA depletion and whole transcriptome random primed reverse transcription (KAPA RNA HyperPrep with Riboerase kit, Roche). Libraries were sequenced using Illumina NovaSeq 6000 reagents and instruments.
Following BCL to FASTQ conversion with bcl2fastq, data were aligned to build 37 of the human reference genomes using BWA-MEM followed by duplicate removal and sorting. Somatic single-nucleotide variants and small indels were identified using Freebayes (Garrison, Erik & Marth, Gabor 2012 arXiv 1207), filtering for variants detected on both strands, a tumor allele frequency 5% or greater, a normal allele frequency less than or equal to 3%, and a tumor:normal allele frequency ratio of 10 or greater. A 2×2 contingency table of reference and alternate alleles in tumor and normal samples was constructed for the remaining variants, and Fisher’s exact test and the Benjamin and Hochberg procedure were used to call somatic variants, targeting a false discovery rate of <1%. Structural variants were detected using Manta, and amplifications and deletions were called using a custom in-house algorithm calculated on a per gene basis based on read count ratios in tumor versus normal samples (amplified: log2 ratio >1; deleted: log2 ratio <−1.35).12 Tumor mutation burden was calculated using a custom in-house algorithm, counting all somatic, protein coding variants predicted to change the amino sequence of the impacted protein in the SnpEff-annotated Freebayes VCF file.
For whole transcriptome analysis, FASTQ files were aligned using STAR. Ashion analysis included fusion detection using STAR-Fusion and FusionInspector, and detection of clinically actionable alternative transcripts (ie, EGFRvIII, ARv7) using a custom algorithm based on detection of unique 32 nucleotide sequences to identify these alternative transcripts. Global transcript quantification and differential expression analysis were performed in the research setting. Briefly, Salmon (V.0.14.1) was run in the quasi-mapping mode using the GRCh38 build to estimate transcript abundance, quantified in transcripts per kilobase million reads values and leveraging transcript IDs to gene names to produce gene level abundance estimates.13 Gene level count files were processed using DESeq2 (Bioconductor).
Statistical analysis
A Student’s t-test was used to evaluate the association between tumor mutational burden (TMB) and clinical benefit. A two-tailed Fisher’s exact test was used to assess enrichment of genomic alterations (categorical value, gene mutated or non-mutated) and clinical benefit (CB, NCB). P values <0.05 were considered significant. Analysis was performed using GraphPad Prism V.8.3.0.
Results
Patient characteristics
Figure 1 outlines the inclusion criteria for the analysis cohorts. Of the 91 patients with mRCC that received genomic profiling as part of standard clinical care, 58 patients were included in the analysis. A total of 17 patients received both immunotherapy and VEGF-TKI therapy sequentially. This resulted in 43 patients in the VEGF-TKI therapy and 32 patients in the immunotherapy-treated cohorts, respectively.
Patient characteristics based on treatment are presented in table 1. Median age was 63.2 (range 31.6–84.0), and 43 (74%) patients were male. Clear cell histology was observed in 47 (81%) patients, while 11 (19%) patients had non-clear cell RCC. Non-clear cell RCC subtypes included six patients with papillary RCC, three patients with chromophobe RCC and two patients with sarcomatoid RCC. The majority (77%) of patients in the VEGF-TKI therapy cohort received treatment in the first-line setting. The most commonly used VEGF-TKI therapies were sunitinib (n=17, 40%), cabozantinib (n=9, 21%) and the combination of lenvatinib/everolimus (n=6, 14%). One-third (n=11, 34%) of the patients included in the immunotherapy cohort received nivolumab/ipilimumab combination in the first-line setting. The remaining patients in the immunotherapy cohort received nivolumab in the second-line or third-line setting.
Patient characteristics and clinical outcomes
In the VEGF-TKI therapy cohort, 2 (5%) patients achieved a CR, 7 (16%) achieved a PR and 28 (65%) patients achieved SD, of which 25 (58%) maintained SD for over 6 months. In the immunotherapy cohort, the best response was CR in 1 (3%) patient, PR in 6 (19%) patients, and SD in 12 (37%) patients, of which 10 (31%) maintained SD for over 6 months. CB rate was 79% in the VEGF-TKI therapy cohort and 53% in the immunotherapy cohort. After a median follow-up of 14.2 months (95% CI 9.5 to 18.9), median progression-free survival was 14.2 (95% CI 9.0 to 18.5) months in the VEGF-TKI therapy cohort and 15.6 (95% CI NR to NR) months in the immunotherapy cohort.
Comprehensive genome analyses
Clinical tumor-normal WES of the overall cohort yielded frequent genomic alterations in the VHL (64%), PBRM1 (38%), SETD2 (24%), KDM5C (17%), MTOR (12%) and TERT (12%) genes. Figure 2 represents the genomic landscape of the study population along with treatment information and CB outcomes. Notably, TERT promoter mutations were found to be mutually exclusive of PBRM1 mutations.

Genomic landscape of the entire cohort included in the analysis. IO, immunotherapy; TT, targeted therapy.
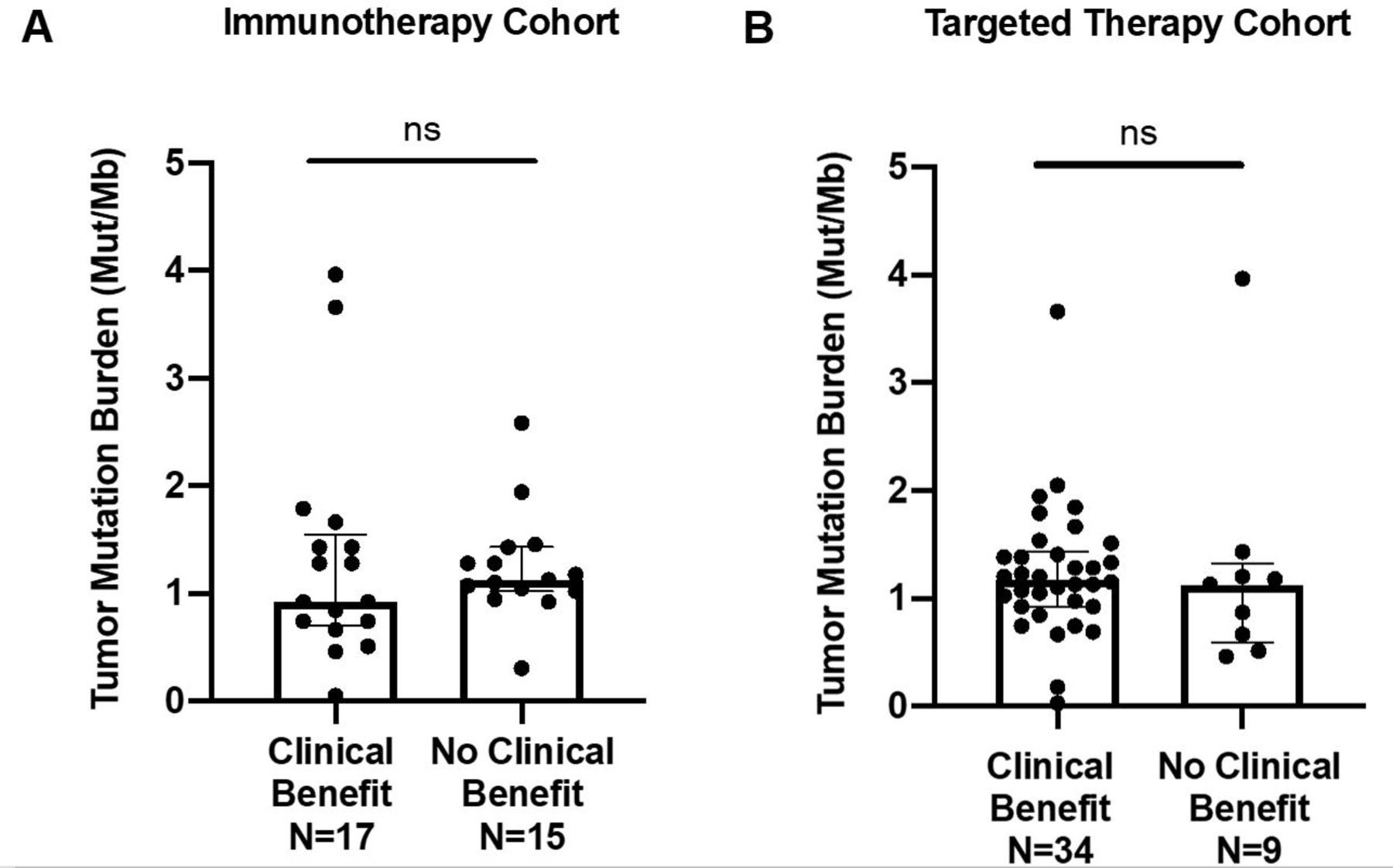
Median TMB of the overall cohort was 1.2 mutations/Mb (range 0.03–4.0). Box plots demonstrating the comparison of TMB between CB and NCB patients in immunotherapy and VEGF-TKI therapy cohorts can be appreciated in figure 3. No statistical significance was observed between CB and NCB patients in either cohort.
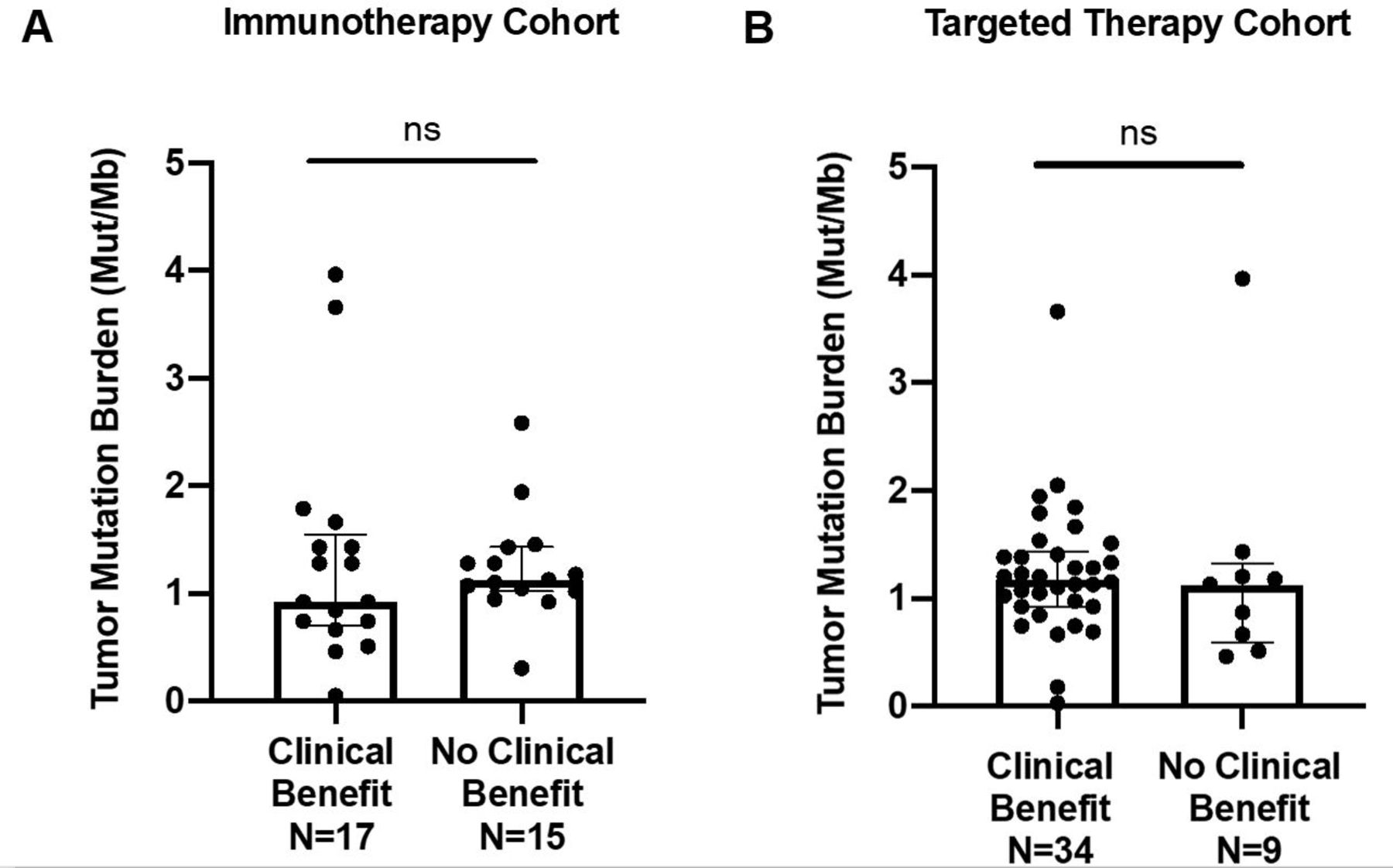
Comparison of tumor mutational burden (TMB) of patients with clinical benefit versus no clinical benefit in (A) immunotherapy (p=0.82) and (B) targeted therapy (p=0.91) cohorts. Individual tumor TMB values are represented in the dot plots, and median TMB with IQRs for each group are presented. ns, not significant.
In the immunotherapy cohort, PBRM1 loss-of-function mutations were more frequent in patients with CB, whereas VHL, SETD2, KDM5C, CACNA1D, and TP53 alterations were more frequent in patients with NCB (figure 4A). However, the differences did not reach statistical significance (p values>0.05). TERT promoter mutations were found to be enriched in patients with NCB in the immunotherapy-treated cohort (p=0.038). Moreover, none of the patients with TERT promoter mutated tumors obtained CB from immunotherapies (figures 2 and 4A). In the VEGF-TKI therapy-treated cohort, no single genes were found to be associated with CB (figure 4B).
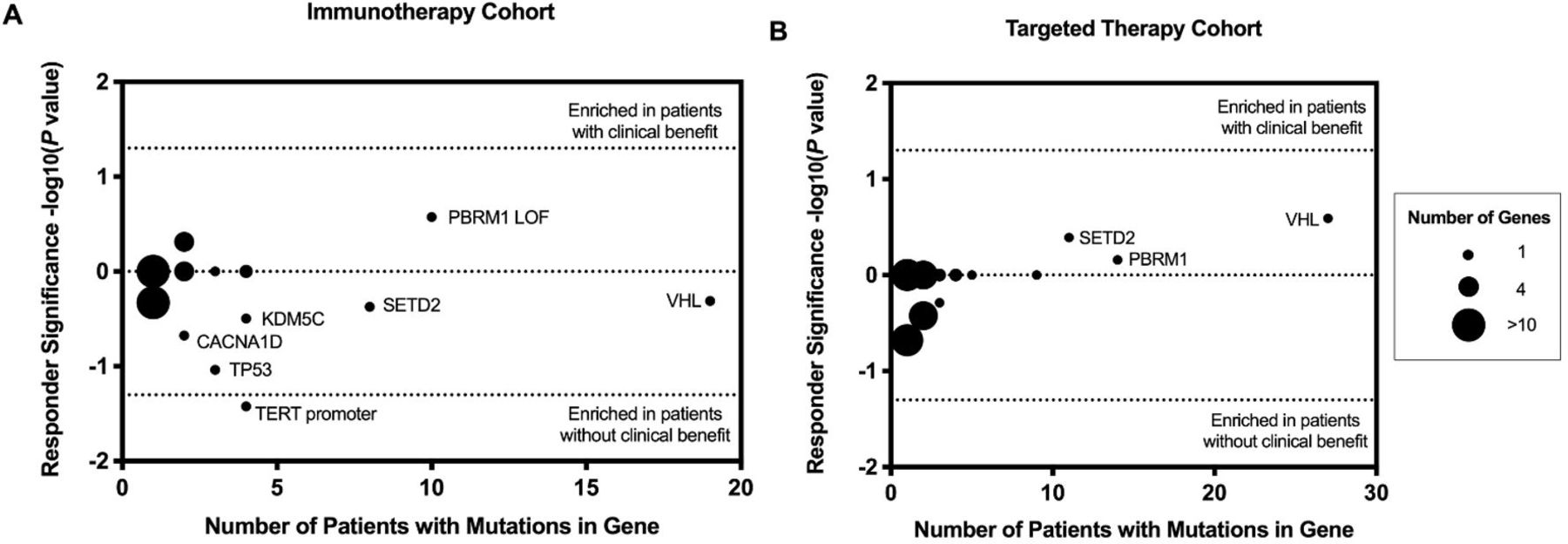
Genomic alterations associated with clinical benefit from (A) immunotherapies and (B) targeted therapies. Enrichment determined by Fisher’s exact test with p value<0.05. Immunotherapy cohort: TERT promoter mutation (p=0.038). Other genes were not significantly associated with clinical benefit (TP53 (p=0.092), CACNA1D (p=0.212), PBRM1 loss-of-function (LOF) mutations (p=0.265), KDM5C (p=0.319), SETD2 (p=0.423), VHL (p=0.491)). No genes were associated with clinical benefit to targeted therapy (VHL (p=0.257), SETD2 (p=0.407), PBRM1 (p=0.693)).
Transcriptome analyses
Transcriptional analysis was then carried out in the IO cohort to determine if there were any significant gene expression signature changes in the TERT promoter-mutated samples that were correlated with NCB compared with the TERT promoter wild-type samples. Of the 32 patient samples who were initially included in the IO cohort for the above-mentioned WES analysis, 28 had RNA-seq data that met the quality control measures of the GEM Extra assay. Of these samples, four (14%) were from TERT promoter mutated with NCB and the remainder were TERT promoter wild type (n=24, 86%). The top differentially expressed genes with higher levels of enrichment in the TERT promoter-mutated group included SST, CYSLTR2, WNK2, and PTGES (online supplementary table S1). Overall, 135 genes were found to be significantly more enriched in the TERT-mutated samples compared with the wild type. Downstream gene set enrichment and pathway analysis with ENRICHR and X2kweb analysis further identified MYC and KAT2A as enriched transcription factor pathways within TERT promoter mutant tumors (online supplementary figure S1). The X2Kweb pathway analysis found that the top enriched kinase pathways were CSNK2A1, CDK1, MAPK14, ATM, and CDK4 (online supplementary figure S2). The final step of the X2K network analysis for the inferred upstream regulatory network integrating results from the transcription factor and kinase pathway analysis with expansion of further predicted targets found that the key kinase nodes were GSK3beta, ATM, CDK4, MAPK14, DNAAPk, and CK2alpha (figure 5).
Supplemental material
Supplemental material
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression2Kinases2 network analysis for genes enriched in TERT-mutated IO samples.
Discussion
Our study suggests that while PBRM1 appears to have a prognostic role, trending toward enrichment among patients with CB from both VEGF-TKI therapy and immunotherapy, it does not appear to be predictive. One novel finding in our series is the identification of TERT promoter mutation as a potential predictor of lack of benefit from immunotherapy. The same association was not identified in patients receiving VEGF-TKI therapy. Whole transcriptome analysis found that TERT-mutated patient samples from IO non-clinical benefit patients were significantly enriched for genes that were transcription factor targets of MYC and KATA2 and kinase targets of CDK4, ATM, and MAPK14.
The TERT gene on chromosome 5 p is expressed at high levels in the vast majority of human cancers.14 The enzymatic product of the TERT gene plays a role in chromosomal elongation. The frequency of TERT alteration in mRCC is less than 10%, but recent series suggest that mutations in the TERT promoter may confer a poor prognosis.15 16 Specifically, Casuscelli et al examined a series of 281 patients with both clear cell and non-clear cell RCC—the presence of TERT promoter mutations was associated with a HR of 2.68 (95%CI 1.19 to 6.01; p=0.013) for cancer-specific survival. From a biological perspective, TERT promoter mutations may be associated with upregulation of ETS-mediated signaling.17 This, in turn, could upregulate hypoxia-inducible genes and confer sensitivity to VEGF-directed agents. This sensitivity may not extend to patients receiving immunotherapy.
Our findings around PBRM1 are not necessarily inconsistent with previous findings. As noted previously, PBRM1 mutation has been linked to benefit with nivolumab in several prior studies.9 10 However, there are also several studies in which PBRM1 mutation has been linked to benefit from VEGF-directed agents. Voss et al explored the role of several genes of interest in the context of the randomized COMPARZ and RECORD-3 trials.18 COMPARZ, a randomized, phase III study comparing sunitinib to pazopanib, was used as a training cohort, while RECORD-3, a randomized, phase II study comparing sunitinib and everolimus, was used as a validation cohort. In this study, mutation in PBRM1 was associated with a significant survival advantage across patients (HR 1.58, 95% CI 1.16 to 2.14; p=0.0035). Taking into consideration the studies cited herein, it is no surprise that our dual analysis of patients treated with VEGF-directed therapies and immunotherapy shows a prognostic, but not predictive, role of PBRM1.
Limitations of this study include its retrospective design and modest sample size. Additionally, tissue for genomic profiling was derived from heterogeneous sites, including both primary tumor and sites of metastases. Patients in this series may have been exposed to multiple types of therapy across multiple lines. Bearing this in mind, if patients received immunotherapy prior to VEGF-directed agents, for instance, there may be some residual benefit from previous lines of therapy. This could cloud our assessment of clinical benefit. Another potential limitation in our study is the inclusion of non-clear cell histologies. This amounted to approximately 20% of our study population, mirroring the frequency of these histologies seen in a real-world setting. Currently, in non-clear cell RCC, the same clinical conundrum exists—namely, whether VEGF-directed therapy or immunotherapy represents a superior option. Response rates to date are quite similar with immunotherapy (eg, pembrolizumab) and VEGF-directed therapy (eg, cabozantinib) at approximately 20%–30% in different series.19 20 These response rates are proportionately lower than response rates observed in clear cell RCC, and we therefore felt it was reasonable to include this subset of patients. Finally, median duration of follow-up in the immunotherapy cohort was somewhat shorter than in the targeted therapy cohort.
Conclusions
Our results confirm previous observations that PBRM1 has a prognostic role in patients with mRCC treated with VEGF-directed therapy and immunotherapy but cannot predict benefit between these classes of agents. However, further studies should be undertaken to investigate whether TERT mutations could have this predictive capability. Until biomarkers are developed to aid in treatment selection, mRCC therapy will remain in a state of equipoise.
Acknowledgments
The authors wish to thank Drs Sheri Skerget, William PD Hendricks, J Jonathan Keats, Nicholas Schork and Thomas Royce for helpful discussions.
References
Footnotes
Twitter @NazliDizman
ND and YL contributed equally.
Contributors Study concept and design: ND, YL, SB, JMT, SP. Acquisition, analysis, or interpretation of data: all authors. Drafting of the manuscript: ND, YL, SB, NS, PGB, JH, SP. Critical revision of the manuscript for important intellectual content: all authors. Statistical analysis: ND, YL, SB, DE, TI. Study supervision: SB, JT, SP.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests SKP reports consulting for Genentech, Aveo, Eisai, Roche, Pfizer, Novartis, Exelixis, Ipsen, BMS and Astellas.
Patient consent for publication Not required.
Ethics approval The City of Hope Comprehensive Cancer Center institutional review board approved this study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement No data are available. Datasets generated and/or analyzed during the current study are not publicly available since data sharing has not been included in the institutional review board approval.