


Abstract
Background/Aim: The furo[2,3-b]chromone derivatives are natural products that have been used as folklore medicines for various diseases. Here, the cytotoxicity of 12 synthesized furo[2,3-b]chromone derivatives was investigated and subjected to quantitative structure–activity relationship (QSAR) analysis. Materials and Methods: Cytotoxicity against three human oral squamous cell carcinoma cell lines and three types of oral normal mesenchymal cells was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. Tumor specificity (TS) was evaluated as the ratio of the mean 50% cytotoxic concentration (CC50) against normal oral cells to that against carcinoma cell lines. Potency-selectivity expression (PSE) was calculated by dividing the TS value by CC50 against tumor cells. Apoptosis induction was evaluated by cell morphology and caspase-3 activation. Morphological changes were monitored under light microscopy. For QSAR analysis, 288 physicochemical, structural and quantum chemical features were calculated from the most stabilized structure optimized using Corina. Results: Four furo[2,3-b]chromone derivatives showed relatively strong tumor selectivity. In particular, the derivatives with phenylethenyl and methoxy groups showed the highest TS, equivalent to that of melphalan, although their PSE values did not reach those of doxorubicin and 5-fluorouracil. Microscopical observation demonstrated that at cytotoxic concentrations, (2R,3aR,9aR)-rac-3a,9a-dihydro-7-methoxy-4-oxo-2-(2-phenylethenyl)-4H-furo[2,3-b][1]benzopyran-3,3(2H)-dicarboxylic acid 3,3-dimethyl ester and (2R,3aR,9aR)-rac-3a,9a-dihydro-7-methoxy-4-oxo-2-(1-propen-1-yl)-4H-furo[2,3-b][1]benzopyran-3,3(2H)-dicarboxylic acid 3,3-dimethyl ester did not produce a population of shrunken cells typical of apoptotic cells, in contrast to cells treated with actinomycin D. Tumor selectivity of furo[2,3-b]chromone derivatives strongly correlated with features related to the number of intramolecular unsaturated bonds, molecular flexibility, molecular density, lipophilicity, molecular size, and molecular shape. Conclusion: Chemical modification of the lead compound may be a potential choice for designing a new type of anticancer drug.
Many furochromone derivatives have been isolated from Ammi visnaga (Umbelliferae), used in folk medicine for different ailments. However, there are very few documentations of their synthetic methodologies and biological activities (1). Sesquiterpene chromones isolated from Ferula fukanensis inhibited nitirc oxide (NO) production in lipopolysaccharide and interferon-γ-stimulated Raw264.7 mouse macrophage-like cells, through inhibition of inducible NO synthase gene expression (2). Sesquiterpenoids isolated from the roots of Ferula mongolica were identified as new class of α-glucosidase inhibitors (3). Four oxepinochromenones (conioxepinols A-D) and one furochromenone (coniofurol A), from an organic solvent extract of solid-substrate fermentation culture of fungus Coniochaeta sp. (Coniochaetaceae) (4), and three sesquiterpene coumarin derivatives and two sesquiterpene chromone derivatives (5) showed moderate cytotoxicity against several human tumor cell lines. However, these previous studies did not investigate cytotoxicity against normal control cells.
We established a simple in vitro assay system for assessing antitumor potential using human oral squamous cell carcinoma (OSCC) cell lines and human normal oral cells (6, 7). Using this system, we have demonstrated that many anticancer drugs showed excellent tumor specificity (8). Among a total of 133 compounds, (E)-3-[2-(4-hydroxyphenyl)ethenyl]-6-methoxy-4H-1-benzopyran-4-one (classified as 3-styrylchromone) (9, 10), (E)-3-[2-(4-chlorophenyl)ethenyl]-7-methoxy-2H-1-benzopyran (classified as 3-styryl-2H-chromenes) (11) showed the highest tumor specificity with the least keratinocyte toxicity (7).
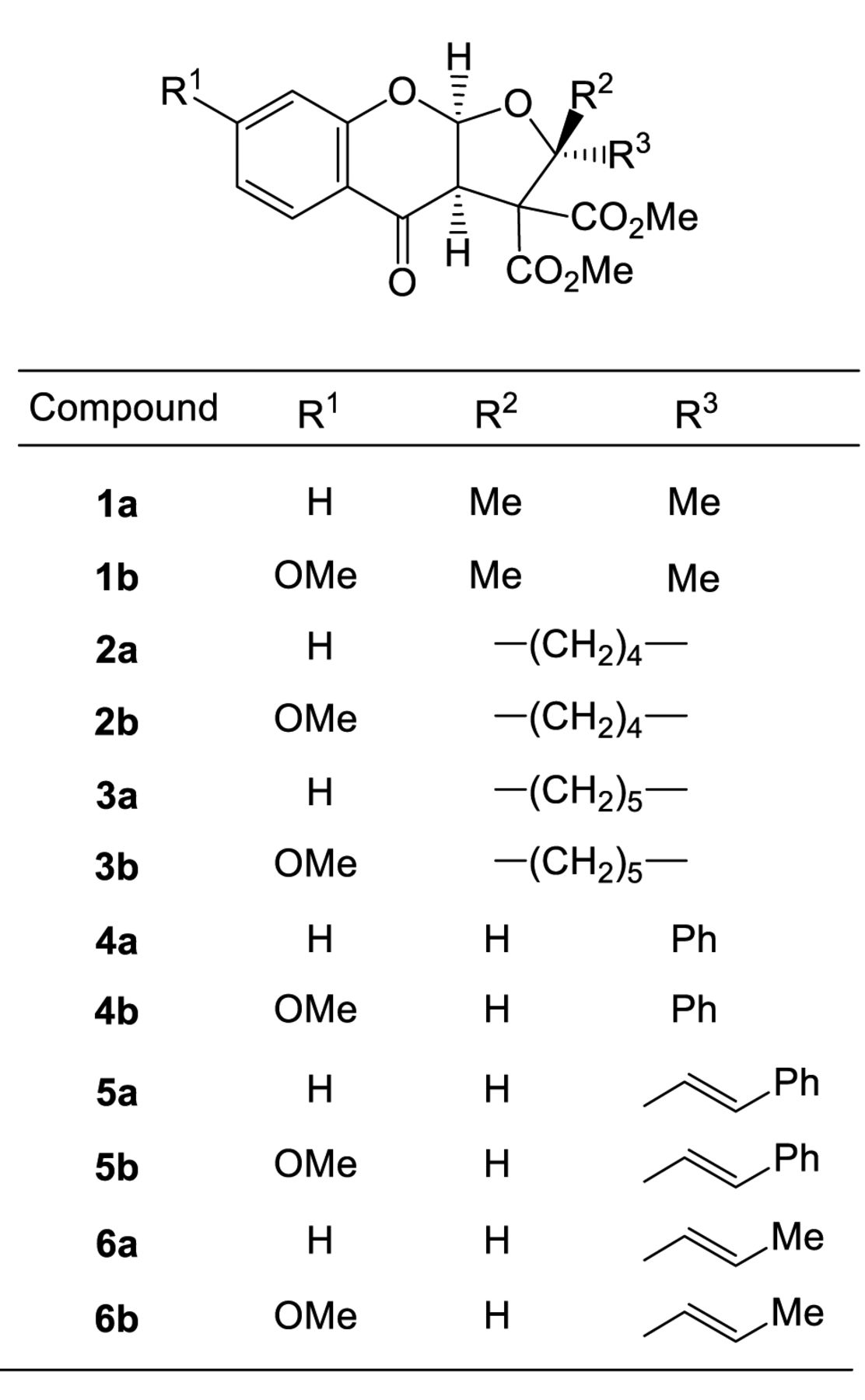
In continuation of the discovery of new antitumor compounds, here 12 furo[2,3-b]chromones (Figure 1) (12) were investigated for their cytotoxicity against three OSCC cell lines and three types of human normal oral cells, and then subjected to quantitative structure–activity relationship (QSAR) analysis.
Materials and Methods
Materials. The following chemicals and reagents were obtained from the indicated companies: Dulbecco's modified Eagle's medium (DMEM), from GIBCO BRL, Grand Island, NY, USA; fetal bovine serum (FBS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), doxorubicin from Sigma-Aldrich Inc., St. Louis, MO, USA; dimethyl sulfoxide (DMSO), culture plastic dishes and plates (96-well) were purchased from Becton Dickinson, Franklin Lakes, NJ, USA.
Synthesis of test compounds. (3aR,9aR)-Rac-3a,9a-dihydro-2,2-dimethyl-4-oxo-4H-furo[2,3-b][1]benzopyran-3,3(2H)-dicarboxylic acid 3,3-dimethyl ester (1a), (3aR,9aR)-rac-3a,9a-dihydro-7-methoxy-2,2-dimethyl-4-oxo-4H-furo[2,3-b][1]benzopyran-3,3(2H)-dicarboxylic acid 3,3-dimethyl ester (1b), (3aR,9aR)-rac-3’a,9’a-dihydro-4’-oxo-spiro[cyclopentane-1,2’(3’H)-[4H]furo[2,3-b][1]benzopyran]-3’,3’-dicarboxylic acid 3’,3’-dimethyl ester (2a), (3aR,9aR)-rac-3’a,9’a-dihydro-7-methoxy-4’-oxo-spiro[cyclopentane-1,2’(3’H)-[4H]furo[2,3-b][1]benzopyran]-3’,3’-dicarboxylic acid 3’,3’-dimethyl ester (2b), (3aR,9aR)-rac-3’a,9’a-dihydro-4’-oxo-spiro[cyclohexane-1,2’(3’H)-[4H]furo[2,3-b][1]benzopyran]-3’,3’-dicarboxylic acid 3’,3’-dimethyl ester (3a), (3aR,9aR)-rac-3’a,9’a-dihydro-7-methoxy-4’-oxo-spiro[cyclohexane-1,2’(3’H)-[4H]furo[2,3-b][1]benzopyran]-3’,3’-dicarboxylic acid 3’,3’-dimethyl ester (3b), (2R,3aR,9aR)-rac-3a,9a-dihydro-4-oxo-2-phenyl-4H-furo[2,3-b][1]benzopyran-3,3(2H)-dicarboxylic acid 3,3-dimethyl ester (4a), (2R,3aR,9aR)-rac-3a,9a-dihydro-7-methoxy-4-oxo-2-phenyl-4H-furo[2,3-b][1]benzopyran-3,3(2H)-dicarboxylic acid 3,3-dimethyl ester (4b), (2R,3aR,9aR)-rac-3a,9a-dihydro-4-oxo-2-(2-phenylethenyl)-4H-furo[2,3-b][1]benzopyran-3,3(2H)-dicarboxylic acid 3,3-dimethyl ester (5a), (2R,3aR,9aR)-rac-3a,9a-dihydro-7-methoxy-4-oxo-2-(2-phenylethenyl)-4H-furo[2,3-b][1]benzopyran-3,3(2H)-dicarboxylic acid 3,3-dimethyl ester (5b), (2R,3aR,9aR)-rac-3a,9a-dihydro-4-oxo-2-(1-propen-1-yl)-4H-furo[2,3-b][1]benzopyran-3,3(2H)-dicarboxylic acid 3,3-dimethyl ester (6a), (2R,3aR,9aR)-rac-3a,9a-dihydro-7-methoxy-4-oxo-2-(1-propen-1-yl)-4H-furo[2,3-b][1]benzopyran-3,3(2H)-dicarboxylic acid 3,3-dimethyl ester (6b) were synthesized by the ring-expansion reactions of methanochromanones with selected aldehydes or ketones, according to previous methods (12). All compounds were dissolved in DMSO at 40 mM and stored at −20°C before use.

Structure of 12 furo[2,3-b]chromones.
Cell culture. Human normal oral mesenchymal cells (human gingival fibroblast, HGF; human periodontal ligament fibroblast, HPLF; human pulp cells, HPC) were established from the first premolar tooth extracted from the lower jaw of a 12-year-old girl (13), and cells at 10-18 population-doubling levels were used in this study. Human OSCC cell lines [Ca9-22 (derived from gingival tissue); HSC-2, HSC-4 (derived from tongue)] were purchased from Riken Cell Bank (Tsukuba, Japan). All of these cells were cultured at 37°C in DMEM supplemented with 10% heat-inactivated FBS, 100 units/ml, penicillin G and 100 μg/ml streptomycin sulfate under a humidified 5% CO2 atmosphere. Cell morphology was checked periodically under the light microscope (EVOSfl; ThermoFisher Scientific, Waltham, MA, USA).
Assay for cytotoxic activity. Cells were inoculated at 2.5×103 cells/0.1 ml in a 96-microwell plate. After 48 h, the medium was replaced with 0.1 ml of fresh medium containing different concentrations of single test compounds. Cells were incubated further for 48 h and the relative viable cell number was then determined by the MTT method (8-11). The relative viable cell number was determined by the absorbance of the cell lysate at 560 nm using a microplate reader (Infinite F50R; TECAN, Kawasaki, Japan). Control cells were treated with the same amounts of DMSO and the cell damage induced by DMSO was subtracted from that induced by test agents. The concentration of compound that reduced the viable cell number by 50% (CC50) was determined from the dose–response curve and the mean value of CC50 for each cell type was calculated from triplicate assays.
Cytotoxic activity of 12 furo[2,3-b]chromones against oral malignant and non-malignant cells. Each value represents the mean of triplicate determinations. Two sets of tumor-specificity (TS) and potency-selectivity expression (PSE) values were determined using all malignant and non-malignant cells, or the pair of the cells derived from the same (gingival) tissue.
Calculation of tumor-selectivity index (TS). TS was calculated using the following equation: TS=mean CC50 against three types of normal oral cells/mean CC50 against for OSCC cell lines [(D/B) in Table I]. Since both Ca9-22 and HGF cells were derived from the gingival tissue (14), the relative sensitivity of these cells was also compared [(C/A) in Table I]. We did not use human normal oral keratinocytes as controls, since many anticancer drugs showed potent cytotoxicity against normal keratinocytes by inducing apoptosis (8).
Calculation of potency-selectivity expression (PSE). PSE was calculated by the following equation: PSE=TS/CC50 against tumor cells ×100 [that is, (D/B2) ×100 (HGF, HPLF, HSC vs. Ca9-22, HSC-3, HSC-4) using all non-malignant and malignant cells, and (C/A2) ×100 (HGF vs. Ca9-22) using the pair of the cells from the same tissue (gingiva) (Table I).
Estimation of CC50 values. Since the CC50 values had a distribution pattern close to a logarithmic normal distribution, we used the pCC50 (i.e., the −log CC50) for the comparison of the cytotoxicity between the compounds. The mean pCC50 values for normal cells and tumor cell lines were defined as N and T, respectively (11).
Calculation of chemical descriptors. The 3D-structure of each chemical structure (drawn by Marvin Sketch ver 16, ChemAxon, Budapest, Hungary, http://www.chemaxon.com) was optimized by CORINA Classic (Molecular Networks GmbH, Nürnberg, Germany) with partial charge calculations (amber-10: EHT) in Molecular Operating Environment (MOE) version 2015.1001 (Chemical Computing Group Inc., Quebec, Canada). The number of structural descriptors calculated from MOE and Dragon 7.0 (Kode srl., Pisa, Italy) after the elimination of overlapped descriptors were 288 and 2532, respectively.
Seven MOE descriptors listed in Table II were significantly correlated with T, N and T-N. Since the use of Dragon descriptors produced heavy overlapping of similar descriptors that showed higher correlation with T, N and T–N, and therefore correlation diagrams were plotted using MOE descriptors.
Statistical treatment. The relationships among cytotoxicity. TS and chemical descriptors was investigated using simple regression analyses by JMP Pro version 12.2.0 (SAS Institute Inc., Cary, NC, USA). The significance level was set at p<0.05.
Results
Cytotoxicity. A total of 12 furo[2,3-b]chromones were synthesized, without (a series) or with (b series) introduction of methoxy group at C-7 of benzopyran ring (Figure 1). Introduction of dimethyl (1a, 1b), spirocyclopentyl (2a, 2b), spirocyclohexyl (3a, 3b) or phenyl group (4a, 4b) at C-2 position showed essentially no cytotoxic activity against human oral carcinoma cell lines (mean CC50=291.8-400 μM) and normal oral cells mean CC50=382.6-400 μM). On the other hand, addition of phenylethenyl (5a, 5b) or propenyl (6a, 6b) group significantly enhanced the cytotoxicity for both oral carcinoma cell lines (mean CC50=37.2-49.3 μM) and normal oral cells (mean CC50=148.1-260.9 μM) (Table I). Representative dose-response curves of compounds (5b, 6b) are shown in Figure 2A, which demonstrated that these compounds were cytotoxic rather than cytostatic.
Explanation of chemical descriptors that correlate with cytotoxicity to tumor cells, normal cells and tumor specificity.
Tumor specificity. TS vales were calculated by dividing the mean CC50 value for three types of normal cells by that for four OSCC cell lines (D/B, Table I). Compounds 1a, 1b, 2a, 2b, 3a, 3b, 4a and 4b showed very low tumor specificity (TS=1.0-1.3). On the other hand, compounds 5a, 5b, 6a and 6b showed much higher tumor specificity (TS=3.6-7.0). In particular, compound 5b (TS=7.0) showed comparable tumor specificity with melphalan (TS=10.9) (Table I).
Considering that HGF is the normal cell type corresponding to cancer cell Ca9-22 (both derived from gingival tissues), TS values were also generated by dividing the average CC50 value for HGF cells by that for Ca9-22 cells (C/A, Table I). Compounds 5a, 5b, 6a and 6b (TS=7.2-13.5) showed slightly higher tumor specificity than melphalan (TS=5.7). Compound 5b had the highest TS value (13.5) (Table I).
PSE value. In order to identify the most promising compounds in terms of both good potency and selective cytotoxicity, PSE values were calculated. Compounds 5a, 5b, 6a and 6b had much higher PSE values than compounds 1a, 1b, 2a, 2b, 3a, 3b, 4a and 4b, while that for 5b gave comparable PSE value with melphalan (Table I).
Compounds 5b and 6b at the cytotoxic concentration (30 and 60 μM) produced spread, round cells, but failed to produce a population of shrunken cells, in contrast to actinomycin D-treated apoptotic cells (Figure 2B). Western blot analysis failed to detect cleaved products of poly (ADP-ribose) polymerase (PARP) and caspase-3, in contrast to actinomycin D (data not shown).
Properties of descriptors that significantly affects the cytotoxicity against tumor cells (T) and normal cells (N), and tumor specificity (T-N).
Computational analysis. We next performed the QSAR analysis of the 12 furo[2,3-b]chromones in regards to their cytotoxicity against tumor cells and normal cells. Among a total of 288 MOE descriptors, seven descriptors correlated well with cytotoxicity and tumor specificity (Table III).
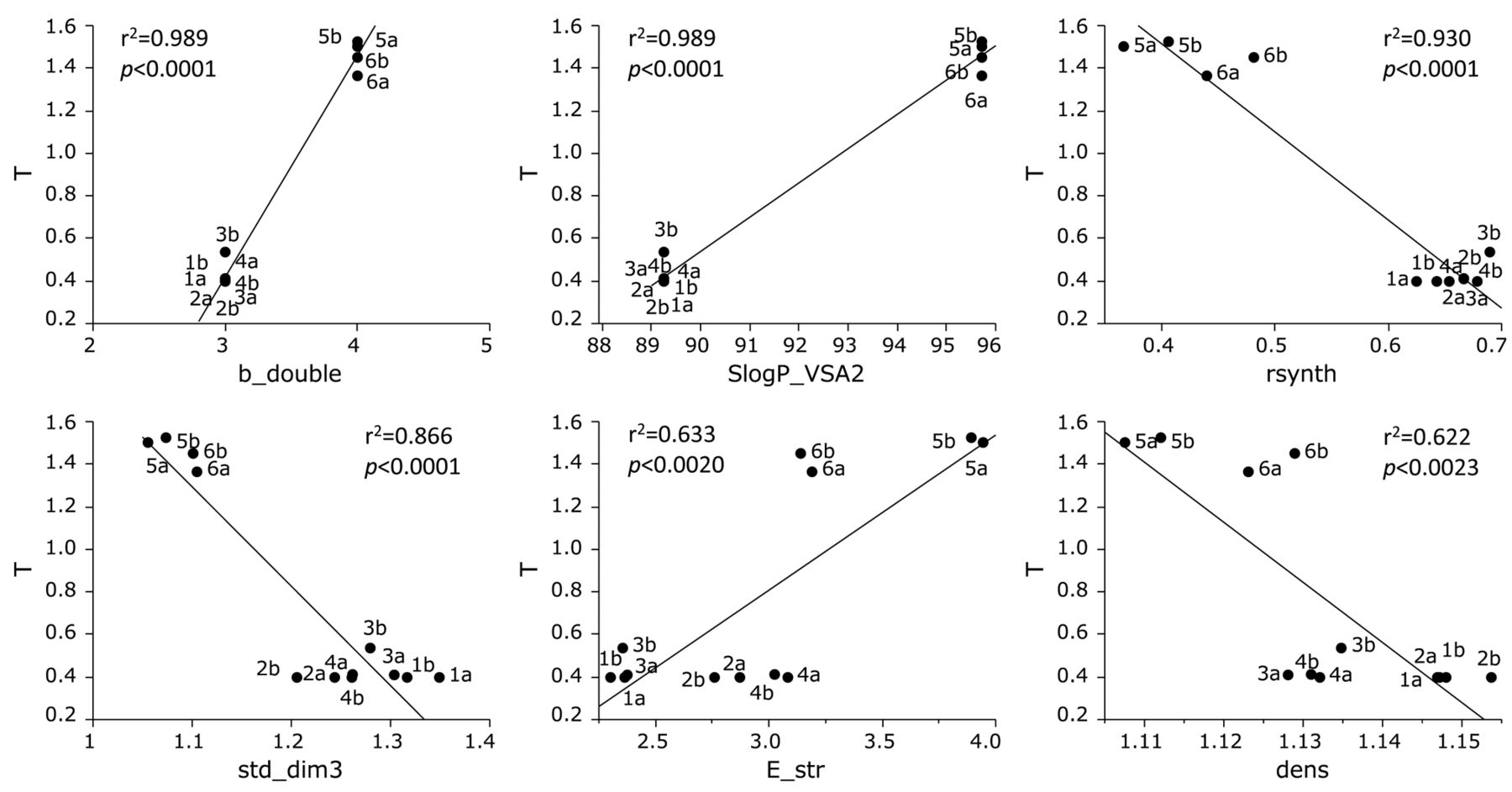
Cytotoxicity of the 12 furo[2,3-b]chromones against human OSCC cell lines was correlated positively with double bond (b_double; r2=0.989, p<0.0001), size and lipophilicity (SlogP_VSA2; r2=0.989, p<0.0001) and bond stretch (E_str; r2=0.633, p=0.002), while negatively with synthetic feasibility (r synth; r2=0.930, p<0.0001), shape (std_dim3; r2=0.866, p<0.0001) and density (dens; r2=0.622, p=0.0023) (Figure 3).
Cytotoxicity of the 12 furo[2,3-b]chromones against human normal oral mesenchymal cells was correlated positively with b_double (r2=0.857, p<0.0001), SlogP_VSA2 (r2=0.857, p<0.0001) and E_str (r2=0.626, p=0.0022), while negatively with rsynth (r2=0.895, p<0.0001), std_dim3 (r2=0.792, p=0.0001) and density (r2=0.622, p=0.0023) (Figure 4).

Dose–response for loss of cellular viability (A) and morphological changes (B) induced by compounds 5b and 6b. A: Ca9-22, HSC-3, HSC-4, human gingival fibroblast (HGF), human periodontal ligament fibroblast (HPLF) and human pulp cell (HPC) were incubated for 48 h with the indicated concentrations of compound 5b or 6b, and cell viability was determined by the MTT method. Each value represents the mean±S.D. of triplicate assays. B: Ca9-22 cells were incubated for 24 h without or with compound 5b or 6b (30 or 60 μM) or 1 μM actinomycin D (Act. D, positive control), and then assessed for morphology under light microscopy (EVOSFL; ThermoFisher Scientific, Waltham, Mass, USA). Bar=100 μm.
TS of the 12 furo[2,3-b]chromones was correlated positively with b_double (r2=0.969, p<0.0001), SlogP_VSA2 (r2=0.969, p<0.0001), flexibility (b_rotR; r2=0.606, p=0.0029) and E_str (r2=0.590, p=0.0035), while negatively with rsynth (r2=0.876, p<0.0001) and std_dim3 (r2=0.832, p<0.0001) (Figure 5).

Determination of correlation coefficient between chemical descriptors and cytotoxicity of 12 furo[2,3-b]chromones against tumor cells (defined as T). The mean values of the negative log of the concentration of compound that reduced the viable tumor cell number by 50% (pCC50) were defined as T. See Table II for explanation of chemical descriptors.
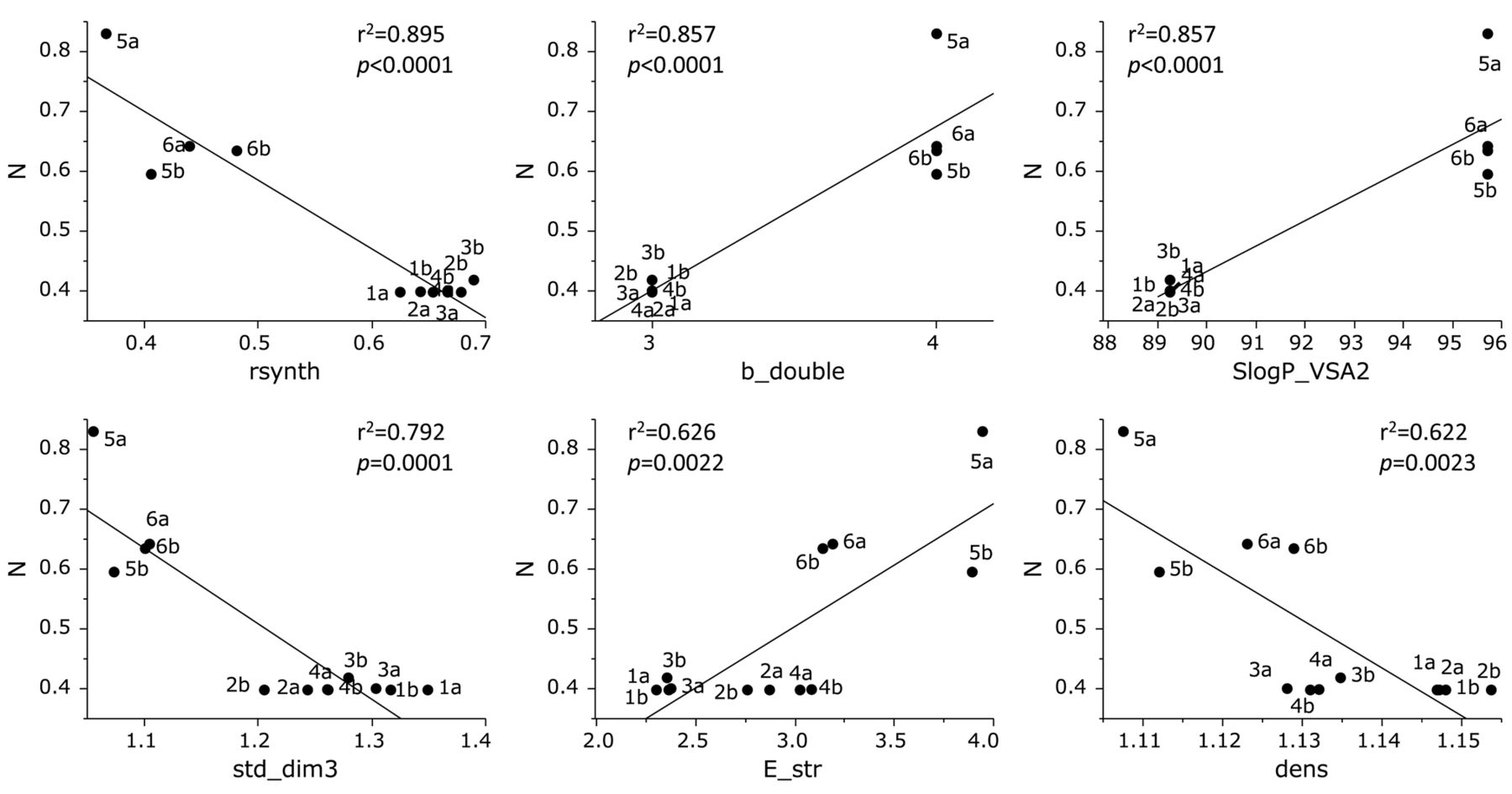
Determination of coefficient between chemical descriptors and cytotoxicity of 12 furo[2,3-b]chromones against normal cells (defined as N). The mean values of the negative log of the concentration of compound that reduced the viable normal cell number by 50% (pCC50) were defined as N. See Table II for explanation of chemical descriptors.
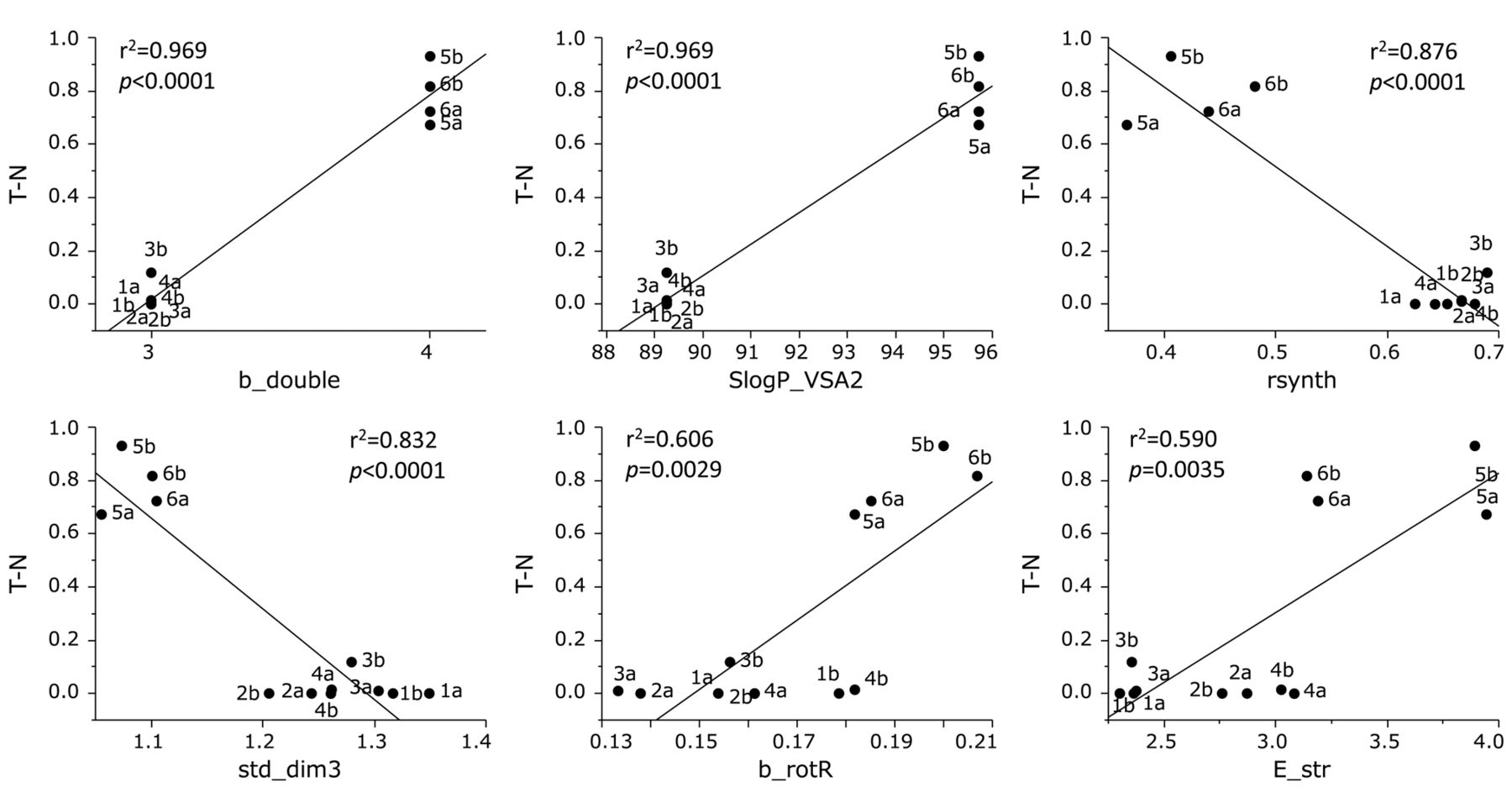
Determination of coefficient between chemical descriptors and tumor specificity of 12 furo[2,3-b]chromones (defined as T-N, that is the difference in mean values for tumor and normal cells of the negative log of the concentration of compound that reduced the viable cell number by 50% (pCC50). See Table II for explanation of chemical descriptors.
Effect of the introduction of a methoxy group (OMe) at C-7 of benzopyran ring on tumor-specificity (TS) and potency-selectivity expression (PSE) values for six sets of furo[2,3-b]chromone derivatives.
Discussion
The present study demonstrated that among 12 furo[2,3-b]chromones, compounds introduced with dimethyl (1a, 1b), spirocyclopentyl (2a, 2b), spirocyclohexyl (3a, 3b) or phenyl group (4a, 4b) at C-2 position showed essentially no cytotoxic activity nor tumor specificity, whereas compounds with phenylethenyl (5a, 5b) or propenyl (6a, 6b) group significantly enhanced the cytotoxicity and tumor specificity. Among the latter group, (5b) showed the greatest tumor specificity (evaluated by TS and PSE value), comparable with that of melphalan, although much weaker than that of 5-fluorouracil (5-FU) and doxorubicin (Table I). We also found that when a methoxy group was introduced at C-7 of benzopyran ring, both TS and PSE values of most of the compounds increased, especially of (5b) (Table IV). It was unexpected that both (5b) and (6b) at cytotoxic concentration would not induce apoptotic cells characterized by cell shrinkage. It is difficult to demonstrate that these compounds actually lack the ability to induce apoptosis. In order to elucidate the mechanism of cell death induction by these compounds, the possibility of the induction of other types of cells and growth arrest in OSCC cells should be tested.
QSAR analysis demonstrated that tumor selectivity of furo[2,3-b]chromone derivatives strongly correlated with intramolecular unsaturated bond number, molecular flexibility, molecular density, lipophilicity, molecular size, and molecular shape. Chemical modification of the lead compound may be a potential choice for designing a new type of anticancer drug.
Acknowledgements
This work was partially supported by KAKENHI from the Japan Society for the Promotion of Science (JSPS) (15K08111, 16K11519).
Footnotes
This article is freely accessible online.
Conflicts of Interest
The Authors wish to confirm that there are no known conflicts of interest associated with this publication and there was no significant financial support for this work that could have influenced its outcome.
- Received April 23, 2018.
- Revision received May 15, 2018.
- Accepted May 16, 2018.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}