


Abstract
Background: Neuroendocrine tumors (NETs) are the second most common digestive malignancy. For advanced NETs, survival is not satisfactory. Vitamin D has emerged as a promising anticancer drug. Materials and Methods: Cell proliferation assay, western blot, flow cytometry, and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assays were applied. Results: We demonstrated that RIN-m cells, neuroendocrine tumor cells, expressed vitamin D receptor (VDR) and VDR expression increased with increasing exposure to 1α,25-dihydroxyvitamin D3 [1α,25(OH)2D3] or MART-10, a 1α,25(OH)2D3 analog. MART-10 had anti-growth effect on RIN-m cells comparable to those of 1α,25(OH)2D3. The growth inhibition of both drugs was mediated by induction of cell-cycle arrest at G0/G1 phase and apoptosis. Western blot assay further revealed that this G0/G1 arrest was due to the up-regulation of p27 and down-regulation of cyclin dependent kinase 4 (CDK4), with MART-10 also reducing CDK6. Apoptosis induction was further supported by increased cleaved caspase-3 expression after treatment. Conclusion: MART-10 appears to be a promising regimen for NET treatment.
Neuroendocrine tumors (NETs) belong to a heterogeneous group of malignancies. Although with rare incidence, NETs still rank as the second most common type of digestive cancer (1, 2). Generally, NETs have a relatively low growth rate and the ability to secret a variety of peptide hormones to induce systemic symptoms (3). Although NETs may develop in almost any organ, most stem from the pancreas, originating from the islets of Langerhans, or the gastrointestinal tract, originating from enterochromaffin cells of the gut. The indolent nature and growth behavior of NETs sometimes mean this disease is not diagnosed until the development of metastases, which mainly involves the liver. For metastatic NETs, current antitumor treatments mainly consist of somatostatin analogs, chemotherapy, liver chemoembolization, interferon and targeted therapies (4), but none of these provide long-term survival. Thus, studies to investigate new regimens for NET treatment are justified.
Since the non-mineral functions of vitamin D were discovered, including antiproliferation, pro-apoptosis, pro-differentiation, anti-angiogenesis etc., abundant studies regarding the application of vitamin D to treated cancer have emerged (5). However, since systemic administration of 1α,25-dihydroxyvitamin D3 [1α,25(OH)2D3]; the active form of vitamin D, would induce hypercalcemia, the clinical practice of using 1α,25(OH)2D3 to treat cancer is hampered. Thus, thousands of 1α,25(OH)2D3 analogs have been created to avoid this drawback while trying to potentiate the antitumor effect.
19 Nor-2α-(3-hydroxypropyl)-1α,25(OH)2D3 (MART-10) (6), a newly synthesized 1α,25(OH)2D3 analog, has been shown by our group to be a promising anticancer drug against a variety of cancer types (7-14). Its in vivo effect and safety have also been shown through application of MART-10 to treat pancreatic cancer in a xenograft animal model (14).
Since there are no effective treatments available to treat advanced NET, in this study, we applied 1α,25(OH)2D3 and MART-10 to treat NETs and evaluated their effects on NET cell growth and related mechanisms.
Materials and Methods
Vitamin D compounds. 1α,25(OH)2D3 was purchased from Sigma (St. Louis, MO, USA). MART-10 was synthesized as previously described (6).
Cell culture. Rat insulinoma cell line, RIN-m, was purchased from (Sigma, St. Louis, MO, USA). RIN-m cells were grown in RPMI 1640 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific). Culture medium was changed three times per week.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. RIN-m cells were grown in 24-well culture plate were treated with 10−6 to 10−8 M 1α,25(OH)2D3 or 10−7 to 10−9 M MART-10 for 7 days. The cell viability was determined by MTT assay as previously described (15).
Cell-cycle analysis by flow cytometry. A FACS Calilbur (BD Biosciences, San Jose, CA, USA) flow cytometer was used for cell-cycle analysis as described previously (10, 14). Briefly, after 4 days of treatment with 10−7 M of 1α,25(OH)2D3 or MART-10, the cells were collected and fixed overnight. The fixed cells were stained in propidium iodide (PI) buffer at 4°C for 1 h before analysis.
Western blot for protein expression. The procedures for protein extraction, blocking, and immunodetection were described previously (10). The primary antibodies used in this study were monoclonal antibodies against VDR (sc-13133; Santa Cruz Biotechonology, Santa Cruz, CA, USA); p21 (#2947), p27 (#2552), cyclin-dependent kinase 4 (CDK4) (#12790), CDK6 (#3136), cyclin D3 (#2936), caspase 3 (#9662), and cleaved caspase 3 (#9664) (all Cell Signaling, Danvers, MA, USA). The secondary antibodies (1:5000) were anti-rabbit (111-035-003; Jackson Immunoresearch, West Grove, PA, USA) or anti-mouse secondary antibodies (M114; Leinco Technologies, Fenton, MO, USA). The blots were detected using enhanced chemiluminescence reagents (WBKLS0500; Millipore, Billerica, MA, USA). Membranes were detected by VersaDoc™ Imaging System (Bio-Rad, Hercules, CA, USA) for analysis.
Apoptosis analysis by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay. Tunnel assay was used to measure DNA fragmentation (16). Briefly, cells were plated on autoclaved glass coverslips in six-well culture plates and treated with 10−7 M of 1α,25(OH)2D3 or MART-10. Cellular DNA was stained with apoptosis detection kits (Millipore), and the assay was performed according to the recommendations from the manufacturer.
Statistical analysis. The data from each group were compared by Student t-test. A p-value of less than 0.05 was considered as indicating a significant difference. SPSS computer software package (version 10.0; Chicago, IL, USA) was employed to conduct the statistics.
Results
VDR expression of RIN-m cells and the effect of 1α,25(OH)2D3 and MART-10 on VDR expression. Figure 1A shows the western blot of VDR expression in RIN-m cell with and without 1α,25(OH)2D3 or MART-10 treatment. The western blot demonstrated that RIN-m cell expresses VDR and 1α,25(OH)2D3 or MART-10 treatment increased VDR expression in these cells. Figure 1B shows the quantitative result of the western blot. 1α,25(OH)2D3 and MART-10 at 10−7 M were able to up-regulate RIN-m cell VDR expression to 2.4±0.2- and 2.5±0.23-fold that of the control.
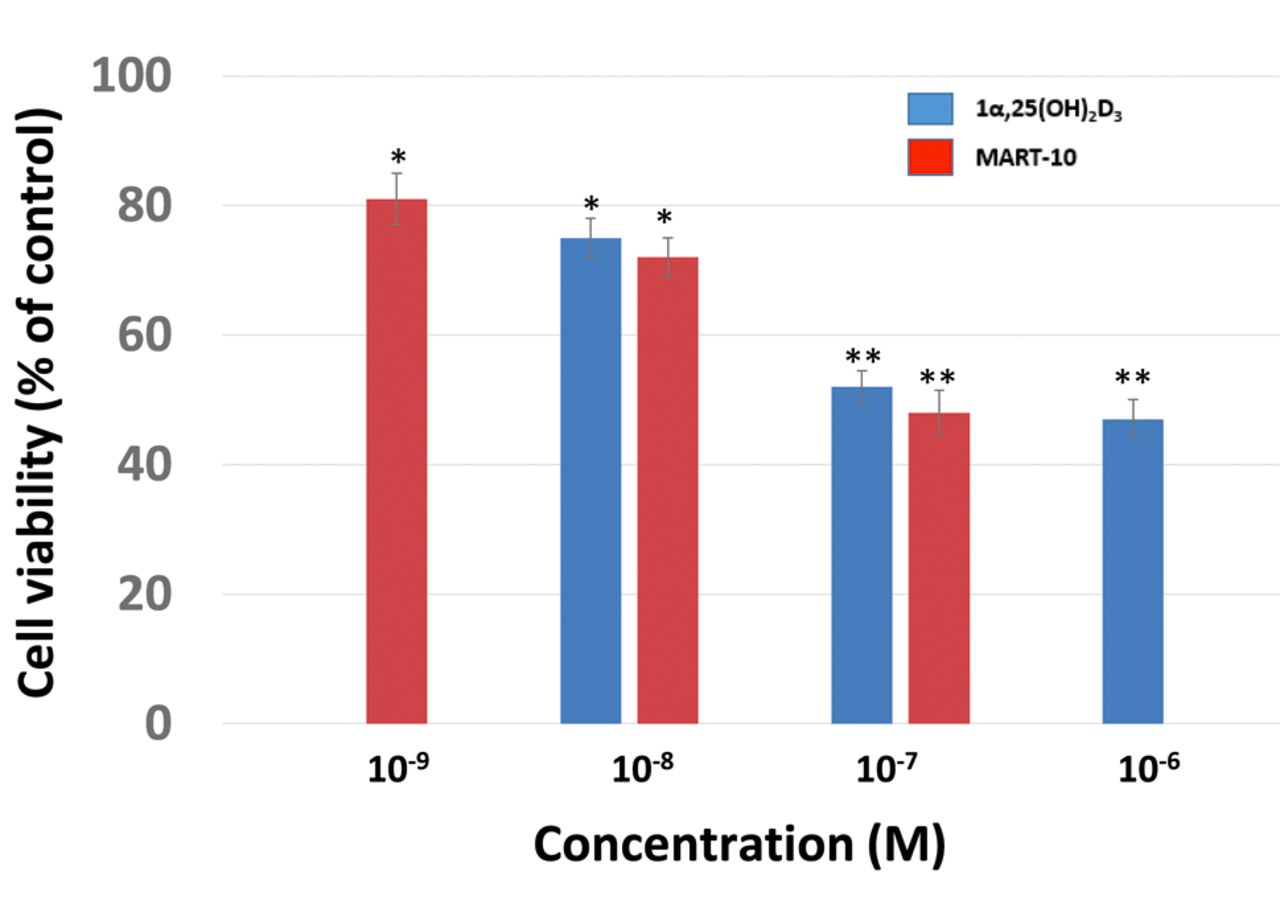
Effect of 1α,25(OH)2D3 and MART-10 on RIN-m cell growth. As shown in Figure 2, the result of MTT assay revealed that after 7 days of treatment, 1α,25(OH)2D3 at concentrations of 10−8 M or higher significantly suppressed RIN-m cell growth compared to the control. MART-10, at concentrations of 10−9 and more also significantly inhibited RIN-m cell growth. Both compounds induced significant growth inhibition on neuroendocrine tumor cell growth in vitro, with MART-10 seeming more potent than 1α,25(OH)2D3 in this respect.
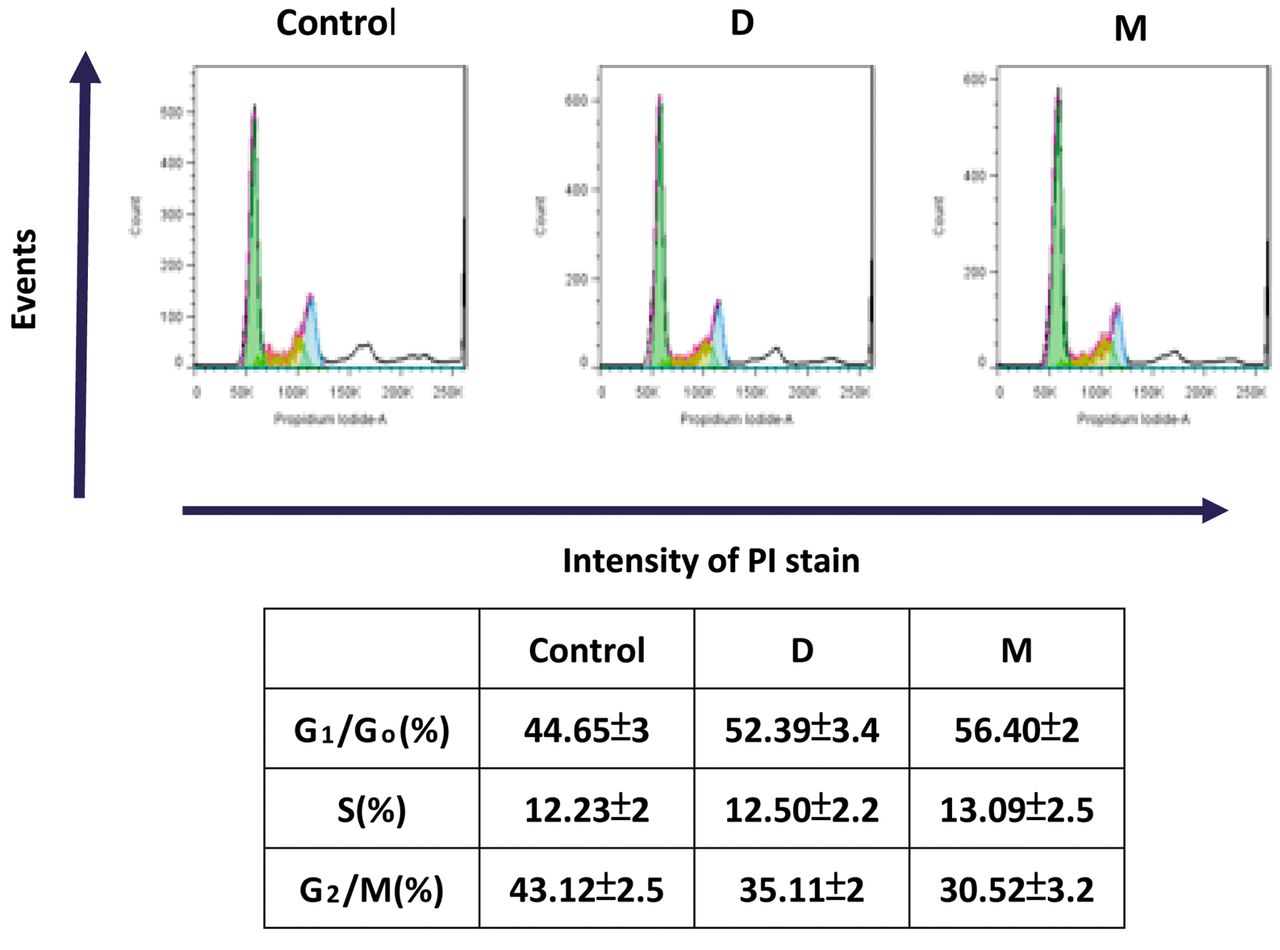
Effect of 1α,25(OH)2D3 and MART-10 on cell-cycle progression in NET cells. To further understand how 1α,25(OH)2D3 and MART-10 influenced RIN-m cell growth, we next analyzed by flow cytometry the cell-cycle distribution of RIN-m cells after 4 days of treatment. Figure 3 indicates that treatment with 10−7 M 1α,25(OH)2D3, and with MART-10 significantly increased the G1 phase cell percentage from 44.65±3% to 52.39±3.4% and 56.4±2%, respectively. Our results reveal that 1α,25(OH)2D3 and MART-10 were able to induce RIN-m cell cycle arrest at the G0/G1 phase, leading to the growth inhibition as demonstrated in Figure 2.
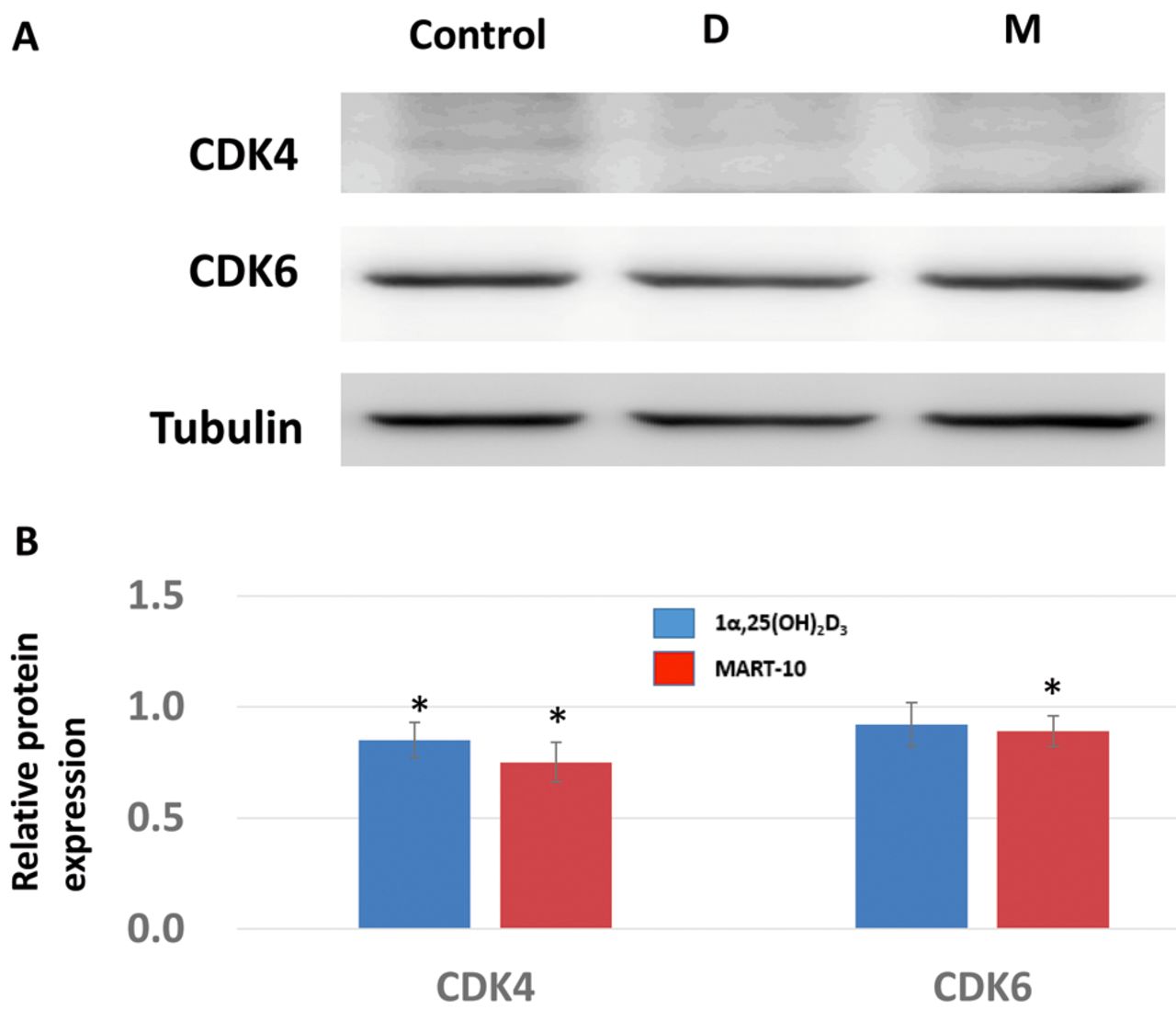
Effect on 1α,25(OH)2D3 and MART-10 on G1/S transition-related CDK inhibitors, CDKs, and cyclins in RIN-m cells. For further investigation of how 1α,25(OH)2D3 and MART-10 induced G0/G1 cell-cycle arrest in RIN-m cells, expression of two main CDK inhibitors responsible for G1/S transition, p21 and p27, were evaluated. As shown in Figure 4, 2 days of exposure to 10−7 M of 1α,25(OH)2D3, and of MART-10 significantly up-regulated p27 expression relative to the control. For p21, neither 1α,25(OH)2D3 nor MART-10 influenced its expression significantly. Two important CDKs, CDK4 and CDK6, were also studied. CDK4 expression was attenuated by both 1α,25(OH)2D3, and MART-10 at 10−7 M. No significant change was found for CDK6 expression in RIN-m cells after treatment (Figure 5). We next examined cyclin D3 expression in RIN-m cells after treatment and found that neither agent had any obvious effect on cyclin D3 expression (Figure 6).

Vitamin D receptor (VDR) expression in RIN-m cells. A: Western blot depicting VDR expression with and without 10−7 M 1α,25(OH)2D3 (D) or 10−7 M MART-10 (M) treatment in RIN-m cells. Tubulin was used as control. B: Quantitative results of western blotting. Two days of treatment with 1α,25(OH)2D3 or MART-10 increased VDR expression in RIN-m cells. Results are presented as a percentage that of the control. Each value is the mean±SD of three to five determinations. *p<0.05 and **p<0.001 versus control.
Effect of 1α,25(OH)2D3 and MART-10 on apoptosis induction in RIN-m cells. After 7 days of treatment, apoptotic cells were determined by TUNEL assay. Figure 7 shows that 1α,25(OH)2D3 and MART-10, at a concentration of 10−7 M, increased the apoptotic cell ratio compared to the control, indicating that both 1α,25(OH)2D3 and MART-10 had the ability to induce apoptosis of RIN-m cells. The finding that 1α,25(OH)2D3 and MART-10 treatment significantly increased cleaved caspase-3 expression in RIN-m cells (Figure 8) supported the result shown in Figure 7.

Dose-dependent antiproliferative effect of 1α,25(OH)2D3 and MART-10 on RIN-m cells. RIN-m cells were treated with 1α,25(OH)2D3 or MART-10 for 1 week at the indicated concentrations. Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Results are presented as a percentage that of the control. Each value is the mean±SD of three to five determinations. *p<0.05 and **p<0.001 versus control.
Discussion
Human VDR, consisting of 427 amino acids, has a molecular weight of 52 kDa. 1α,25(OH)2D3 needs to enter the nucleus to bind with VDR, which further interacts with retinoid X receptor (RXR) to form a heterodimer in order to modulate gene expression (17). Since the discovery of VDR in a variety of human tissues, it is not surprising that 1α,25(OH)2D3 has been found to play a vital role in many important physiological functions. As long as genes have vitamin D response elements (VDREs) within the promoter regions, their expressions is subject to vitamin D modulation (18). Using Chip-seq assay and genome-wide association studies, Ramagopalan et al. demonstrated that there are at least 2776 1α,25(OH)2D3–VDR–RXR complex binding sites in the human genome and expression of 229 genes is significantly changed after 1α,25(OH)2D3 stimulation, with most of the affected genes being associated with autoimmune disease and cancer (19), suggesting the possible close relation between cancer and 1α,25(OH)2D3.
Our study shows that RIN-m cells express VDR (Figure 1). After 1α,25(OH)2D3 or MART-10 treatment, VDR expression in RIN-m cells increased (Figure 1), implying RIN-m cells are vitamin D responsive. As shown in Figure 2, both 10−6 M to 10−8 M 1α,25(OH)2D3, and 10−7 M to 10−9 M MART-10 effectively inhibited RIN-m cell growth, consistent with the result shown in Figure 1. Since MART-10 has been shown to be active and safe in vivo, our finding suggests that MART-10 could be further applied in vivo as a regimen for NET treatment.

Cell-cycle distribution for RIN-m cells after treatment with 10−7 M 1α25(OH)2D3 (D) or MART-10 (M) as analyzed by flow cytometry. A: Representative DNA histogram of RIN-m cells with and without 1α,25(OH)2D3 or MART-10 treatment for 4 days. B: Distribution of RIN-m cells at G1/G0, S and G2/M phase after treatment. PI: Propidium iodide.
In order for cell proliferation to proceed, cells must pass through cell-cycle progression. The cell cycle consists of four phases, namely G1, S, G2, and M phases.During the S phase, cells conduct DNA replication; while in the M phase, cells begin to separate into two daughter cells. Between S and M phases is a “gap phase”, which is G2. Before S phase is G1 phase which separate G0 and S phases. When cells exit the cell cycle, they remain in a quiescent status, which is the G0 phase. Most cells in the human body lie in the G0 phase and only a few cells are proliferating actively, such as epithelium cells or bone marrow (20). Human cancer cells have unscheduled proliferation, mainly due to constitutive mitogenic signaling, leading to cell-cycle deregulation (21), and thus targeting cell-cycle progression is deemed a promising way to control tumor growth. Figure 3 clearly demonstrates that both 1α,25(OH)2D3 and MART-10 treatment increased the percentage of G1/G0 RIN-m cells, indicating both agents can induce cell-cycle arrest at G0/G1, resulting in the inhibition of cell proliferation as shown in Figure 2.
An important restriction point, which is deemed as the initial control for cells to enter the cell cycle, is located in the mid-to-late G1 phase (22). To pass through this restriction point to proceed to proliferation, cells first need CDK4 and CDK6 to bind with cyclin D, which further phosphorylate retinoblastoma (RB) to a inactive status and release transcriptional factor E2F from the E2F–RB complex to trigger expression of S phase genes (23, 24). As shown in Figure 5, both 1α,25(OH)2D3 and MART-10 suppressed CDK4 expression in RIN-m cells significantly, with CDK6 only being inhibited by MART-10, resulting in the G0/G1 cell-cycle arrest shown in Figure 3. As for cyclin D3, neither 1α,25(OH)2D3 nor MART-10 inhibited its expression significantly (Figure 6). Of note, the activity of CDK4 and CDK6 are regulated by endogenous CDK inhibitors, mainly p21 and p27 (25). We found both 1α,25(OH)2D3 and MART-10 up-regulated p27 expression in RIN-m cells but had no effect on p21 (Figure 4). Collectively, based on our results, we conclude that 1α,25(OH)2D3 and MART-10 are both effective at suppressing RIN-m cell growth through induction of G0/G1 cell-cycle arrest by down-regulation of CDK4 (CDK6 only repressed by MART-10) and up-regulation of p27.
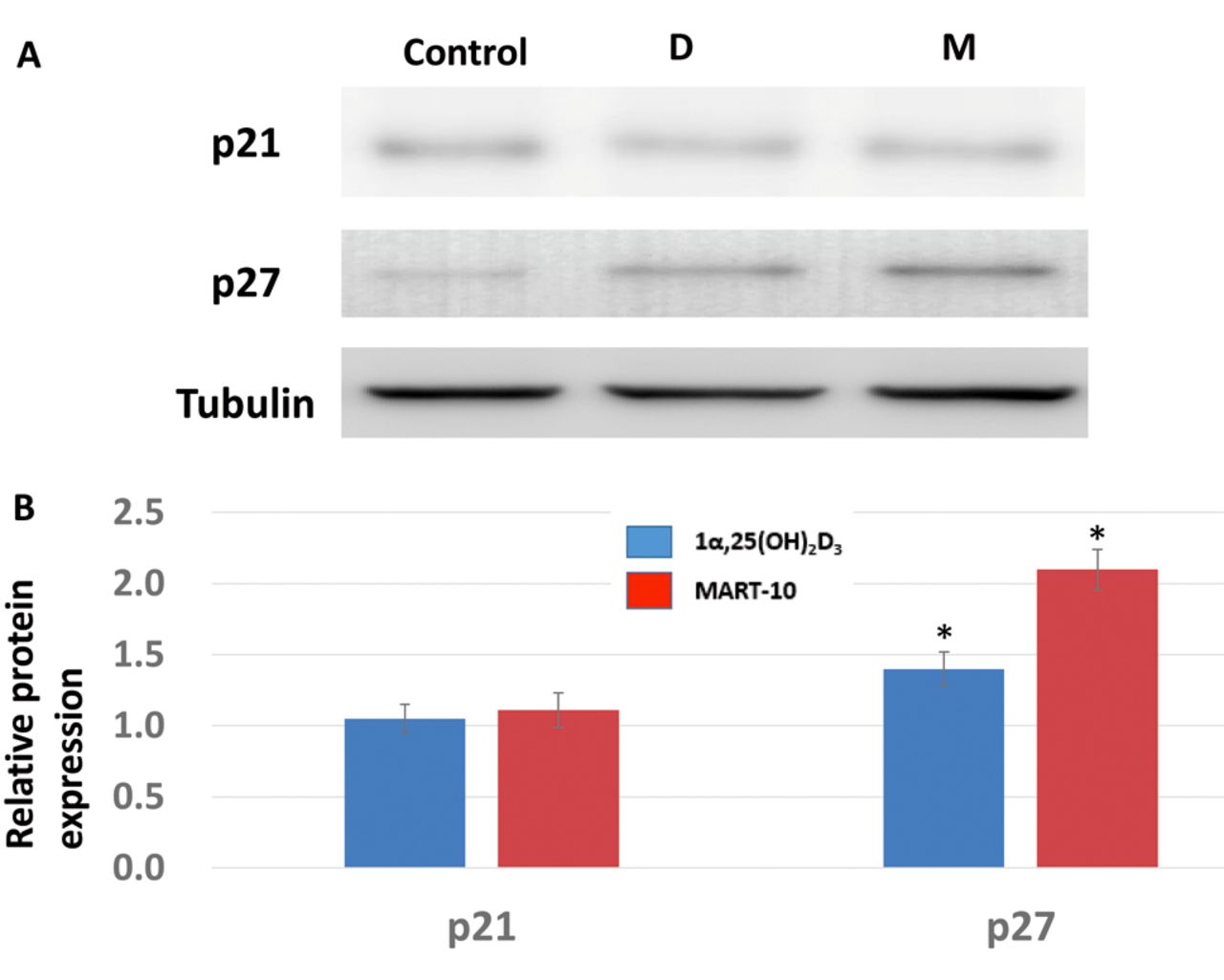
Western blot analysis for the expression of p21 and p27 relative to those of the control after treating RIN-m cells with 10−7 M 1α, 25(OH)2D3 (D) or MART-10 (M). A: Western blot depicting p21 and p27 protein expression in RIN-m cells after treatment for 2 days with and without 10−7 M 1α, 25(OH)2D3 or MART-10. Tubulin was used as the loading control. B: The quantitative result of the western blot. Both 1α,25(OH)2D3 and MART-10 increased p27 expression in RIN-m cells. Each value is the mean±SD of three independent determinations. *p<0.05 and **p<0.001 versus control.

Effects of 1α,25(OH)2D3 (D) and MART-10 (M) on cyclin-dependent kinase 4 (CDK4) and -6 expression in RIN-m cells. A: Western blot depicting CDK4 and CDK6 protein expression in RIN-m cells treated with and without 10−7 M 1α, 25(OH)2D3 or MART-10 treatment for 2 days. Tubulin was used as the loading control. B: Quantitative results of western blotting. Both 1α,25(OH)2D3 and MART-10 reduced CDK4 expression in RIN-m cells, while CDK6 expression was attenuated only by MART-10. Each value is the mean±SD of three independent determinations. *p<0.05 and **p<0.001 versus control.

Effects of 1α,25(OH)2D3 (D) and MART-10 (M) on cyclin D3 expression in RIN-m cells. A: Western blot depicting cyclin D3 expression in RIN-m cells with and without 10−7 M 1α, 25(OH)2D3 or MART-10 treatment for 2 days. Tubulin was used as the loading control. B: Quantitative results of western blotting. Neither 1α,25(OH)2D3 nor MART-10 affected cyclin D3 expression in RIN-m cells. Each value is the mean±SD of three independent determinations. *p<0.05 and **p<0.001 versus control.

Effects of 1α,25(OH)2D3 and MART-10 on apoptosis of RIN-m cells as analyzed by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay. The apoptotic effects on RIN-m cells induced by 10−7 M 1α,25(OH)2D3 and MART-10 treatment for 7 days were analyzed by TUNEL assay to measure the extent of DNA fragmentation visualized by fluorescence microscopy. The relative apoptotic index as compared to the control is shown. Each value represents the average of three determinations±SD. *p<0.05 versus control.
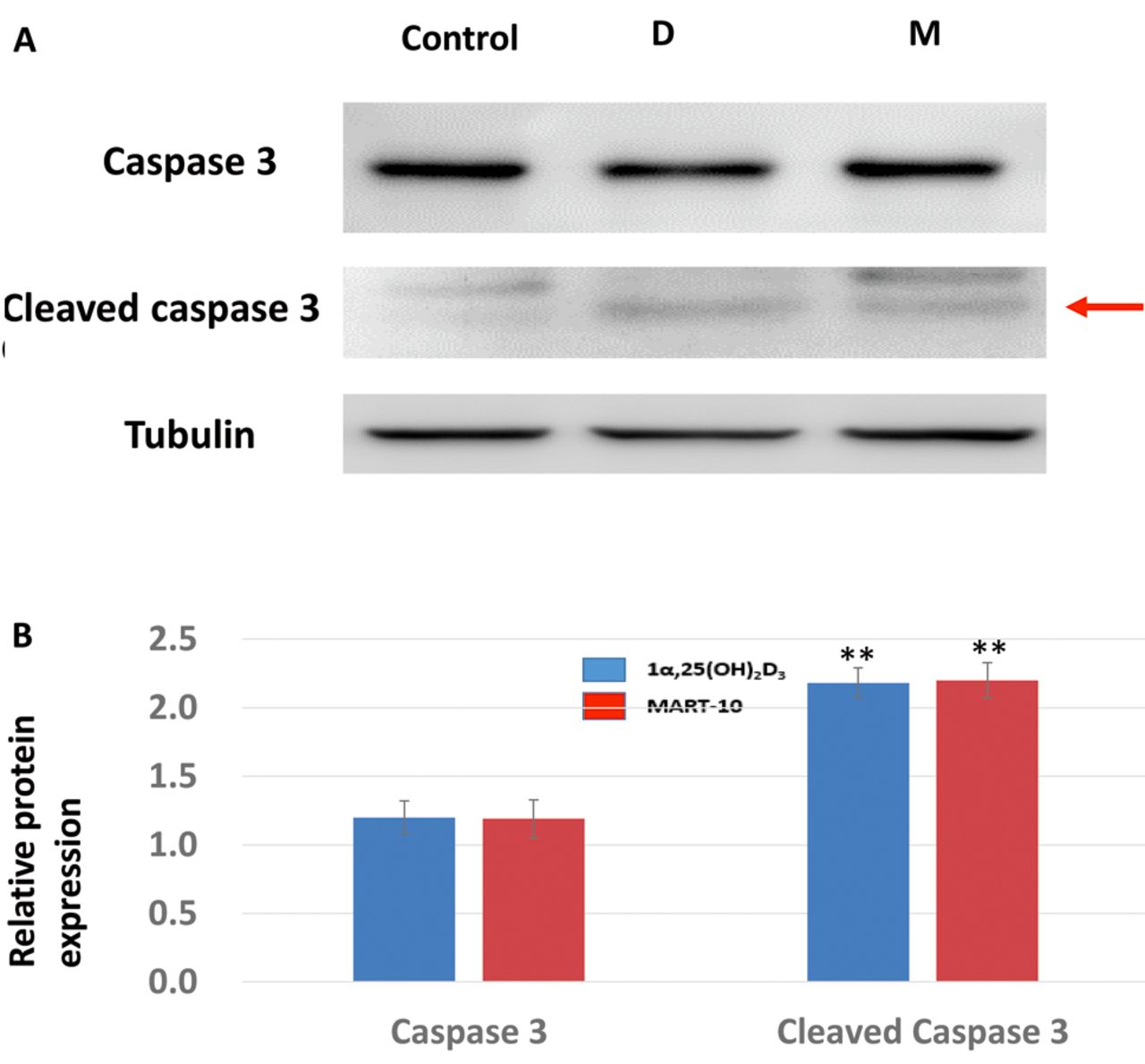
Effects of 1α,25(OH)2D3 (D) and MART-10 (M) on caspase 3 and cleaved caspase 3 expression in RIN-m cells. A: Western blot analysis of caspase 3 and cleaved caspase 3 expression in RIN-m cells after either 10−7 M of 1α,25(OH)2D3 or MART-10 treatment for 7 days. Tubulin was used as the loading control. B: Quantitative analysis ofwestern blotting. Both 1α,25(OH)2D3 and MART-10 increased cleaved caspase 3 expression in RIN-m cells. Mildly increased expression of caspase 3 was also noted while not reaching statistical significance. **p<0.01 versus control.
Apoptosis or programmed cell death, type 1 cell death, is an important physiological mechanism for eliminating harmful or unnecessary cells and controlling cell proliferation, thus maintaining tissue homeostasis (26). Therefore, intact apoptotic signaling and function are fundamentally important. Thus, escape of apoptosis regulation is deemed a hallmark of cancer (27). We therefore investigated the effect of 1α,25(OH)2D3 and MART-10 on apoptosis induction in RIN-m cells. As evaluated by TUNEL assay (Figure 7), both 1α,25(OH)2D3 and MART-10 treatment induced apoptosis of RIN-m cells, which was supported by the finding that cleaved caspase 3 expression was increased after 1α,25(OH)2D3 and MART-10 treatment (Figure 8).
Conclusion
The newly synthesized 1α,25(OH)2D3 analog MART-10 has a growth-inhibitory effect on RIN-m cells comparable to that of 1α,25(OH)2D3. The anti-growth mechanisms of 1α,25(OH)2D3 and MART-10 against RIN-m cells are mediated by cell-cycle arrest at G0/G1 and apoptosis induction. Since there are no effective treatments available for advanced NET and MART-10 has been shown to be active and safe in vivo, further studies applying MART-10 to treating NETs in animal studies or even clinical trials are warranted.
Acknowledgements
This work is supported by grants 103-2314-B-182A-085 and 104-2314-B-182A-017 (Ministry of Science and Technology) to Kun-Chun Chiang and CMRPG2E0211-2 (Chang Gung Memorial Hospital) to Po-Jen Hsieh.
Footnotes
↵* Co-first Authors.
This article is freely accessible online.
Conflicts of Interests
All Authors declare no conflict of interest in regard to this study.
- Received April 15, 2016.
- Revision received May 24, 2016.
- Accepted May 25, 2016.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}