


Abstract
Background: Heat-shock proteins (HSPs) as well as microRNAs have been identified to orchestrate crucial mechanisms in prostate cancer (PCa) progression and treatment resistance. Due to cytoprotective properties of HSPB1 we analyzed molecular mechanisms of drug resistance in PCa cell culture systems, and notably found HSPB1 functionality linked to microRNA miR-1 activities. Materials and Methods: HSPB1 and miR-1 levels were genetically modified in PCa cell lines and alterations in molecular and cellular responses were assessed by quantitative reverse transcription/polymerase chain reaction, western blotting, and proliferation assays. Results: Our data provided for the first time evidence that HSPB1 regulates miR-1 expression, and subsequently restores oncogenic signaling pathways of androgen receptor (AR) and transforming growth factor β1 (TGFB1). Conclusion: Our data point towards HSPB1 and miR-1 involvement in development of castration-resistant PCa and therefore represent promising targets for anticancer therapy of advanced PCa.
The involvement of heat-shock proteins (HSPs) during initiation and progress of prostate cancer (PCa) has been shown. Large HSP complexes consisting of primary chaperones (e.g. HSPA1, HSPAA1/HSPAB1, HSPB1) and accessory co-chaperones (e.g. DnaJ homolog subfamily B member 1; DNAJB1, stress-induced phosphoprotein 1; STIP1) participate in protein binding and release, and therefore control half-life and localization of client proteins. Thus, HSPs orchestrate diverse metabolic and regulatory pathways (1). In PCa progression, HSPs are tightly up-regulated and frequently induced during radiotherapy and drug administration (2, 3). In PCa tissue, the small chaperone HSPB1 is highly expressed and plays a pivotal role in tumor cell growth and survival. HSPB1 function is linked to cytoskeleton remodeling, cell motility, androgen receptor (AR)-driven proliferation, and treatment resistance (4-7). Consistent with its multi-functional properties, HSPB1 is appropriate for targeted-therapy (8).
Although various studies have gained new insights into HSPB1 functions and interactions in PCa cells, molecular mechanisms by which HSPB1 triggers chemoresistance have not been fully-elucidated. Since we started examining anti-oncogenic properties of the tumor-suppressor microRNA miR-1 in a PCa cell model system, most notably, we found basal protein levels of HSPB1 to be negatively correlated with the expression of miR-1. The most interesting aspect of this observation is that microRNAs often regulate signaling and effector cascades of chemoresistance in PCa (9, 10). Thus, malignant cells may benefit from HSPB1-driven suppression of miR-1 as a cellular mechanism which may contribute to the drug sensitivity of cancer cells.
In the present study, we identified HSPB1 as cellular inhibitor of miR-1 expression. Furthermore, we focused on the characterization of miR-1 functionality in the context of cellular proliferation and found miR-1 to target the AR and transforming growth factor-β (TGFB) signaling, and probably further signal cascades for HSPB1-driven enhancement of tumor progression and chemoresistance.
Materials and Methods
Cell culture. PCa cell lines LNCaP and PC-3 (Cell Lines Service, Heidelberg, Germany) were propagated in RPMI-1640 medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (PAN Biotech, Aidenbach, Germany) in an atmosphere with 5% CO2 and at 37°C. PC-3-HSPB1 cells stably-overexpressing HSPB1 were generated as described elsewhere (7) and selected with 400 μg/ml G418 (Carl Roth, Karlsruhe, Germany).

Heat-shock protein (HSP) HSPB1 suppresses tumor suppressor miR-1 expression in LNCaP and PC-3 prostate cancer (PCa) cells. A: Equal amounts of total cell lysates were separated and immunochemically analyzed with antibodies directed against HSPB1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), with GAPDH signals served as loading control. Western blot examinations were performed with unmodified LNCaP cells (column 1), PC-3 cells (column 2), PC-3 cells stably overexpressing HSPB1 (column 3) and PC-3 cells transiently transfected with pcDNA3.1-HSPB1 vector encoding for HSPB1 (control transfection: column 4; transient transfection: column 5). B: High concentrations of HSPB1 as shown in panel A correlated with low miR-1 expression levels detected by quantitative reverse transcription and polymerase chain reaction (qRT-PCR). MicroRNA preparations of samples described in panel A (columns 1 to 5) were conducted for miR-1 quantification using miR-1-specific primers and standardized to U6 RNA expression levels. Data are given as the mean±SD, statistically analyzed by Student's t-test. *p≤0.05; **p≤0.01; *** p≤0.001.
Transfection experiments. One day prior to transfection, cells were plated into 6-well (150,000 cells/well) or 24-well (30,000 cells/well) cell culture plates coated with 0.01% poly-L-lysine (Sigma-Aldrich, Deisenhofen, Germany) for 10 min. For overexpression of miR-1 and HSPB1, 1 μg DNA (24-well) and 3 μg DNA (6-well) per well were transferred into cells using Lipofectamin 2000 reagent (Invitrogen, Karlsruhe, Germany). miR-1-specific inhibitor molecules Anti-hsa-miR-1 miScript miRNA Inhibitor (Qiagen, Hilden, Germany) were applied by transfection using siLentFect (BioRad, München, Germany). The eucaryotic expression vector pHSP27 was kindly provided by C. Kubisch (Munich, Germany). Empty vectors pcDNA3.1 (Invitrogen) and pSuperior (OligoEngine, Seattle, WA, USA) were used for control transfections.
Cloning of the miR-1 mimicking expression vector pmiR-1. A DNA plasmid encoding for the cDNA sequence of mature miR-1 mimicking RNA (pmiR-1) was constructed using the pSuperior system from OligoEngine. Two oligonucleotides (miR-1 forward 5’-GATCCCCTGGAATGTAAAGAAGTATGTATTTCAAGAGAATACATACTTCTTTACATTCCATTTTTA-3’ and miR-1 reverse 5’-TCGAT A A A A AT G G A AT G TA A AG A AG TAT G TAT T C T C T T G A AATACATACTTCTTTACATTCCAGGG-3’) were hybridized by a temperature gradient from 95°C to 4°C for 40 min. Due to the asymmetrical design of both complementary oligonucleotides, the hybridization products formed defined single-stranded 5’ overhangs, which became ligated into the BglII/XhoI (Thermo Scientific, Waltham, MA, USA) double-digested pSuperior vector. After ligation (T4 DNA Ligase; Thermo Scientific), positively-selected clones were verified by restriction analysis and sequencing.
Western blotting. Evaluation of protein levels was carried out with cells plated at a density of 150,000 cells/well in a 6-well cell culture plate and harvested after an incubation of 24 h to 72 h. Cells were prepared in lysis buffer [50 mM Tris (pH 7.5), 150 mM NaCl, 10 mM K2HPO4, 5 mM EDTA, 10% glycerol, 1% Triton X-100, 0.05% sodium dodecysulfate, 1 mM Na3VO4, 20 mM NaF, 0.1 mM phenyl-methylsulfonyl fluoride, 20 mM 2-phosphoglycerate] supplemented by Complete Protease Inhibitor Cocktail (Roche Applied Science, Mannheim, Germany). Total protein was determined using Bradford Reagent (BioRad) and equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Subsequently, proteins were transferred onto a nitrocellulose membrane (GE Healthcare Europe, Freiburg, Germany) and blocked utilizing Rotiblock (Carl Roth). Proteins of interest were detected by overnight incubation with specific antibodies directed against HSPB1, AR and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; all Cell Signaling Technology, Danvers, MA, USA). Western blot membranes were incubated with peroxidase-coupled secondary antibodies (Cell Signaling Technology) for 1 h and protein signals were visualized by using SuperSignal West Dura Chemiluminescent Substrate (Thermo Scientific) and a ChemiDoc system (BioRad). Quantification of protein signals was carried out by Image Lab 3.0 software (BioRad) using GAPDH-specific signals as loading control.
MicroRNA quantification by reverse transcription and real-time polymerase chain reaction (qRT-PCR). Total small RNA was extracted using the mirPremier microRNA Isolation Kit (Sigma-Aldrich) and 2 μg of total microRNA were applied in reverse transcription by using the miScript II RT Kit (Qiagen). Subsequently, quantification was performed by real-time PCR (miScript SYBR Green PCR Kit; Qiagen) with the CFX96 Real-Time PCR Detection System (BioRad) and the primer Hs_miR-1_1 miScript Primer Assay (Qiagen). For quantification, miR-1 signals were standardized to U6 RNA (Hs_RNU6-2_1 miScript Primer Assay; Qiagen) as reference.
mRNA quantification by qRT-PCR. Total RNA was prepared by peqGOLD TriFast (Peqlab, Erlangen, Germany) according to the supplier's descriptions and quantified using a Nanodrop ND-2000 UV/vis spectrophotometer (Peqlab). Reverse transcription was carried out with 1 μg of total RNA, the Superscript III First-Strand Synthesis System (Invitrogen) and an oligo dT primer. Real-time PCR was carried out using a fluorescence dye (SensiMix SYBR Kit; Bioline, London, UK) and the CFX96 Real-Time PCR Detection System (BioRad) with the following target-specific primer pairs: AR forward: 5’-TGCCTGATCTGTGGAGATGA-3’, AR reverse: 5’-CGAAGACGACAAGATGGACA-3’, TGFB1 forward: 5’-GCCCTGGACACCAACTATTG-3’, TGFB1 reverse: 5’-CGTGTCCAGGCTCCAAATG-3’, kallikrein-3 (KLK3) forward: 5’-CCGGAGAGCTGTGTCACCAT-3’, KLK3 reverse: 5’-GTGCAGCACCAATCCACGTC-3’, and ribosomal protein large P0 (RPLP0) forward: 5’-CAATGGCAGCATCTACAACC-3’, RPLP0 reverse: 5’-ACTCTTCCTTGGCTTCAACC-3’ as reference gene.
Generation of LNCaP cells stably-overexpressing miR-1-mimicking RNA. LNCaP cells were transfected with the vector pmiR-1 as described above, passaged once per week and selected with 0.3 μg/ml puromycin (Invitrogen). Cell clones stably overexpressing miR-1 mimicking RNA molecules were identified by qRT-PCR and compared to maternal LNCaP cells. Subsequent cultivation of LNCaP-miR-1 cells was performed in the presence of 0.3 μg/ml puromycin.
Proliferation assay. Cellular proliferation was examined by cell counting utilizing a CASY Cell Counter and Analyzer Model TT (Roche Applied Science, Mannheim, Germany). Therefore, 30,000 cells/well were plated in 24-well cell culture plates and treated for 96 h or 144 h. At indicated time points, adherent cells were detached by trypsin treatment, suspended in 10 ml CASYton solution (Roche Applied Science), and the number of living cells was determined in 400 μl cell suspension in triplicates.
Statistical analysis. Results of at least three independent experiments were statistically analyzed, using the unpaired Student's t-test, and expressed as the mean±SD compared to control cells. Differences at p≤0.05 were considered significant.
Results
HSPB1 diminishes microRNA miR-1 expression in PCa cells. The basal expression of miR-1 in PCa cell lines LNCaP and PC-3 was negatively correlated to the intracellular HSPB1 concentration. The higher the level of HSPB1 protein in LNCaP cells (Figure 1A), the lower the expression of miR-1 was (defined as 1.0; Figure 1B), whereas the low abundance of HSPB1 protein in PC-3 cells (Figure 1A) was related to a significantly elevated level of miR-1 (71.9-fold, p=0.0014; Figure 1B). From there, we assumed a regulatory connection between HSPB1 and miR-1 and we conducted further experiments to validate our hypothesis.
As shown by qRT-PCR, stable overexpression of HSPB1 (Figure 1A) reduced high expression levels of miR-1 from 71.9-fold to 13.2-fold (p=0.0028; Figure 1B) compared to LNCaP cells. These observations were additionally confirmed by transient transfections, with transient up-regulation of HSPB1 (Figure 1A) revealing a clear reduction of miR-1 level from 63.9-fold (Figure 1B) to 15.3-fold (p=0.0001; Figure 1B) compared to basal miR-1 expression in LNCaP cells.

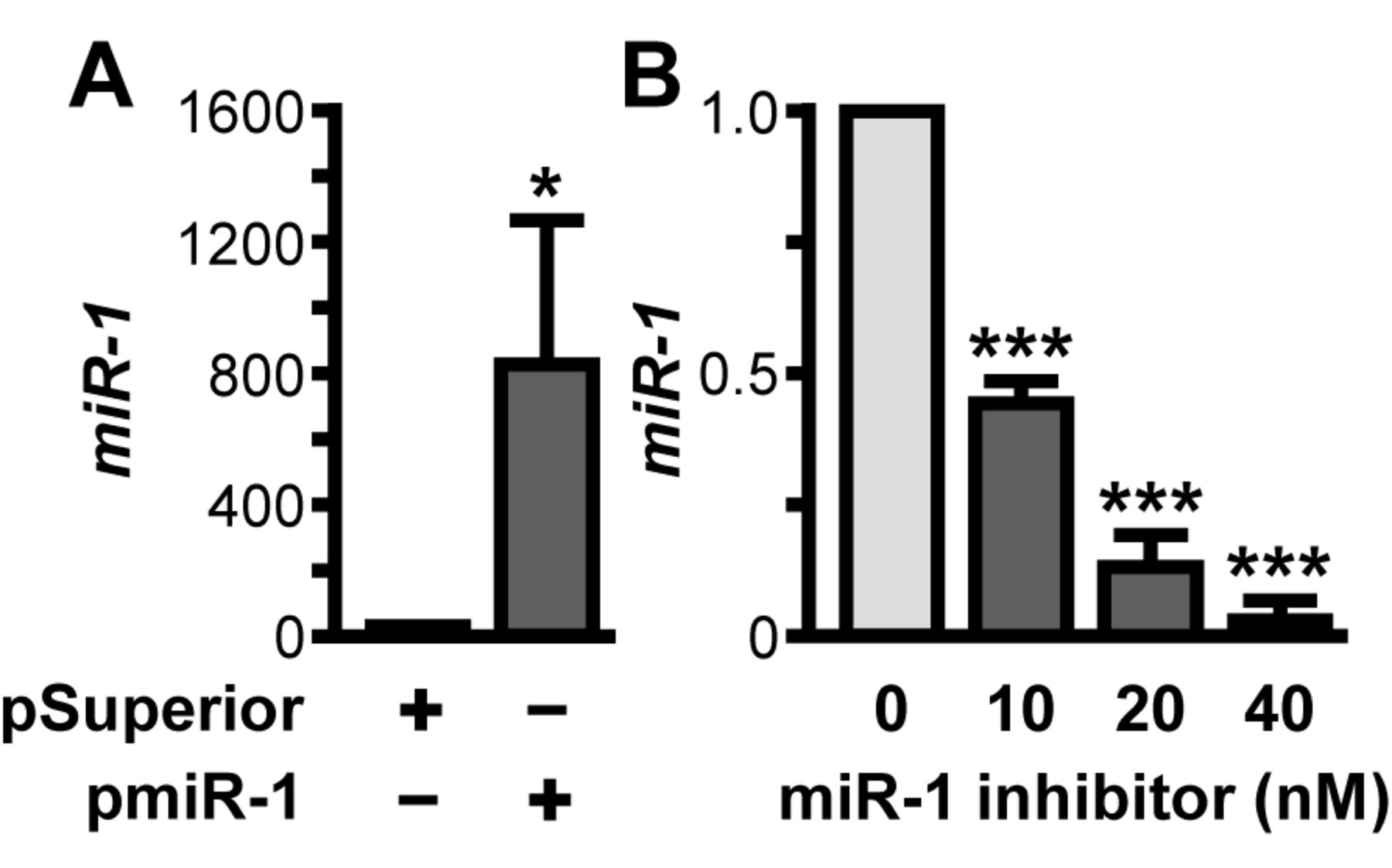
Experimental modulation of cellular miR-1 expression using the plasmid pmiR-1 and a commercially available miR-1-specific inhibitor. A: LNCaP cells with a low level of miR-1 expression were transiently transfected with pmiR-1, incubated and subsequently analyzed for miR-1 expression, standardized to U6 RNA expression levels, by quantitative reverse transcription and polymerase chain reaction (qRT-PCR) using miR-1-specific primers. B: PC-3 cells with a high level of basal miR-1 expression were treated with Anti-hsa-miR-1 miScript miRNA Inhibitor (Qiagen). miR-1 levels in total microRNA preparations were assessed by qRT-PCR using miR-1-specific primers and standardized to U6 RNA expression levels. Data are given as the mean±SD, statistically analyzed by Student's t-test. *p≤0.05; **p≤0.01; ***p≤0.001.
miR-1 overexpression attenuates PCa cell proliferation. These data prompted us to study whether the identified suppression of miR-1 may be part of HSPB1-driven resistance pathways potentially counteracting the antiproliferative efficacy of cancer drugs. Therefore, the DNA plasmid pmiR-1 encoding for an miR-1 cDNA sequence was constructed, showing greatly enhanced overexpression of miR-1-mimicking RNA, resulting in a 810.7-fold increase of miR-1 72 h after transfection (p=0.0455; Figure 2A). miR-1 analysis after DNAse treatment of microRNA preparations, as well as control reactions using the DNA plasmid pmiR-1 as PCR template were negative for miR-1 signals (data not shown). To assess knock-down of miR-1, a commercially available miR-1-specific inhibitor was used, which led to significant and concentration-dependent suppression of miR-1 expression of 2.2-fold at 10 nM (p<0.0001), 6.3-fold at 20 nM (p<0.0001), and 26.1-fold at 40 nM (p<0.0001), as shown in Figure 2B. Following miR-1 inhibition, experiments were carried out applying an miR-1 inhibitor at a concentration of 40 nM.
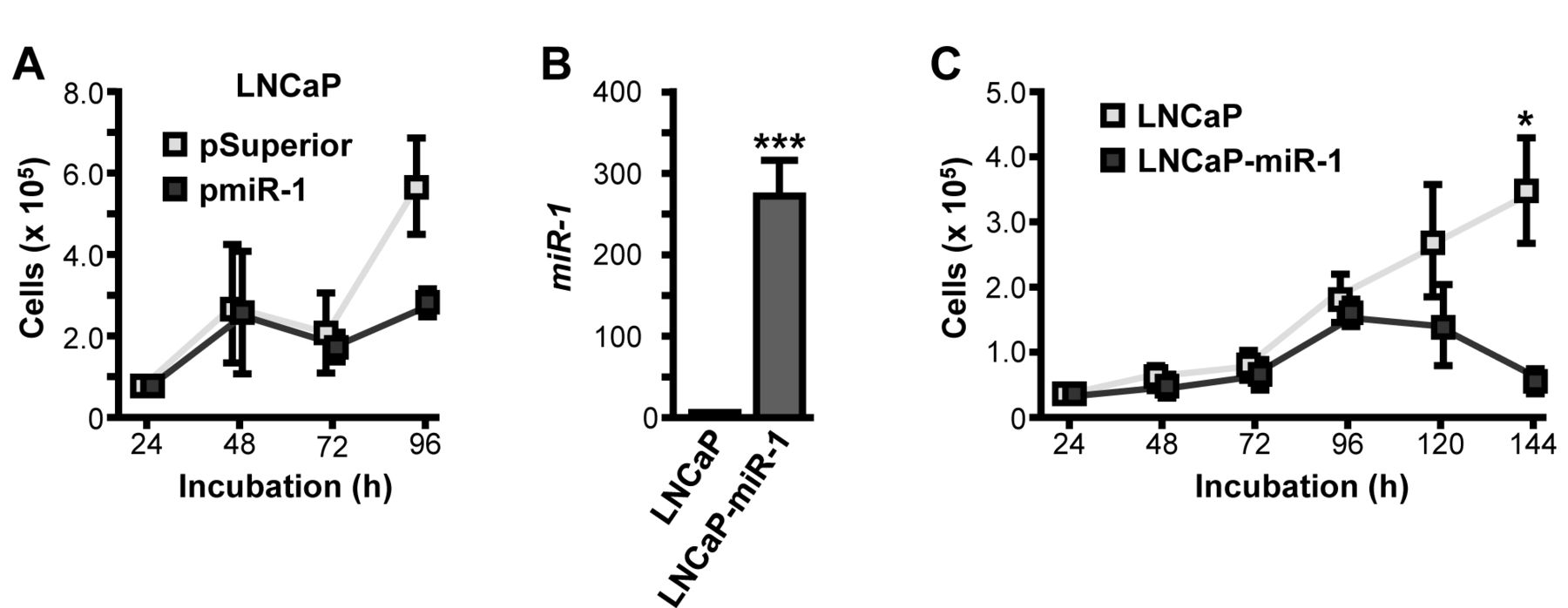
In several cancer entities, miR-1 properties are linked to cellular growth inhibition and therefore we conducted growth kinetics assays with cells overexpressing miR-1 molecules. In LNCaP cells transiently transfected with pmiR-1, elevated miR-1 levels were confirmed by qRT-PCR (data not shown). Cellular growth analysis tended to show a 2.0-fold but statistically insignificant decrease of cell numbers within 96 h (p=0.1454; Figure 3A). These data were confirmed by experiments using LNCaP cells stably overexpressing miR-1 (LNCaP-miR-1) which exhibited an intense increase of basal miR-1 expression compared to maternal LNCaP cells (278-fold, p=0.0003; Figure 3B). Subsequent growth kinetic assays showed proliferative characteristics comparable to those on transient miR-1 transfection, with a 7.1-fold reduction of cellular growth after 144 h following miR-1 expression (p=0.0243; Figure 3C).

Growth-inhibitory properties of miR-1 in LNCaP cells transiently, and stably, transfetced, respectively, with the miR-1 encoding plasmid pmiR-1. A: LNCaP cells were transiently transfected with pmiR-1 and incubated for 96 h. Cellular proliferation was assessed at different time points utilizing a CASY Cell Counter and Analyzer Model TT (Roche Applied Science). B: Overexpression of miR-1 in the newly-generated stable cell line LNCaP-miR-1 detected by quantitative reverse transcription and polymerase chain reaction using miR-1-specific primers and standardized to U6 RNA expression levels. C: LNCaP-miR-1 cells stably overexpressing miR-1 were incubated for 144 h. Cellular proliferation was assessed at different time points utilizing a CASY Cell Counter and Analyzer Model TT (Roche Applied Science). Data are given as the mean±SD, statistically analyzed by Student's t-test. *p≤0.05; **p≤0.01; ***p≤0.001.
miR-1-driven growth inhibition is obligated by AR and TGFB pathway suppression. From the blockade of cellular growth processes, we concluded that miR-1 has selective effects on proliferative pathways in PCa. By modulating miR-1 expression, we found the AR to be suppressed by miR-1 on the mRNA (2.0-fold, p=0.0364; Figure 4A) and protein (1.5-fold, p=0.0047; Figure 4B) levels, as well as its transcriptional activity (7.0-fold, p<0.0001; Figure 4C), the latter assessed as reduced transcription of the AR target gene KLK3. Besides down-regulating proliferative AR signals, we additionally identified TGFB1 signaling to be targeted by miR-1. Transient overexpression of miR-1 in LNCaP cells expressing a low level of miR-1 led to significant reduction of TGFB1 (2.0-fold, p=0.0253; Figure 4D), whereas the inhibition of miR-1 expression in PC-3 cells expressing high levels of miR-1 caused an increase of transcriptional rates of TGFB1 mRNA (2.7-fold, p=0.0043; Figure 4E).
Discussion
Our current findings significantly expand the understanding over HSP-driven chemoresistance and qualify the HSPB1-regulated microRNA miR-1 as a target structure for development of DNA-based drugs and furthermore as an appropriate molecule for indirect targeting by HSPB1 inhibition. We found evidence that HSPB1 regulates miR-1, which for the first time provides evidence that an HSP operates as a regulator of microRNA expression. Our observations were supported by histological studies which showed correlations of elevated HSPB1 expression (11, 12) and diminished miR-1 levels (13) in primary PCa tissue.
Since 2007, approximately 80 microRNA species have been identified and partially distinguished by pro- and anti-oncogenic properties in PCa progression (14-16). One of these is miR-1, however, relatively little is known about the cellular role of miR-1 in PCa. Formerly, miR-1 was thought to be specific for muscle cells in skeletal muscle and heart tissue, with they being very low or nearly undetectable levels in other organs (17). Even though reduced miR-1 expression in PCa tissue samples was described in 2008 (13), to our knowledge only four publications exist specifying miR-1 functionality in PCa cells. Thus, miR-1 was linked to decreased proliferation, enhanced motility and epithelial-mesenchymal transition of PCa cells (18-20), indicating miR-1 functionality as a tumor suppressor and corroborate the findings of the data presented here.
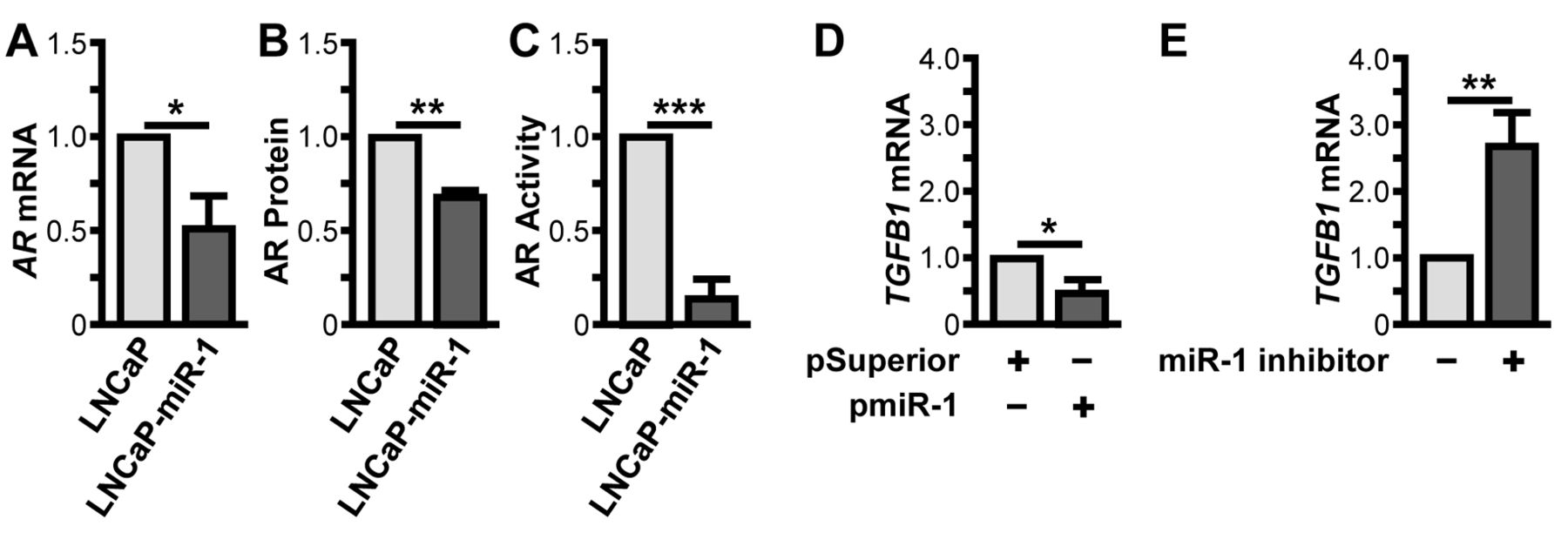
Suppression of oncogenic androgen receptor (AR) and transforming growth factor-β1 (TGFB1) signaling pathways in prostate (PCa) cancer cells. A. LNCaP-miR-1 cells stably overexpressing miR-1 were analyzed for basal AR mRNA levels by quantitative reverse transcription and polymerase chain reaction (qRT-PCR) using AR-specific primers and standardized to ribosomal protein large P0 (RPLP0) mRNA expression levels. AR mRNA levels of LNCaP-miR-1 cells were compared to basal AR mRNA levels of maternal LNCaP cells. B: LNCaP-miR-1 cells stably overexpressing miR-1 were immunochemically analyzed by western blotting. Equal amounts of total cell lysates were separated and immunochemically analyzed with antibodies directed against AR and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), with GAPDH signals served as loading control. AR levels of LNCaP-miR-1 cells were compared to basal AR levels of maternal LNCaP cells. C: LNCaP-miR-1 cells stably overexpressing miR-1 were analyzed for AR transcriptional activity by qRT-PCR analysis of the AR target gene kallikrein-3 mRNA (KLK3). KLK3 mRNA levels of LNCaP-miR-1 cells were compared to basal KLK3 mRNA levels of maternal LNCaP cells. D: LNCaP cells with low miR-1 levels transiently overexpressing miR-1 were analyzed for TGFB1 mRNA levels by qRT-PCR using TGFB1-specific primers and standardized to RPLP0 mRNA expression levels. TGFB1 mRNA levels of transfected LNCaP cells were compared to basal levels of control LNCaP cells. E: PC-3 cells with high basal miR-1 expression were treated with Anti-hsa-miR-1 miScript miRNA Inhibitor (Qiagen) and analyzed for TGFB1 mRNA levels by qRT-PCR using TGFB1-specific primers and standardized to RPLP0 mRNA expression levels. TGFB1 mRNA levels of treated PC-3 cells were compared to basal levels of control PC-3 cells. Data are given as the mean±SD, statistically analyzed by Student's t-test. *p≤0.05; **p≤0.01; ***p≤0.001.
Very interestingly, based on AR and TGFB1 analysis after miR-1 modulation, our study shows that miR-1 is an inhibitor of AR and TGFB1 cascades. Both signaling pathways are known key regulators of oncogenesis and tumor proliferation, and thus, promote PCa progression (21, 22). From this it follows that anti-therapeutic activity of frequently up-regulated HSPB1 suppresses the anticancer properties of miR-1 and subsequently re-activates tumor-promoting cascades of AR and TGFB1 signaling. Overexpression of miR-1-mimicking RNA in LNCaP cells led to down-regulation of AR expression (Figure 4A to C), which may contribute, at least, partly to HSPB1-mediated induction of AR expression (7). Interestingly, inhibiting miR-1 synthesis in AR-negative PC-3 cells caused an increase of AR mRNA, however, AR protein re-expression was not detectable (data not shown). Previous experiments of our group demonstrated that miR-1 also diminishes biosynthesis of shortened, constitutively active AR isoforms (23). Besides AR signaling, miR-1 suppresses the multifunctional growth factor TGFB1 and, to our knowledge, we identified for the first time HSPB1-driven control of TGFB1 signal transduction in PCa cells. This is very important due to the dual role of TGFB1 signaling in cancer cells. The multi-functional nature of TGFB1 is reflected in its opposing pro-oncogenic effects, e.g. proliferation, metastasis, immunosuppression, and remodeling of the extracellular matrix, as well as anti-oncogenic effects on cellular growth inhibition and induction of apoptosis (21, 24). Moreover, it has been shown that TGFB1 pathways are linked to AR signaling (25) and therefore may facilitate ligand-independent growth of castration-resistant PCa. Thus, HSPB1-driven suppression of miR-1 restores AR and TGFB1 signaling, which may represent an important step in enabling PCa cell growth in a low androgen environment and in the presence of anticancer drugs.
In conclusion, our findings improve the understanding of survival mechanisms in drug-resistant PCa cells. Besides inhibition of apoptotic pathways (26, 27), chemotherapy-induced HSPB1 expression initiates the inhibition of miR-1 and subsequently the restoration of pro-oncogenic AR and TGFB1 signals. Moreover, our data point to promising new targets for alternative therapy approaches: HSPB1 inhibition and miR-1-mimicking nucleic acid compounds may reduce HSPB1-miR-1-driven treatment resistance in PCa therapy.
Acknowledgements
The Authors thank Anne Brandenburg and Katja Wittig for excellent technical assistance.
- Received March 18, 2014.
- Revision received May 16, 2014.
- Accepted May 19, 2014.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Reinforcement of the Tumor Suppressing Properties of microRNA-1 by Substitution at the C2' Position of Varying Ribose Residues in Chemically Synthesized microRNA-1 Molecules
- Physiological and Genetically Engineered Expression Modulation Methods Do Not Affect Cellular Levels of the Heat Shock Protein HSP60 in Prostate Cancer Cells
- Overexpression of MicroRNA-1 in Prostate Cancer Cells Modulates the Blood Vessel System of an In Vivo Hen's Egg Test-Chorioallantoic Membrane Model
- Functionality of the Tumor Suppressor microRNA-1 in Malignant Tissue and Cell Line Cells of Uterine Leiomyosarcoma
- MicroRNA-1 and MicroRNA-21 Individually Regulate Cellular Growth of Non-malignant and Malignant Renal Cells
- The Tumor Suppressor MicroRNA-1 Exhibits Restricted Inhibition of Proliferation of Ovarian Cancer Cells