


Abstract
We have previously reported that sodium 5,6-benzylidene-L-ascorbate (SBA) induced dramatic antitumor activity in inoperable cancer patients, but induced only marginal tumor specificity in vitro. Here the tumor specificity and type of cell death induced by benzaldehyde (BA), a degradation product of SBA, was investigated, using human tumor cell lines (oral squamous cell carcinoma [OSCC], glioblastoma, myelogenous leukemia) and human normal oral cells (gingival fibroblast, pulp cell, periodontal ligament fibroblast). BA showed much higher tumor-specific cytotoxicity than SBA. BA induced the formation of autophagosomes, the destruction of mitochondrial structure and digestion of broken organelles, without any apparent induction of internucleosomal DNA fragmentation and caspase activation in an OSCC cell line HSC-2, in a similar manner to SBA. However, pretreatment with 3-methyladenine or bafilomycin A1, autophagy inhibitors, did not completely rescue the cells from the cytotoxicity induced by BA. The study suggests that BA may play an important role in the induction of antitumor activity of SBA in vivo, although the autophagic phenotypes induced by BA may be involved in both cell death and survival.
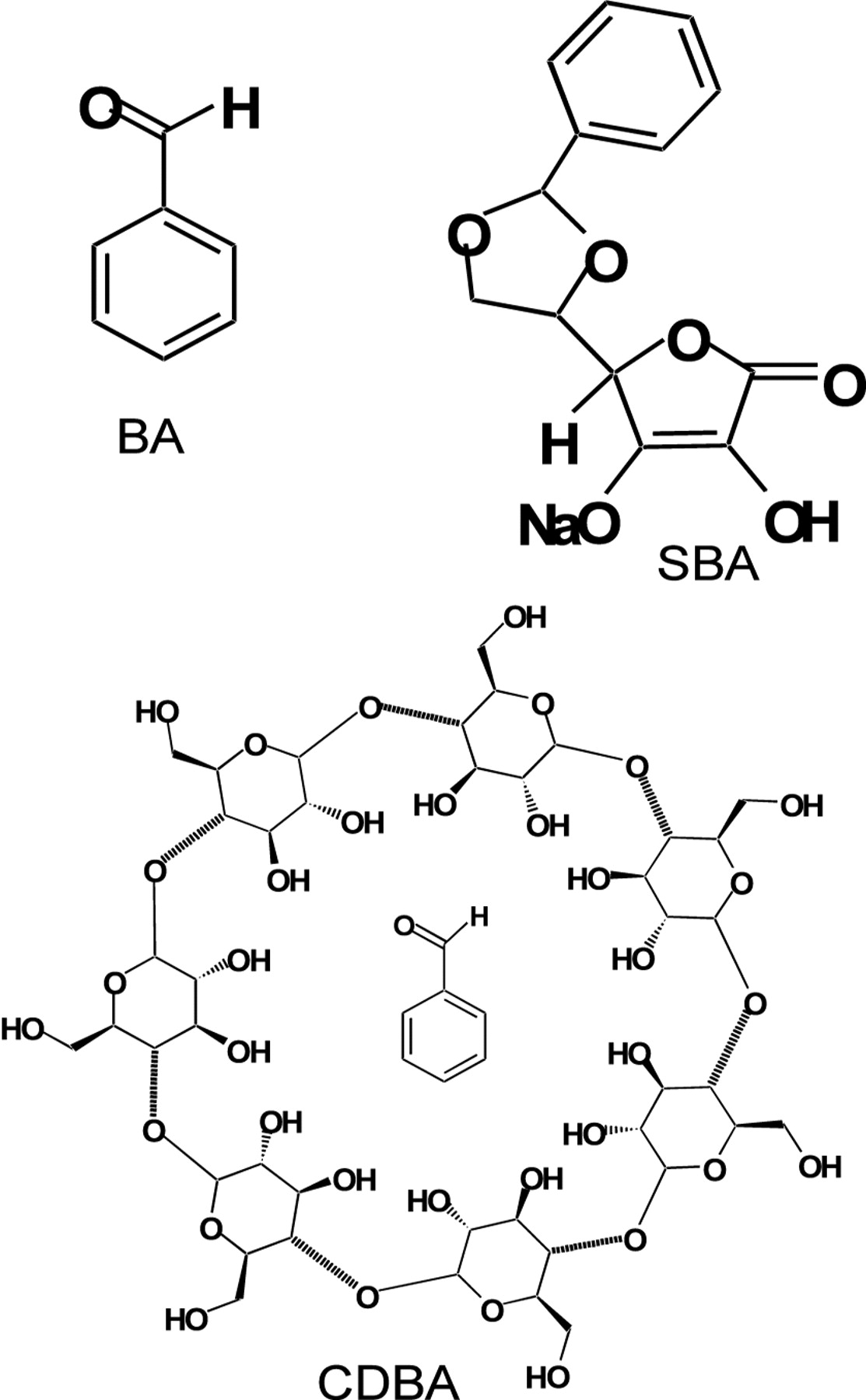
Benzaldehyde (BA) (structure shown in Figure 1), a volatile fraction of figs (1) and its derivatives have shown antitumor activity against implanted Ehrlich carcinoma, adenocarcinoma and colon cancer, but were inactive against several other tumor types in mice (2). The administration of BA derivatives, such as β-cyclodextrin benzaldehyde inclusion compound (CDBA), 5,6-benzylidene-L-ascorbate (SBA) and 4, 6-O-benzylidene-D-glucopyranose (BG) to patients with advanced, inoperable carcinomas has induced remarkable antitumor activity (3-7). We have previously reported that the intravenous administration of SBA induced the degeneration of rat chemically induced hepatocellular carcinomas (8) and colon tumors (9) and the antitumor activity of SBA was not induced via a host-mediated mechanism (8). It has been reported that the cytotoxicity and protein synthesis inhibition by the deuterated benzaldehyde derivative zilascorb (2H) (sodium 5,6-benzylidene-d1-L-ascorbic acid sodium salt) was modified by aminotriazole, suggesting an important role of H2O2 in SBA-mediated cytotoxicity (10). In support of this, an experiment using peroxyoxalate chemiluminescensce technique demonstrated that SBA produced H2O2 in quantities necessary for cell death induction (11). We have previously reported that SBA induced rapid mitochondrial changes (i.e., the disassembly of cristae and decrease in the electron density at non-cytotoxic concentrations, and the swelling and vacuolization at cytotoxic concentrations) in a human submandibular gland carcinoma cell line (HSG) while the nuclear architecture (the profile and the ratio of heterochromatin and euchromatin and thickness of the nuclear membrane) remained intact (12). This suggested that the mitochondria, not the nucleus, may be the target organelle of SBA. However, the mechanism inducing this antitumor activity by SBA has been unclear.
Our recent study with human normal and tumor cells demonstrated that CDBA and SBA showed disappointingly lower tumor-specificity (13, 14), in contrast to their outstanding clinical effects. A degradation study of SBA using high-performance liquid chromatography (HPLC) (15) demonstrated that SBA was unstable and spontaneously produced BA and ascorbic acid by cleavage of acetal under acidic conditions (16). Here whether or not BA, a degradation product of SBA, shows higher tumor-specificity than SBA was investigated, using human normal (gingival fibroblast [HGF], pulp cell [HPC], periodontal ligament fibroblast [HPLF]) and tumor cells (oral squamous cell carcinoma, OSCC, [HSC-2, HSC-3, HSC-4], glioblastoma [T98G, U87MG], myelogenous leukemia [HL-60, ML-1, KG-1]).

Structure of benzaldehyde (BA), sodium 5,6-benzylidene-L-ascorbate (SBA) and β-cyclodextrin benzaldehyde inclusion compound (CDBA).
There are at least three types of cell death: apoptosis (type I programmed cell death characterized by blebbing, chromatin condensation, internucleosomal DNA fragmentation and the loss of cell surface microvilli), autophagy (type II programmed cell death characterized by the formation of autophagosomes and autophagolysosomes engulfing the broken organelles) and necrosis (17, 18). Therefore, the type of cell death induced by BA in HSC-2 cells was also investigated.
Materials and Methods
Materials. The following chemicals and reagents were obtained from the indicated companies: Dulbecco's modified Eagle's medium (DMEM) (Gibco BRL, Grand Island, NY, USA); fetal bovine serum (FBS) (JRH Bioscience, Lenexa, KS, USA); RPMI-1640, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma Chem. Co., St. Louis, MO, USA) and BA (Wako Pure Chemical Ind. Ltd., Osaka, Japan). CDBA and SBA were provided by Ichijokai Hospital, Chiba, Japan.
Cell culture. The HL-60 cells were provided by Professor K. Nakaya, Showa University. The ML-1 and KG-1 cells were provided by Professor K. Takeda, Tokyo University of Science. The HSC-2, HSC-3 and HSC-4 cells were obtained from Professor M. Nagumo and the T98G and U87MG cells were provided by Dr. M. Iida, both of Showa University, Japan. The normal oral cells (HGF, HPC, HPLF) were prepared from periodontal tissues, according to the guideline of the Intramural Ethic Committee (No. A0808), after obtaining the informed consent from the patients. Since normal oral cells have a limited lifespan of about 20 population doubling levels (PDL) (19), they were used at 5-9 PDL. The HL-60, ML-1 and KG-1 cells were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated FBS in a humidified 5% CO2 atmosphere. The other cells were cultured in DMEM supplemented with 10% heat-inactivated FBS. The normal cells were detached by 0.25% trypsin-0.025% EDTA-2Na in phosphate-buffered saline without Mg2+ and Ca2+ (PBS(−)) and subcultured at a 1:4 split ratio once a week. The five adherent tumor cell lines were similarly trypsinized and subcultured.
Assay for cytotoxic activity. Cells (3×103) were seeded in 96-microwell plates (Falcon; Becton Dickinson and Company, Franklin Lakes, NJ, USA) and incubated for 48 hours to allow cell attachment. Near-confluent cells were treated for 24 or 48 hours with various concentrations of the test compounds in fresh medium. The relative viable cell number of adherent cells was then determined by the MTT method. In brief, the BA-treated cells were washed once with PBS(−), and incubated for 4 hours with 0.2 mg/ml of MTT in the culture medium. After removing the medium, the reaction product, formazan, was extracted with dimethyl sulfoxide and the absorbance (the relative viable cell number) was measured at 540 nm by a microplate reader (Multiskan Bichromatic Labsystems, Helsinki, Finland). The viability of the suspended cells, i.e., HL-60, ML-1 and KG-1 was determined by cell counting with a hemocytometer after staining with 0.15% trypan blue PBS(−). The 50% cytotoxic concentration (CC50) was determined from the dose–response curve. The tumor-specificity index (TS) was calculated by the following equation: TS=mean CC50 (normal cells) / mean CC50 (all tumor cell lines or OSCC).
Assay for DNA fragmentation. HSC-2 cells (6×104) were inoculated onto a 6-well plate (Falcon) and incubated for 48 hours. The cells were treated for 6 or 24 hours with 1, 2 or 4 mM BA. After washing once with PBS(−), the cells were collected by scraping with a rubber policeman on ice. They were lysed with 50 μl lysis buffer (50 mM Tris-HCl, pH 7.8, 10 mM EDTA, 0.5% [w/v] sodium N-lauroylsarcosinate) and incubated for 2 hours at 50°C with 0.4 mg/ml RNase A and 0.8 mg/ml proteinase K. DNA was extracted with 50 μl NaI solution (40 mM Tris-HCl, pH 8.0, 7.6 M NaI, 20 mM EDTA-2Na) and then 250 μl of ethanol was added. After centrifugation for 20 min at 20,000 ×g, the precipitate was washed with 1 ml of 70% ethanol. The DNA was dissolved in TE buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA) and applied to 2% agarose gel electrophoresis in TBE buffer (89 mM Tris-HCl, pH 8.0, 89 mM boric acid, 2 mM EDTA). A DNA molecular marker (Bayou Biolabs, Harahan, LA, USA) and DNA from apoptotic HL-60 cells induced by UV irradiation (6 J/m2/min, 1 min), followed by 6 hours incubation in regular culture medium, was run in parallel as a positive control (20). After staining with ethidium bromide, the DNA was visualized by UV irradiation, and photographed by a (charge coupled device) camera (Bio Doc-It, UVP, Inc., Upland, CA, USA).
Cytotoxic activity of BA against human normal cells and tumor cell lines.
Assay for caspase activation. HSC-2 cells (6×105) were inoculated onto 100-mm dishes (Falcon), incubated for 48 hours and then treated for 6, or 24 hours with 0, 1, 2 or 4 mM BA. The cells were washed twice with PBS(−) and lysed in lysis solution (50 mM Tris-HCl, pH 7.5, 0.3% NP-40, 1 mM DTT). After standing for 10 min on ice and centrifugation for 20 min at 15,000 ×g, the supernatant was collected. Lysate (50 μl, equivalent to 150 μg protein) was incubated with 50 μl lysis solution containing substrates for caspase-3 (DEVD-p-nitroanilide [pNA], caspase-8 (IETD-pNA) or caspase-9 (LEHD-pNA) for 4 hours at 37°C. The absorbance of the liberated chromophore pNA was measured at 405 nm using a microplate reader (21). Apoptotic HL-60 cells incubated for 6 hours with 1 μg/ml actinomycin D were used as positive control.
Assay for LC3 accumulation in autophagosomes. Autophagy marker Light Chain 3 (LC3) is known to exist in two forms. LC3-I is found in the cytoplasm. Upon the induction of autophagy, the C-terminal glycine of LC3-I is conjugated to phosphatidylethanolamine, resulting in the formation of autophagosome membrane-bound LC3-II. The autophagosome subsequently fuses with a lysosome (secondary lysosome), where enclosed/engulfed materials, including LC3-II, are degraded. The level of LC3-II generally correlates with the number of autophagosomes.
cDNA encoding LC3 was subcloned into the EcoRI site of pAcGFP1-C2, a green fluorescent protein (GFP) fusion protein expression vector (Clontech Laboratories Inc., Mountain View, CA, USA). The plasmid constructs were verified by DNA sequencing using an Applied Biosystems 310 DNA sequencer (21). HSC-2 cells were transfected with a mixture of 1.6 μg of plasmid DNA and 4 μl FuGene HD (Roch Diagnostics GmbH, Mannheim, Germany). After transfection for 18 hours, the cells were treated for 4 hours with 0 or 2 mM BA. Mock transfection was performed using an empty pAcGFP1-C2 expression vector. The GFP-LC3 accumulation was visualized by confocal laser scanning microscopy LSM510 (Carl Zeiss Inc., Ltd.), using band-pass filters: excitation at 488 nm and emission at 505-530 nm, as described previously (21).
Electron microscopy. HSC-2 cells were incubated for 0 (control, without BA), 3, 6, 9 or 24 hours with 2 mM BA, then washed once with PBS(−), and scraped into 2% glutaraldehyde. The fixed cells were postfixed in 1% osmium tetraoxide-0.1 M cacodylate buffer (pH 7.4) at 4°C, dehydrated and embedded in Araldite 502 (Ciba-Geigy, Basel, Switzerland). Fine sections were stained with uranyl acetate and lead citrate, prior to being analyzed under a JEM-1210 transmission electron microscope (JEOL, Tokyo, Japan) at an accelerating voltage of 100 kV (21).
Autophagy inhibition. Two autophagy inhibitors, 3-methyladenine (a phosphatidylinositol 3 kinase inhibitor that inhibits the formation of autophagosomes) (22) and bafilomycin A1 (a V type proton pump that inhibits the formation of secondary lysosomes) (23) were used. HSC-2 cells (0.1×104) were inoculated onto 96-microwell plates and incubated for 48 hours. The cells were pretreated for 60 min with 3-methyladenine (final concentration 5 or 10 mM) or bafilomycin A1 (final concentration 0.2 or 1 μM), and then incubated for up to 24 hours with various concentrations of BA. The cells were then lysed with 100 μl of Caspase-Glo™ 3/7 Reagent (Promega, Madison, WI, USA). After incubation for 30 min, the luminescence was measured by a luminescence reader (Micro Lumat, LB96P, EG & G Berthold, Bad Wildbad, Germany). The possibility of autophagy induction was further tested by Western blot analysis.

Failure of BA to induce apoptotic cell death in HSC-2 cells. A: DNA from incubated cells was extracted and applied to agarose gel electrophoresis. M: DNA marker, UV: DNA from apoptotic HL-60 cells induced by UV-irradiation. Similar data were obtained in another two independent experiments. B: Caspase activity as measured at 405 nm of the cleaved product for each substrate. Each value represents the mean±S.D. from 3 in the dependent experiments. Positive control: actinomycin D (Act. D) treated HL-60 cells.
Western blot analysis. Immunoblotting analysis was carried out as follows. Cells in each well were washed twice with PBS(−) and lysed by sodium dodecyl sulfate (SDS) sample buffer and boiling for 5 min. The cell lysates were separated by SDS-10-20 % polyacrylamide gel and transferred onto polyvinylidene difluoride membranes. Anti-LC3 (1:1,000; Medical & Biological Laboratories Co. Ltd., Nagoya, Japan) and anti-actin (1:2,000; Sigma Chemical Co.) antibodies were used as primary antibodies. The bound antibodies were visualized with horseradish peroxidase-conjugated secondary antibodies and Western Lighting™ Chemiluminescence Reagent Plus (PerkinElmer Life Sciences, Boston, MA, USA).
Statistical analysis. Values were expressed as the mean±standard deviation (SD). Statistical analysis was performed by using the Student's t-test. A p-value <0.05 was considered to be significant.
Results
Tumor-specific cytotoxic action of BA. BA showed higher tumor-specificity (TS=7.4-8.8) than SBA (TS=1.9-2.1) (14) and CDBA (TS=2.2-2.4) (13). BA showed higher tumor-specificity against OSCC (TS=6.6 [24 hours], 8.8 [48 hours]) than against all the other tumor cells (TS=3.5 [24 hours], 7.4 [48 hours]) (Table I).
The cells investigated in this study showed considerable variation in sensitivity to BA (Table I). Myelogenous leukemia cells were the most sensitive (mean CC50=0.43 mM), followed by OSCC (mean CC50=2.1 mM), glioblastoma (mean CC50=6.4 mM) and normal cells (the most resistant) (mean CC50= 19 mM). BA showed comparable tumor-specificity, regardless of incubation time (either 24 or 48 hours), and therefore the incubation time of 24 hours or less was used for the following experiments.
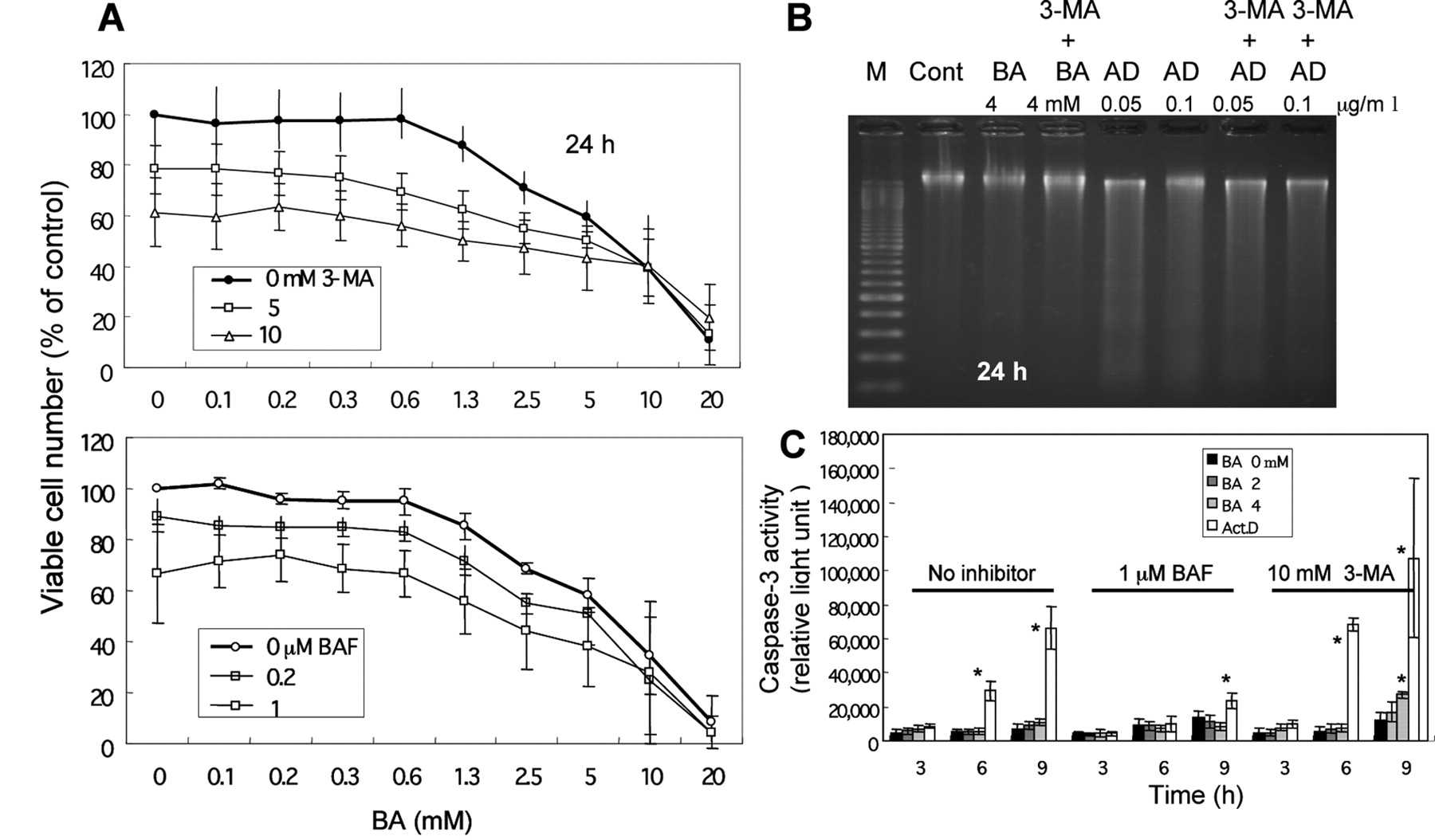
Induction of non-apoptotic cell death by BA. BA did not induce internucleosomal DNA fragmentation, producing a trace of smear pattern of DNA fragmentation at higher concentrations (2 and 4 mM) after 6 hours. Prolonged treatment with BA (24 hours) induced a similar trace smear pattern of DNA fragmentation at a lower concentration (1 mM) (Figure 2A). BA induced little or no activation of caspase-3, caspase-8 and caspase-9 in the HSC-2 cells after 6 or 24 hours, in contrast to the induction of higher caspase activity in the control apoptotic HL-60 cells (Figure 2B). These data suggested that BA induced cell death other than by apoptosis or necrosis.
Autophagy induction. BA induced the production of autophagosomes, as judged by the accumulation of GFP-LC3 (Figure 3A). The fine cell structure of the HSC-2 cells treated with 2 mM BA is shown in Figure 3B. In the cytoplasm of the HSC-2 cells cultured with BA for 3 hours and longer, many vaculolated mitochondria with electron-lucent matrices were found (b-e), whereas no abnormality was found in the control cells (a). In the cells cultured for 6 hours (c), secondary lysosomes were prominent in the cytoplasm, suggesting the digestion of damaged cell organelles. No morphological changes were recognized in the nuclei of the HSC-2 cells cultured with BA. These findings suggested that BA induced the dysfunction of mitochondria. As shown by Western blot (Figure 3C), bafilomycin A1 treatment inhibited acidification (by inhibiting the proton pump) and reduced lysosomal degradation, resulted in the accumulation of band II LC3 in the HSC-2 cells, as expected. The 3-methyladenine treatment which inhibited the formation of autophagosomes, resulted in an increase of band I LC3. Treatment with BA (2, 4 mM) for 3 hours induced the accumulation of band II as well as Band I, suggesting the induction of autophagy. Prolonged incubation (6 h) slightly reduced the band II accumulation, but the level remained higher than that of the control (Figure 3C).

Induction of autophagy by BA. A: GFP-LC3 accumulation. GFP-LC3 transfected HSC-2 cells were incubated for 4 hours without (a) or with 2 mM BA (b), and GFP-LC3 accumulation was visualized by confocal microscopy. B: Induction of secondary lysosomes by BA. Fine cell structure of HSC-2 cells, control without BA treatment (a) or incubated for 3 (b), 6 (c), 9 (d) or 24 (e) h with 2 mM BA, observed under transmission electron microscopy. C: Production of LC3-I and II by BA. HSC-2 cells were incubated for 3 or 6 hours with BA. LC3-I and −II were detected by Western blot analysis. Cells were also treated for 4 hours with 3-methyladenine (3-MA, 10 mM) or bafilomycin A1 (BAF, 1 μM). Similar results were obtained in another independent experiment.
In order to investigate the role of autophagy, the cells were pretreated with autophagy inhibitors before the treatment with BA. Both the inhibitors were slightly cytotoxic (Figure 4A). 3-Methyladenine was found to inhibit the BA-induced cell death only slightly after correction for its cytotoxicity (Figure 4A, upper panel). 3-Methyladenine stimulated the caspase-3 activity (Figure 4C) slightly but significantly (p<0.05), after 9 hour incubation with 4 mM BA, without induction of internucleosomal DNA fragmentation (Figure 4B). On the other hand, bafilomycin A1 did not inhibit the BA-induced cell death (Figure 4A, lower panel), nor did it induce caspase-3 activation (Figure 4C). These results suggested that the autophagy induced by BA may not be solely involved in cell death, but may also contribute cell survival.

Effect of autophagy inhibitors on BA-induced cytotoxicity and apoptotic markers. HSC-2 cells were pretreated for 60 min with the indicated final concentrations of 3-methyladenine (3-MA) or bafilomycin A1 (BAF) before incubation with BA. A: Cell viability (determined by MTT method). B: DNA fragmentation (by agarose gel electrophoresis). AD: Actinomycin D. C: Caspase-3 activity (by substrate cleavage assay). *p<0.05 versus control without BA.
Discussion
The present study demonstrated for the first time that BA showed much higher tumor-specificity (TS=8.8) than SBA (TS=2.1) or CDBA (TS=2.4) (Table I) and sodium ascorbate (TS=2.8) (24). The higher tumor-specificity of BA was due to its lower cytotoxicity to normal cells (CC50=19 mM BA vs. 4.8 mM SBA, 2.1 mM CDBA) (Table I). It remains to be investigated whether the permeability of BA through the cell surface membrane differs between normal and tumor cells.
The present study demonstrated that BA induced autophagy markers, such as the formation of autophagosomes (detected by GFP-LC3 fluorescence and LC3 band-II modification) (Figure 3A and C) and secondary lysosomes (detected by transmission electron microscopy) (Figure 3B), without induction of apoptosis markers (internucleosomal DNA fragmentation, caspase activation) (Figure 2A and B). We have reported previously that CDBA (13) and SBA (14) induced similar morphological changes in OSCC cell lines (i.e., mitochondrial damage, production of secondary lysosomes, without induction of internucleosomal DNA fragmentation and caspase activation). Judging from the similarity of the type of cell death inducted by BA and SBA, it is highly probable that BA is responsible for the antitumor action of SBA. Under acidic conditions, SBA is cleaved to produce BA and ascorbic acid, and it may be that BA attacks the tumor cells. Such conversion from SBA to BA may be possible in vivo, but not occur in cultured cells.
If BA-induced autophagy is involved in the cell death, its cytotoxicity should be completely blocked by autophagy inhibitors. In contrast to our expectation, this was not the case, suggesting that BA-induced autophagy may be involved in both cell death and cell survival.
Recent studies have suggested that autophagy may be a cellular response to protect cells from cell death (25), and the prevention of autophagy by inhibitor treatments may provoke a switch to another type of cell death, either necrosis or apoptosis (26-28). However, in the present system with HSC-2 cells, the transition from autophagy to apoptosis or necrosis was very little. This may have been due to a lack of apoptosis/necrosis-inducing machinery in HSC-2 cells, favoring the commitment towards autophagy. Recently, we have found that SBA induced oxidative stress, autophagy and growth arrest in human colon cancer HT-29 cells (29). There is a possibility that this cell line may also not express apoptosis/necrosis-inducing machinery.
Another factor that may affect the type of cell death is the chemical structure. We have found that many α,β-unsaturated ketones induced non-apoptosis or autophagy (30), accompanied by mitochondrial damage or size reduction, suggesting the mitochondrial target of autophagy. The cytotoxicity of α,β-unsaturated ketones is generated by the interaction between the β-position of the α,β-unsaturated carbonyl moiety and the SH group of any targeted molecules (the so-called non-sterically hindered Michael acceptor), since it was inhibited by N-acetyl-L-cysteine. BA has an aldehyde group attached to a benzene ring, a structural similarity to α,β-unsaturated ketones.
In conclusion, the present study suggests the possibility that BA may play an important role in the antitumor activity of SBA in cancer patients. A combination study of BA and popular antitumor agents is underway.
Acknowledgements
This study was supported in part by a Grant-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan (Sakagami, No.19592156). The Authors acknowledge Drs. Kunii and Kanda for their technical assistance.
- Received July 30, 2010.
- Revision received November 6, 2010.
- Accepted November 8, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}