


Abstract
Triple-negative breast cancer (TNBC) lacks expression of estrogen receptor (ER), progesterone receptor (PR) and HER2 gene. It comprises approximately 15-20% of breast cancers (BCs). Unfortunately, TNBC's treatment continues to be a clinical problem because of its relatively poor prognosis, its aggressiveness and the lack of targeted therapies, leaving chemotherapy as the mainstay of treatment. It is essential to find new therapies against TNBC, in order to surpass the resistance and the invasiveness of already existing therapies. Given the fact that epigenetic processes control both the initiation and progression of TNBC, there is an increasing interest in the mechanisms, molecules and signaling pathways that participate at the epigenetic modulation of genes expressed in carcinogenesis. The acetylation of histone proteins provokes the transcription of genes involved in cell growth, and the expression of histone deacetylases (HDACs) is frequently up-regulated in many malignancies. Unfortunately, in the field of BC, HDAC inhibitors have shown limited effect as single agents. Nevertheless, their use in combination with kinase inhibitors, autophagy inhibitors, ionizing radiation, or two HDAC inhibitors together is currently being evaluated. HDAC inhibitors such as suberoylanilidehydroxamic acid (SAHA), sodium butyrate, mocetinostat, panobinostat, entinostat, YCW1 and N-(2-hydroxyphenyl)-2-propylpentanamide have shown promising therapeutic outcomes against TNBC, especially when they are used in combination with other anticancer agents. More studies concerning HDAC inhibitors in breast carcinomas along with a more accurate understanding of the TNBC's pathobiology are required for the possible identification of new therapeutic strategies.
According to the World Health Organization, breast cancer (BC) is the most frequently diagnosed cancer and the second leading cause of cancer-related death, among women worldwide. Interestingly, according to the American Cancer Society about 12% of women in U.S.A. will develop BC during their lifetime (1-4). There is a variety of risk factors that are associated with BC such as gender, age, ethnicity or lifestyle, but it is now clear that the most dangerous factor for the development of BC is heredity. In particular, in high-risk families, there have been reported more than 1.000 mutations of breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes (1).
BC is a very heterogeneous disease given that its invasive process is associated with a variety of molecular alterations. These changes lead to different subtypes of the disease. The current classification divides BC subtypes into Luminal A, Luminal B, HER2-positive and TNBC (5, 6). A better explanation of BC classification and the corresponding cell lines are presented in Table I (7, 8).
First of all, Luminal A and B represent from 65% to 80% of total BC disease. More specifically they account 50-60% and 15-20% respectively (9). These subtypes are generally correlated to a good prognosis. They are known to express the estrogen receptor (ER-positive) that participates in cell proliferation, viability, and invasion of BC cells. We should mention that patients with luminal B tumors frequently present a worse prognosis than Luminal A patients. This could be explained as in Luminal B tumors HER2 gene, which is associated with potent proliferation, can be amplified (10). The substantial difference though, between Luminal A and B patients is the cell proliferation rate which is higher in the latter.
Secondly, there is the subtype of HER2-positive patients who amplify the oncogene HER2. These tumors are ER negative, so they are different from Luminal B cancers. Unfortunately, hormonotherapy is inefficient against HER2-positive tumors. During the last 20 years the development of novel drugs targeting HER2 (e.g. trastuzumab, lapatinib, pertuzumab) has enhanced the clinical outcomes (1).
Moreover, another severe subtype of BC is triple negative breast cancer (TNBC). It is associated with poor prognosis. TNBC does not express either estrogen, or progesterone receptors, or HER2 gene. These tumors can be further classified in several subtypes. The first subgroup is basal-like, where tumors express some characteristics of breast myoepithelial cells. Basal-like tumors are highly proliferative and are associated with very poor prognosis. Another subgroup is claudin-low, which presents epithelial to mesenchymal transition (EMT) and stem cell-like or/and tumor initiating cell features (11). This subtype is also associated with poor prognosis.
Neoadjuvant anthracycline/taxane-based chemotherapies (e.g., docetaxel, doxorubicin, cyclophosphamide) have been tested in TNBC patients with poor prognosis (5).
Classification of breast cancers and their relation to breast cancer cell lines.
HDACs and their Mechanism of Action
Today, several BC subtypes are treated with many different primary therapeutic protocols, but none of them uses epigenetic drugs, despite the fact that the interest for epigenetics in BC tumorgenesis is increasing (12, 13).
Gene regulation is affected by nucleosome packaging. Nucleosome is the organizing DNA structure that is consisted of about a 200 bp DNA coiled around an octameric core, consisted of histone proteins (14).
N-terminal tails of histones extend outward from the nucleosomal core and are modified by the covalent addition of groups to the side chains of certain amino acids. These modifications, including methylation, acetylation, phosphorylation and the reverse processes, can change the secondary DNA structure. As a result, gene promoter regions become accessible or inaccessible to transcription factors (15).
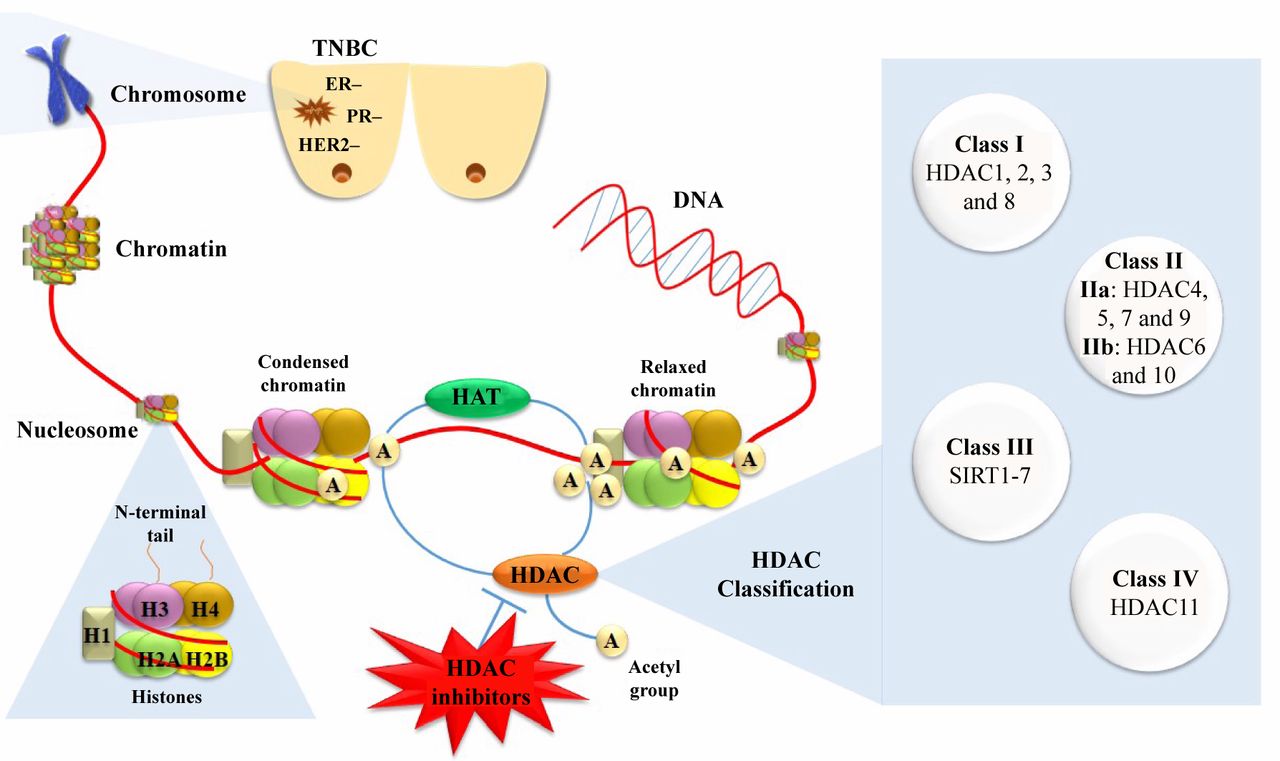
Histone acetyltransferases (HATs) catalyze the reversible process of lysine acetylation at the ε-amino group of proteinogenic lysine residues. Histone acetylation neutralizes the positive charge of lysine residues, therefore is correlated to chromatin relaxation and active gene transcription (17). On the other hand, functional antagonists of HATs, histone deacetylases (HDACs) remove the acetyl groups (16), thus leading to compressed chromatin structure (heterochromatin), and subsequently suppressing gene transcription (18) (Figure 1). Except for the direct effect of acetylation on chromatin structure, gene regulation through acetylation is based on synergistic actions. The specific acetylation patterns on histone tails recruit further chromatin modulators that form co-repressor or co-activator complexes.
Several human HDACs have been identified; recently HDACs have been classified into four classes in accordance to functional criteria and homology to yeast proteins (19). In general, HDACs can be divided into Zn2+-dependent classes (class I, II and IV) and NAD-dependent classes (class III). Class I is consisted of HDACs 1, 2, 3 and 8. Class II can be divided further into class IIa (HDAC4, 5, 7, and 9) and class IIb (HDAC6 and 10). Class III members, or sirtuins as they are often called since they are homologous to silent information regulator 2 (SIR2) of Saccharomyces cerevisiae, consist of SIRT1-SIRT7. Finally, class IV has only one member, HDAC11 (20) (Figure 1).

Therapeutic strategy targeting histone deacetylases against triple-negative breast cancer and histone deacetylase inhibitors classification. TNBC: Triple-negative breast cancer; HAT: histone acetyltransferases; HDAC: histone deacetylases.
HDAC Inhibitors as Anti-cancer Agents
HDAC inhibitors can be classified into four classes according to their chemical structure: a) hydroxamates (e.g. suberanilohydroxamic acid (SAHA)), b) benzamides (e.g. MS-275), c) cyclic peptides (e.g. romidepsin) and d) aliphatic acids (e.g. valproic acid). Alternatively, HDAC inhibitors can be classified according to their specificity for HDAC subtypes or classes. For example, SAHA and trichostatin A are pan-HDAC inhibitors, while MS-275 and romidepsin inhibit class I and valproic acid inhibits class I and IIa HDACs (21).
It is clear that both histone acetylation and deacetylation affect chromatin remodeling as strong epigenetic mechanisms. Interestingly, evidence from several reports indicates that HDAC levels are increased in certain cancer types (22-24). In addition, HDAC inhibitors have been reported to enhance the acetylation of histones, in tumor cells (25). Unlike other cytostatic-type compounds, HDAC inhibitors have been reported to exert much lower cytoxicity on normal cells, than on cancer cells. In general, HDAC inhibitors induce the inhibition of tumor growth and the apoptosis of cancer cells.
Clinical trials (phases I and II) have also demonstrated that HDAC inhibitors result in minor adverse effects in patients (15, 26-30). Their mechanism of action involves binding of their hydroxamate group to the zinc cation (Zn2+) located in the HDAC cavity (31). Several clinical trials seemed to have a beneficial result. For instance, the US Food and Drug Administration has approved SAHA and romidepsin as regimen of cutaneous T-cell lymphoma (29, 31) and peripheral T-cell lymphoma (27) respectively. Moreover, panobinostat treatment is reported to be clinically successful against multiple myeloma (28).
In total, advanced stages of clinical trials have studied several HDAC inhibitors against many cancer types. However, concerning the TNBC field, studies have shown that, in general, HDAC inhibitors succeed clinically beneficial results as complementary treatment (e.g. SAHA and VPA), or in combination with cytotoxic drugs and ionizing radiation (Table II).
SAHA. SAHA or vorinostat is a pan-HDAC inhibitor that induces apoptosis in several types of haematological and solid tumor cells (32). Clinical investigations show that SAHA is a potent inhibitor against tumor types at doses that were well tolerated by patients (33).
According to a study in human TNBC cell lines, MDA-MB-231 and inBT-549, SAHA significantly promotes in vitro motility via activation of epithelial–mesenchymal transition (EMT) phenotype (34). This phenotype is known as the first and critical point for metastatic dissemination of cancer cells. Vimentin up-regulation and E-cadherin down-regulation are considered as markers of EMT (35, 36).
Histone deacetylase inhibitors and their action against triple-negative breast cancer.
Wu et al. found that SAHA treatment induces the expression of the mesenchymal markers N-Cadherin, Vimentin and Fibronectin, while decreases the expression of epithelial marker E-Cadherin. Nevertheless, SAHA treatment does not change neither the expression nor the nuclear translocation of transcription factors related to EMT. These factors were zinc finger proteins SNAI1 (SNAIL) and SNAI2 (SLUG), basic helix-loop-helix transcription factor Twist-related protein (TWIST) and Zinc finger E-box-binding homeobox (ZEB). As a result it seemed that these epithelial–mesenchymal transition related transcription factors are not involved in this process. Moreover, SAHA treatment decreased mRNA and protein expression of forkhead-box protein A 1 (FOXA1) (34). FOXA1, as a growth factor, mediates the hormonal response in BC (37, 38). When FOXA1 is over-expressed, SAHA induced EMT of TNBC cells is limited.
Furthermore, in MDA-MB-231 and BT-549 cells, SAHA-induced down-regulation of FOXA1 transcription and up-regulation of N-cadherin and vimentin were attenuated by silencing of HDAC8, but not HDAC6 (34). Previous studies have suggested that HDAC6 and HDAC8 mediate the SAHA effects in BC cells (39, 40). HDAC8 lacks the conserved C-terminal domain (41), so it is a unique class I HDAC and it has a different expression profile from those of HDACs 1-3 (42). HDAC8 silencing does not change histone acetylation (43), but it has been assumed that it could act at certain promoter sites of FOXA1. Based on this evidence, researchers claimed that HDAC8/FOXA1 signals motivated by SAHA treatment induce the EMT of TNBC cells (34).
As it is known, HDACs participate at the homologous recombination repair pathway of DNA, given the fact that they regulate the expression of genes related to this procedure (15). Studies have shown that ADP-ribose (poly) polymerase (PARP) inhibitors, such as olaparid, could be used as anti-cancer treatment). Min et al. have searched whether SAHA treatment enhances the anti-tumor effect of poly (ADP-ribose) polymerase (PARP) inhibitors in TNBC cells by blocking the homologous recombination repair pathway (46). Investigators used MDA-MB-157, -231, -453, -468 and BT-549 human cells and found that combinational treatment with olaparid and SAHA suppressed the proliferative signaling pathway and promoted inhibition of tumor growth, in comparison to olaparid or SAHA treatment alone. An in vivo study of a MDA-MB-231 xenograft model confirmed this evidence. Moreover, this combination seemed to induce both apoptotic and autophagic cell death, thus enhanced the cytotoxic effects of these inhibitors (46). Further studies could examine more combined treatments of PARP inhibitors with other HDAC inhibitors in order to improve therapeutic approaches for TNBC patients.
Furthermore, it is known that the majority of deaths in TNBC patients are not due to the primary tumors, but to metastases. An in vitro study on MDA-MB-231 and 4T1 (a mouse TNBC cell line) cell lines for the investigation of the effects of ionizing radiation (IR) and SAHA co-treatment, indicated that the combination was therapeutically more effective in a statistically significant way, than IR or SAHA alone. Tumor growth was also inhibited, in vivo, in an orthotopic BC mouse model (47). In addition, this combination increased DNA damage through the inhibition of DNA repair proteins. Concerning SAHA treatment in 4T1 cells, it led to inhibition of BC cell migration and invasion, via the inhibition of matrix metalloproteinase-9 (MMP-9) activity.
In general, evidence shows, in several cancer types, MMPs mediate the invasion and metastasis (48). MMP-9 participates at the degradation of type IV collagen and its over-expression has been found to be associated with invasion and metastatic potential in many types of carcinomas (49-53). In total, evidence indicates that SAHA could serve as a radiosensitizer against TNBC (47).
Also, in the same study, an in vivo experimental metastasis mouse model was used and showed that SAHA treatment inhibited lung metastasis and enhanced radiosensitivity (47). Concerning metastases, another study of an in vivo model of TNBC has reported that SAHA treatment is involved in the induction of DNA double-strand breaks suppressing brain metastatic colonization by 62% (54). Further studies that combine SAHA with DNA active drugs or radiation are needed in order to investigate thoroughly their actions against metastases.
Carlisi et al. studied the combination of SAHA and parthenolide (PN) (55). The principal bioactive sesquiterpene lactone component of feverfew Tanacetum parthenium, PN, was found to induce tumor inhibition on many cancer types, such as osteosarcoma, melanoma (56) and prostatic (57), pancreatic (58), colorectal (59) cancers. At the same time it showed low toxicity against normal cells. PN acts cytotoxically on MDA-MB231 cells, but at low doses it is not effective (60, 61). Investigators have combined PN and SAHA in order to overcome the PN's deficiency (55).
They claimed that pre-treatment of MDA-MB231 cells with SAHA sensitized the cells to the cytotoxic effect of PN (62), so PN and SAHA acted synergistically in order to fight against TNBC cells (55). Moreover, they demonstrated that SAHA treatment leads to acetylation of lysine residues in the NH2-terminal tails of histones H3 and H4 and these findings were in accordance to other studies (63, 64). Furthermore, investigators claimed that there is an association between this histone hyperacetylation provoked by SAHA or SAHA and PN treatment and the up-regulation of tumor suppressor factors p21 and p27, as well as down-regulation of fundamental survival proteins Bcl-2 (55). These results have also been confirmed by another study (65). This evidence demonstrates that epigenetic actions could induce the alterations in the expression of these proteins.
Investigators have also studied the production of reactive oxygen species (ROS) and the autophagic process and they have found that SAHA treatment urges both processes. Both ROS generation and autophagy are reduced by two NADPH oxidase family (NOX) inhibitors - Nox enzymes are major ROS producers-, apocynin and diphenyleneiodonium chloride (DPI). Therefore, it was proposed that SAHA primarily induced NOX to increase the production of ROS, which led to activation of autophagy (55). Another study has shown that SAHA stimulates autophagy in tumor cells (66). When they added PN, SAHA's results on ROS generation and autophagic process were diminished, while they observed a potent activation of apoptosis.
Finally, it has been found that SAHA and PN combined therapy induced a decrease of the expression levels of beclin-1, a protein involved in autophagosome formation (55). Previous studies have shown that beclin-1 is cleaved by caspases (67). N-terminal cleavage fragment of beclin-1 suppresses autophagy, while its C-terminal cleavage fragment promotes the release of cytochrome C from mitochondria (68). As an extension to the above mentioned finding, investigators have claimed that SAHA/PN combination treatment relegates beclin-1 by activating caspase-3. This event could inhibit SAHA-induced autophagy and the release of cytochrome C (55). Taken together this evidence, it could be implied that SAHA/PN co-treatment could be a candidate for TNBC therapy.
A recent study on MDA-MB-231 cells used SAHA in comparison to a kinase inhibitor and found that this co-treatment induces cell death. An important fact was that cell death was induced by both autophagy and apoptosis (69). These results were confirmed by Zhang et al., who used SAHA and the antiangiogenic kinase inhibitor Sorafenib in epithelial tumor cell types and found that this treatment suppressed the expression of anti-apoptotic proteins and increased the activation of CD95 extrinsic apoptotic and lysosomal protease pathways. So this combination has been found to interact synergistically against liver, kidney, and pancreatic tumor cells in vitro (70). These studies indicate that the combination of a kinase and an HDAC inhibitor is a promising treatment modality for the prevention or therapy of aggressive cancer types, especially TNBC.
The effects of HDAC inhibitors (VPA and SAHA) and cisplatin (CDDP), a chemotherapeutic agent, co-treatment against several types of BC cells have been explored using an isobolographic method (71,72). This combination achieved to reduce the doses of the compounds and to be therapeutically beneficial for head and neck squamous cell carcinomas (26, 73) and BC in vitro (74, 75). This study also showed that in MDA-MB231 cells, the combination of the CDDP and VPA at the ratio of 1:1 induced antagonistic interactions, while CDDP and SAHA co-treatment induced synergistic interactions (71). According to the investigators, this could be explained by the fact that SAHA is a potent inhibitor of the HDAC6 activity compared to VPA (26). Many nuclear proteins, like the transcription factor forkhead boxp3 (FOXP3) and the DNA repair factor KU70, are substrates of HDAC6, which is recruited by RNA polymerase II to chromatin in order to suppress the cycles of acetylation/deacetylation. This procedure enables the transcription and transfers cytotoxic poly-ubiquitinylated proteins into autophagosomes (76). All these mechanisms of HDAC6 action and its potential inhibition by SAHA could explain the hypothesis.
Moreover, it has been reported that CDDP/HDAC inhibitor co-treatment induces apoptosis and cell-cycle arrest of TNBC cells (71). Further research in vivo is necessary in order to use this promising combination therapy in TNBC clinical trials.
A recent study has described a new family of hybrid compounds that combine particular structural motifs of tamoxifen (TAM), ferrocifen (FcTAM) and SAHA (77). Ferrocenyl group (Fc) is reported to act as a redox antenna for phenol oxidation via an intra-molecular mechanism (78-80). They have found that ferrocene derivatives are extremely more active than organic analogs in MDA-MB-231 cells and hormone-dependent MCF-7 BC cell lines. FcTAM improved its inhibitory activity by replacing its 3-(dimethylamino) propan-1oxy group with an 8-hydroxyamino-8-oxooctanamido or an 8-amino-8-oxooctanamido group. In MDA-MB-231 BC cells, FcTAM–SAHA treatment exerted better anti-proliferative effects than SAHA treatment alone (77). Interestingly, the organometallic compounds induced the expression of p21waf1/cip1 gene in MCF-7 cells in accordance with their antiproliferative activity (77). Further research on the field of organometallic moiety could contribute to a more proper treatment against TNBC.
Sodium Butyrate and SAHA. Sodium butyrate (NaB) is shown to exhibit beneficial anticancer ability (81). It has the potential to relax the chromatin structure, allowing easier access to transcription-related proteins. These abilities render NaB a potent treatment against several types of solid tumor (82, 83). A study in MDA-MB-231 and BT-549 cell lines using various concentrations of NaB and SAHA for 24 h revealed that these inhibitors suppress cell proliferation, arrest cell cycle at G0/G1 phase, and finally are involved in mitochondrial-related apoptosis. Moreover, both SAHA and NaB treatment decreased the phosphorylation, and also the mRNA and protein levels of mutant p53 (mtp53), while neither of these compounds had anti-proliferative effects on wild type p53 (wtp53) via time and dose dependent manners (84). p53 gene has been reported as the most frequently mutated gene in BC (85). Approximately, 62% of the basal-like TNBC and 43% of the non-basal-like TNBC have mutations in the mtp53 (86).
These data urged the investigators to report that the inhibition occurs during the transcription, given that the transcription of the precursor p53 is down-regulated more rapidly (less than 2h) and aggressively than that of mature p53. Also, the transcription of mtp53 is inhibited by the silencing of HDAC8, while not of HDAC6 (84). Both HDAC6 and HDAC8 are previously reported to mediate the down-regulation of p53 induced by HDAC inhibitors (40, 87).
Moreover, both SAHA and NaB reduced the binding of transcription factor Yin Yang 1 (YY1) with the -102 to -96 position of human p53 promoter. Interestingly, silencing of YY1 significantly inhibits the mtp53 transcription (83). YY1 is suggested to be over-expressed in BC cells and to be a key molecule in BC progression. Knockdown of YY1 led to suppression of the clonogenicity, migration, invasion and tumor formation of BC cells (88).
Based on the above, researchers have found that both SAHA and NaB reduced the association of HDAC8 and YY1. As a result, acetylation of residues 170–200 of YY1 was enhanced, leading to decreased YY1 transcription and finally, inhibition of the YY1-induced p53 transcription (84). In total, this study showed that not only SAHA and NaB treatment could be very promising for TNBC patients, but also HDAC8/YY1/mtp53 signals could become an important target for TNBC therapy.
Trichostatin A and NaB. Trichostatin A (TSA) is an antifungal antibiotic, found in cultured mammalian cells and, at low nanomolar concentrations, in fractionated cell nuclear extracts. In general, as an HDAC inhibitor, TSA not only suppresses HDAC activity, but also leads to cell-cycle arrest in G1 and G2 phase, induces cell differentiation as well as the reversion of transformed cells in vitro (89).
A study of the effect of NaB and TSA on viability of non-tumorgenic MCF10A cells and of MCF-7, T-47D and MDA-MB-231 cell lines (90) has found that MCF10A cells were minimum affected by elevated doses of these HDAC inhibitors while the other tested cell lines were extremely sensitive to treatment with both NaB and TSA. Concerning the TNBC cell line (MDA-MB-231), NaB treatment significantly enhanced the activity of pyruvate kinase, but it neither induced an attenuation of glycolysis, nor enhanced the activity of lactate dehydrogenase, as it happened in T-47 D cells. Furthermore, in both T-47D and MDA-MB-231 cells, the rate-limiting enzyme of the pentose phosphate pathway, Glucose-6phosphate dehydrogenase (G6PDH) was activated by NaB treatment, while in MCF-7 cells, G6PDH activity was inhibited. Finally, both NaB and TSA in MDA-MB-231 and T-47D cells significantly raised the oxygen consumption. These data show that mitochondria might occupy a key role in metastasis (90). Further research is required in order to understand the pathways of action of these inhibitors and then target the proper signals to suppress the specific BC type.
Mocetinostat. Mocetinostat is a class I HDAC inhibitor that is associated with the reversion of cardiac fibrosis (91). Recently, mocetinostat was reported to have promising antitumor activities against various types of cancer cell lines and tumor xenografts in nude mice, as it is related to apoptosis (92-94). A study showed that mocetinostat inhibits the growth of colon cancer cells via the up-regulation of WNT ligand DKK-1 expression (95). Also, in B-cell chronic lymphocytic leukemia cells, it has been shown to lead to apoptosis, as it decreases the expression of Mcl-1 protein and it induces Bax translocation to mitochondria (96). Another study has shown that mocetinostat inhibits the proliferation of prostate cancer cells (93).
Borbely et al. studied the combination of mocetinostat and JQ1 in MDA-MB-231, BT549, MCF-7 and T47-D cells (97). JQ1 is a bromodomain and extra-C terminal (BET) inhibitor and is reported to participate in the expression of oncogenes and tumor suppressor genes. JQ1 is in pre-clinical testing for the treatment of hematological malignancies and neuroblastoma (98-100). Researchers found that either JQ1 or mocetinostat treatment alone, inhibited the expression of genes involved in cell cycle regulation. Interestingly, the combination of JQ1 and mocetinostat decreased even more cell viability as the compounds acted synergistically.
Furthermore, combination treatment was associated with a sharp increase in the expression of many members of the ubiquitin–specific protease 17 (USP17) family of deubiquitinating enzymes (97). Previous studies have reported that USP17 regulates Ras/MAPK signaling partly through the regulation of the RCE1 (101). It is known that the Ras/MAPK pathway is involved in BC progression (102). The activity of this pathway has been tested and it was found that the expression of USP17L5 protein was increased, when at the same time Ras activity was decreased. In other words, USP17 enzymes attenuated the Ras/MAPK pathway and cell viability was suppressed. On the other hand siRNA-mediated depletion of USP17, decreased the cytoxicity, indicating that the Ras/MAPK signaling pathway is involved in the synergistic effect of the combinational treatment (97). All these data show that the combination of HDAC and BET inhibitors could be used as potential therapeutic strategy for BC, given that it reduces the viability of TNBC cells, through induction of USP17.
Panobinostat. Panobinostat (LBH589) is a pan-HDAC inhibitor. It has the ability to block several pathways related to cancer and reverse epigenetic events related to cancer progression (103). Its anti-cancer effects have been observed in hematologic, lung, breast, ovarian, thyroid and prostate malignancies (104). Recently panobinostat was approved for treatment of multiple myeloma with tumor progression after immunomodulating agents and bortezomib (105).
Interestingly, a study has reported significant action of panobinostat against the TNBC, as it exerted anti-proliferative action, provoked apoptosis and inhibited the primary tumor volumes of TNBC xenografts (106). Also in the same study, in vitro and in vivo results have shown that treatment with panobinostat increases significantly the epithelial cell marker CDH1. In general, the expression of EMT markers, such as epithelial-cadherin (CDH1), neuronal-cadherin (CDH2), vimentin (VIM), zinc finger E-box binding homeobox 1 (ZEB1) and 2 (ZEB2), has been associated with poor prognosis of BC patients (107-109). As a result, these markers have become attractive targets for the confrontation of metastasis.
In a recent study it has been tested whether panobinostat has the ability to inhibit the migration, invasion, and metastasis of MDA-MB-231 and BT-549 cell lines, by suppressing EMT (110). In TNBC cells, panobinostat has been reported to inhibit the expression of genes associated to EMT and to induce the expression of CDH1, while these results were not found in luminal A or basal BC cell lines (106). This study has also reported very important results about panobinostat effects on MDA-MB-231 and BT-549 cells, as it augmented histone acetylation, while suppressed the cell proliferation and viability. It led to cell cycle arrest at G2/M phase with a simultaneous decrease in S phase. Moreover, panobinostat induced apoptosis at 24 h. In addition, treatment of ER-negative breast cancer cells (MDA-MB-231 and MDA-MB-435) with panobinostat for 24 h resulted in restoration of ERα mRNA and protein expression. However, reactivation of ERα expression was not due to changes in the methylation profile of CpG island within ER promoter region. Interestingly, after at least 96 h from termination of panobinostat treatment, the expression of ERα mRNA was maintained. Panobinostat treatment released DNA (cytosine-5)-methyltransferase 1 (DNMT1), HDAC1, and the H3 lysine 9 (H3-K9) methyltransferase SUV39H1 from the ER promoter. Investigators claimed that these changes altered the chromatin state to active, characterized by increased the levels of acetylated H3 and H4, decreased methylated H3-K9 and false binding of heterochromatin protein 1 (HP1 alpha) at the promoter (111).
Furthermore, Rhodes et al. compared panobinostat to other HDAC inhibitors (SAHA, TMP269) and found that panobinostat has much more potent anti-EMT effects than the other inhibitors (110). In vitro research showed that changes in EMT-related gene expression were associated with the suppression of cell migration and invasion. Also, in a xenograft model these alterations have induced a statistically significant inhibition of metastasis of TNBC cells to both brain and lung. Except for CDH1expression, panobinostat has induced the suppression of multiple predominant pro-EMT genes including ZEB1 and ZEB2. In addition, when investigators forced exogenous expression of ZEB1 or ZEB2, the inhibitory effects of panobinostat on cell migration, invasion, and CDH1 expression were reversed (110).
It has been recently reported that ZEBs recruit Class I HDACs (48, 112, 113). Given the fact that panobinostat can act against Class I, II and IV HDACs, in addition to inhibition of ZEB expression, panobinostat has the ability to prevent ZEB-mediated repression of CDH1.
However, it is probably the repression of ZEB2 that regulates the effects of panobinostat on tumor genesis, as ZEB1 expression alone was not sufficient to overcome the actions of panobinostat on CDH1 expression (110). To sum up, it is critical to study panobinostat as a novel therapeutic to target the aggressive and metastatic nature of TNBC, as panobinostat is claimed to inhibit both TNBC primary tumor genesis and the metastasis of TNBC cells.
Another study has confirmed these findings, given that it was found that in MDA-MB231 cells independent of estrogen receptor expression, panobinostat induced CDH1 expression (114). Panobinostat was reported to induce the expression of CDH1 and reduce the migration and invasion of TNBC cells. Researchers claimed that panobinostat by-passed the CDH1 transcriptional repressors (Snail, Slug), while the ERa expression and pathway were not affected.
Another combination that has been studied on TNBC cell lines (HCC1937 and MDA-MB-231) is co-treatment of panobinostat and salinomycin (115). Salinomycin is an antibiotic that has been used in farm animals (116) and it has been associated with the inhibition of BC stem cells in TNBC (117, 118). Also, studies in glioblastoma have shown that co-treatment of salomycin and HDAC inhibitors induced the death of stem-like glioblastoma cells (119). Kai et al. have reported that panobinostat and salinomycin acted synergistically and inhibited the TNBC cell proliferation (115).
HDAC inhibitors have been found to be beneficial in BC therapy, as they target BC stem cells, have anti-proliferative effects on aldehyde dehydrogenase 1 (ALDH1)-positive cells, and suppress the formation of BC mammospheres (120). Also, ALDH1 has been associated with TNBC, so ALDH1 can be used as a BC stem cell marker in TNBC (121, 122). Based on these facts, Kai et al. determined the activity of panobinostat on TNBC cells with BC stem cell properties (115). Co-treatment of panobinostat and salinomycin inhibited the BC stem cell properties, as the self-renewal capacity and ALDH1 activity were suppressed. Moreover, this combinational treatment inhibited growth of ALDH1-positive cells, induced apoptosis, arrested cell cycle and regulated EMT in BC stem cells (115). We can assume that further research is needed, given that co-treatment of panobinostat with salinomycin in TNBC is claimed to provide an effective targeted therapy for patients with TNBC.
A study on MDA-MB-231cells investigated the in vitro and in vivo effect of co-treatment of panobinostat with the autophagy inhibitor chloroquine (123). First of all, panobinostat treatment disrupted the hsp90/HDAC6/HSF1/p97 complex (124, 125) and induced a heat shock response mediated by heat shock factor protein 1 (HSF1). This abruption of the HSF1 from the multiprotein comlex is reported to lead to HSF1 phosphorylation, trimerization and nuclear translocation (125, 126). Moreover, panobinostat treatment induced endoplasmic reticulum stress response and autophagy (123), evidence also supported by previous research (127-129). Combinational treatment of panobinostat with chloroquine was found to significantly enhance both the in vivo and the in vitro effect of panobinostat, given the fact that it inhibited tumor growth and increased viability of MDA-MB-231 xenografts (123).
Therefore, panobinostat could be used for treatment of aggressive BC that is resistant to hormonal therapy. We propose that panobinostat should be tested either in monotherapy, or in combination with known anti-cancer agents, like trastuzumab, capecitabine, lapatinib, or paclitaxel.
Entinostat. Previous studies have reported that retinoic acid and its products, such as all-trans retinoic acid (ATRA), urge the differentiation of multiple types of stem cells, including the stem cells that exist in BC (130, 131). On the other hand, ATRA has been used in a clinical trial and its efficacy was limited (132), possibly because of the epigenetic knock-out of the retinoic acid receptor (RAR)-β (133). Recent studies have shown that HDAC inhibitors have the ability to re-express RAR-β, so they restore the sensitivity of the cells to treatment (134-136). It has been reported that co-treatment of HDAC inhibitors with either doxorubicin or retinoids increased the cytoxicity against cancer cells (75, 137).
A novel study has examined the effects of co-treatment with entinostat (MS-275), which is a selective inhibitor of class I HDACs, ATRA, and doxorubicin and reported that TNBC cell growth was inhibited in xenografts and killed tumor cells and in cell cultures (138). Given the fact that direct binding of topoisomerase II-β (TopoII-β) to RAR-β promoter has been reported in acute pro-myelocytic leukemia cells (139), investigators have searched that path and found that entinostat and doxorubicin treatment suppressed the expression of Topo II-β. As a result, RAR-β expression was silenced. It was demonstrated that this co-treatment achieved significant tumor suppression in TNBC, as entinostat urges the cytoxicity mediated by doxorubicin and the differentiation mediated by retinoid (138).
As we mentioned, in metastatic cell lines and invasive BCs E-cadherin is silenced epigenetically and EMT phenotype is reported to be the critical step in the beginning of metastasis (140-142). In MDA-MB-231 and Hs578T cells, entinostat treatment has been reported to reverse the EMT phenotype (143). These cell lines have lost the expression of E-cadherin, while they highly express mesenchymal markers such as N-cadherin and vimentin along with transcriptional repressors such as Twist and Snail (144-147). Treatment with entinostat increased the transcription of E-cadherin, while the mRNA expression of N-cadherin was decreased. Chromatin immunoprecipitation analysis demonstrated that E-cadherin's promoter was activated due to a decrease in the association of TWIST and SNAIL with E-cadherin promoter (143). In a previous study, it has been reported that HDAC1 activity is required for the SNAIL-mediated repression of E-cadherin (145), so investigators studied the activity of HDAC1 and found that entinostat inhibits HDAC1, thus the repression of E-cadherin is reversed. An in vitro study has shown that entinostat inhibited cell migration. Moreover, there was observed an increase of vimentin phosphorylation levels, as well as changes in vimentin filaments. Interestingly, the increased phosphorylation of vimentin led to suppression of the formation of microtentacles based on tubulin (143), which are known to help floating cells attach to other surfaces (148).
We should also mention that a subset of cells within a breast tumor may cause escape from the primary site. These cells are named as tumor-initiating cells (TICs). TICs produce progenitor cells, which cannot self-renew, but they may comprise the greater part of the tumor (149). Entinostat treatment on TNBC cells has been shown to decrease the level of TICs, therefore entinostat might be used to prevent development of metastasis (150).
It is apparent that the ability of entinostat to reverse the EMT phenotype and reduce the migration of TNBC cells, as it reduces the attachment of floating cancer cells to new surfaces, render it a promising therapeutic agent against metastasis.
YCW1. The octanedioic acid [3-(2-(5-methoxy-1H-indol-1yl)ethoxy)phenyl]-amide N-hydroxyamide (YCW1) is a novel HDAC inhibitor that has been developped by structure-based analyses (151, 152). Researchers have tested the co-treatment with YCW1 and ionizing radiation (IR) in a murine and a human TNBC cell line, 4 T1, and MDA-MB-231, respectively. This combination was shown to increase autophagy and endoplasmic reticulum stress, so these compounds acted as a cytotoxic. Interistingly, in comparison to the effects of SAHA, YCW1 significantly enhanced toxicity. Study in an orthotopic BC mouse model confirmed these results (153). Therefore, co-treatment with IR and YCW1 induced autophagic cell death.
N-(2-Hydroxyphenyl)-2propylpentanamide. N-(2-Hydroxyphenyl)-2propylpentanamide (Compound 2) is an aryl derivative of valproic acid. These derivatives are produced by combining valproic acid and the arylamine core of SAHA with different substituents at its carboxyl group. Compound 2 has exerted the most promising results between other derivatives that were submitted to docking simulations. In vitro studies against HeLa, rhabdomyosarcoma and BC cells lines have shown that compound 2 was the best anticancer agent (154). Interestingly, it was effective against TNBC cells. It seemed that compound 2 targeted HDAC8. In addition, in vitro studies have shown that compound 2 inhibited cancer cell proliferation at a much lower concentration, compared to valproic acid (154).
Uncommon Results
Unfortunately, despite the promising results in several pre-clinical trials, when HDAC inhibitors are used as single agents, they failed to be clinically beneficial against solid tumors (33). For instance, in the field of metastatic BC, SAHA treatment in 14 patients failed to achieve adequate single-agent activity (155). Moreover, de Cremoux et al. have used different HDAC inhibitors (SAHA, panobinostat, abexinostat) in MDA-MB231, Hs 578T and SUM149 human cell lines and found that despite the cytotoxicity of both of these inhibitors Estrogen receptor 1 (ESR1) and estrogen receptor 2 (ESR2) genes were not re-expressed in vitro (156). Xenograft studies with abexinostat treatment for three consecutive days did not induce the expression of ESR1/ESR1 related genes or of ERa protein. In addition, intra-tumor H3 acetylation was observed (156). Another study in MDA-MB231 and SKBR3 lines treated with SAHA or valproic acid for 48 h, showed minimal or no alterations in ERa or in ERb mRNa and protein (157). Also, different HDAC inhibitors have been shown to cause different effects on luminal and mesenchymal-like breast cancer cells, either elevating histone acetylation or showing no effects (158).
Conclusion
TNBC is a serious subtype of BC that is correlated with poor prognosis due to the high proliferation rate. New therapeutic strategies are needed in order to cure TNBC. Epigenetic mutations are associated with repression of BC development and therapy resistance.
Between all the epigenetic treatments, HDAC inhibitors represent the first successful anti-cancer epigenetic therapy. Studies show that they affect positively the therapy of subtypes of hematological malignancies. However, it is still unclear how effective they are against solid tumors (159).
Nevertheless, the majority of studies in the field of TNBC therapy tend to combine HDAC inhibitors with kinase inhibitors, autophagy inhibitors, antibiotics, chemotherapy, IR, or even two HDAC inhibitors treatment in order to enhance their efficacy against TNBC (47, 69, 71, 84, 115, 123, 153). In the majority of the studies, co-treatment of an HDAC inhibitor with another compound induced the inhibition of tumor growth and showed anti-proliferative effects (46, 47, 77, 84, 115, 123, 138, 154). Also, several HDAC inhibitors are assosiated not only with autophagic cell death, but also apoptosis (44, 55, 70, 71, 84, 97, 153). Interestingly, many studies have found that HDAC inhibitors have the ability to convert ER-negative tumors to ER-positive tumors (111, 160, 161). Finally, a recent study showed that entinostat treatment can prevent the metastasis (143).
However, as we mentioned above, we should not forget that there are several studies in the pertinent literature with contradictory results (155-157). These unexpected studies reveal the necessity for further investigation in the field of HDAC inhibitors against TNBC cancer. We should understand the nature of the molecular basis of the selectivity of HDAC inhibitors and which are the proper combining treatments in order to find more clinically beneficial treatment against TNBC.
Acknowledgements
Christos Damaskos would like to express his gratitude to his teacher, Spyridon Garmpis who not only selflessly taught him, but also his actions inspired him to learn more, do more and become better.
Footnotes
↵* These Authors contributed equally to this study.
This article is freely accessible online.
Conflicts of Interest
The Authors declare that they do not have any conflict of interest.
- Received May 26, 2017.
- Revision received July 17, 2017.
- Accepted July 19, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- Epigenetic Modulation of SPCA2 Reverses Epithelial to Mesenchymal Transition in Breast Cancer Cells
- Novel Imidazo[2,1-b]oxazole Derivatives Inhibit Epithelial Cell Transformation and Triple Negative Breast Cancer Tumorigenesis
- Molecular Classification and Future Therapeutic Challenges of Triple-negative Breast Cancer
- Temozolomide-resistant Glioblastoma Depends on HDAC6 Activity Through Regulation of DNA Mismatch Repair
- Triple-Negative Breast Cancer: The Progress of Targeted Therapies and Future Tendencies
- Histone Deacetylase Inhibitors and Anaplastic Thyroid Carcinoma
- The Role of Histone Deacetylase Inhibitors in Uveal Melanoma: Current Evidence
- Combination Treatment of Polo-Like Kinase 1 and Tankyrase-1 Inhibitors Enhances Anticancer Effect in Triple-negative Breast Cancer Cells
- Histone Deacetylase Inhibitors as a Novel Targeted Therapy Against Non-small Cell Lung Cancer: Where Are We Now and What Should We Expect?
- Tumor Suppression Efficacy of Heat Shock Protein 90 Inhibitor 17AAG in a Liposarcoma Mouse Model