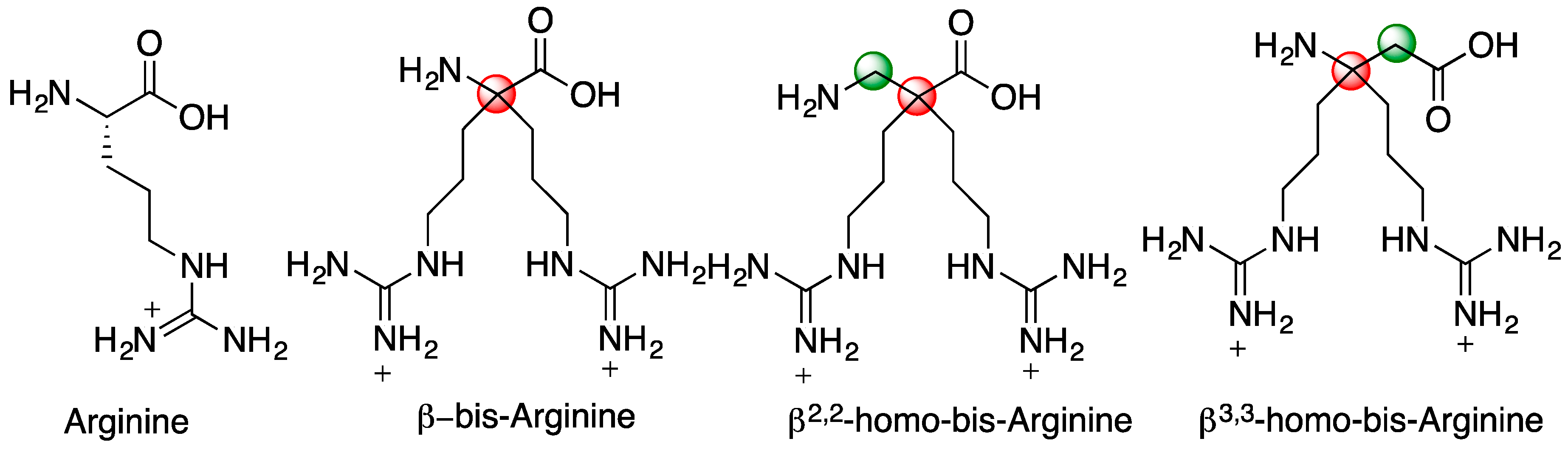
4.3. Product Characterisation
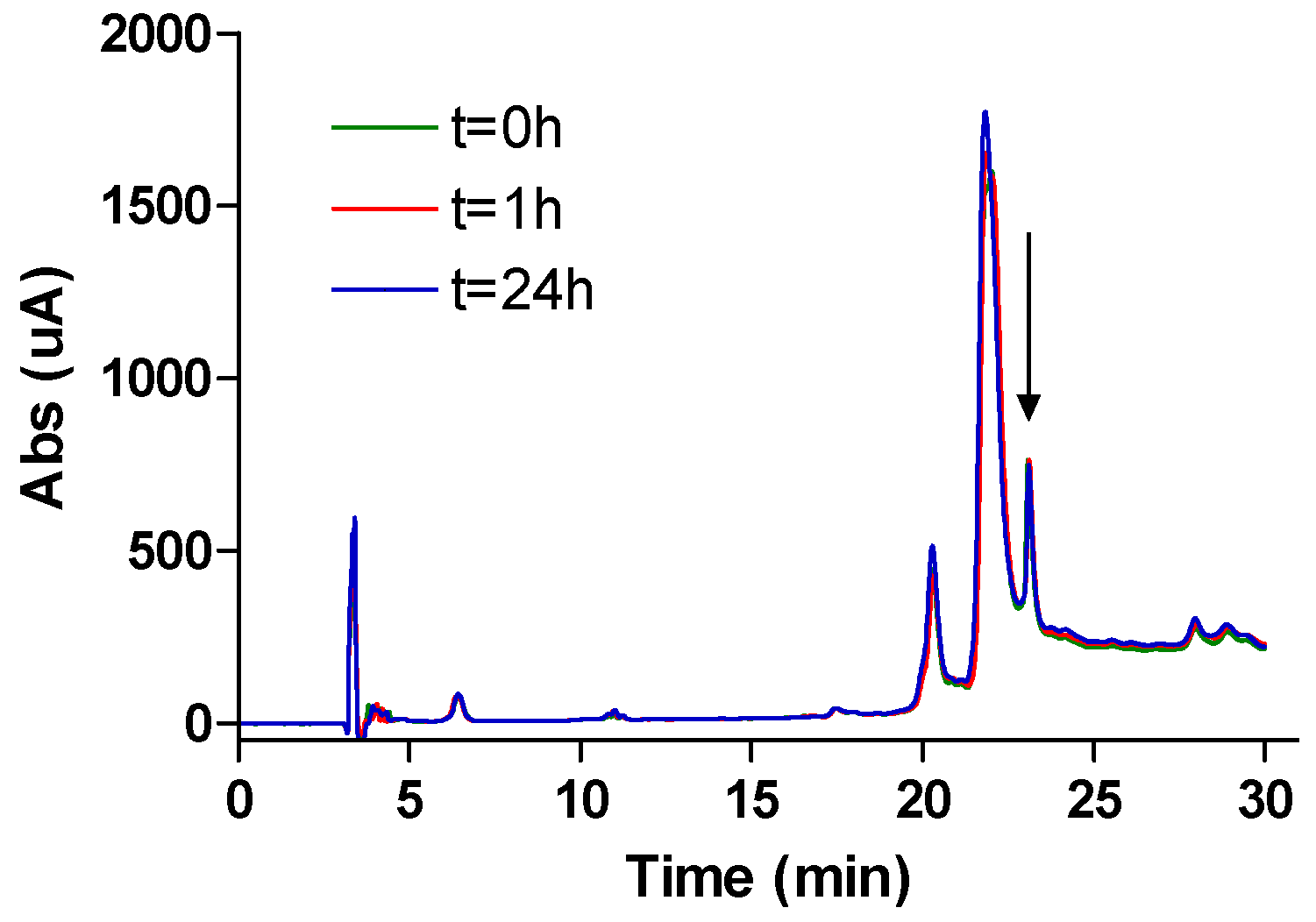
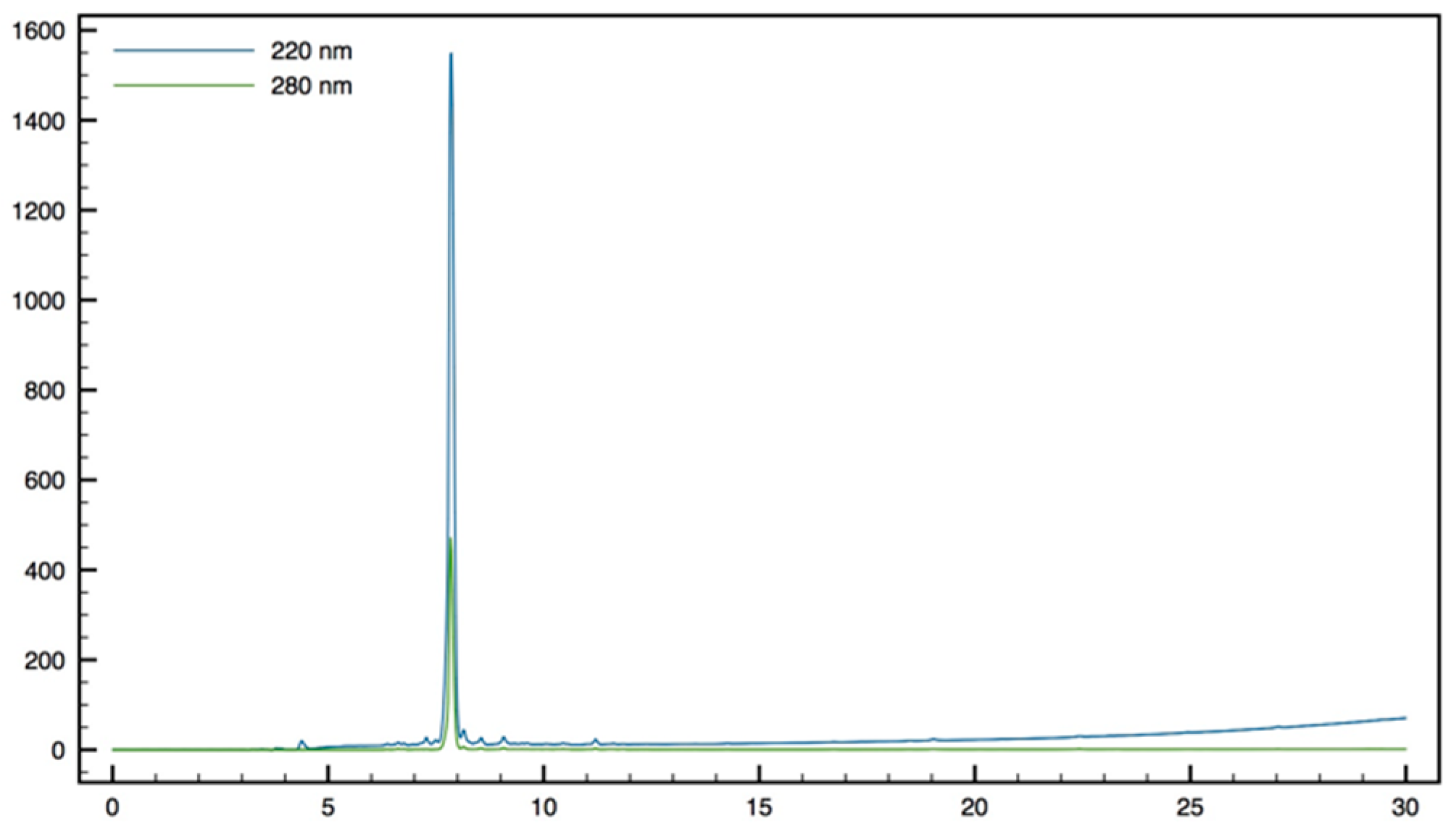
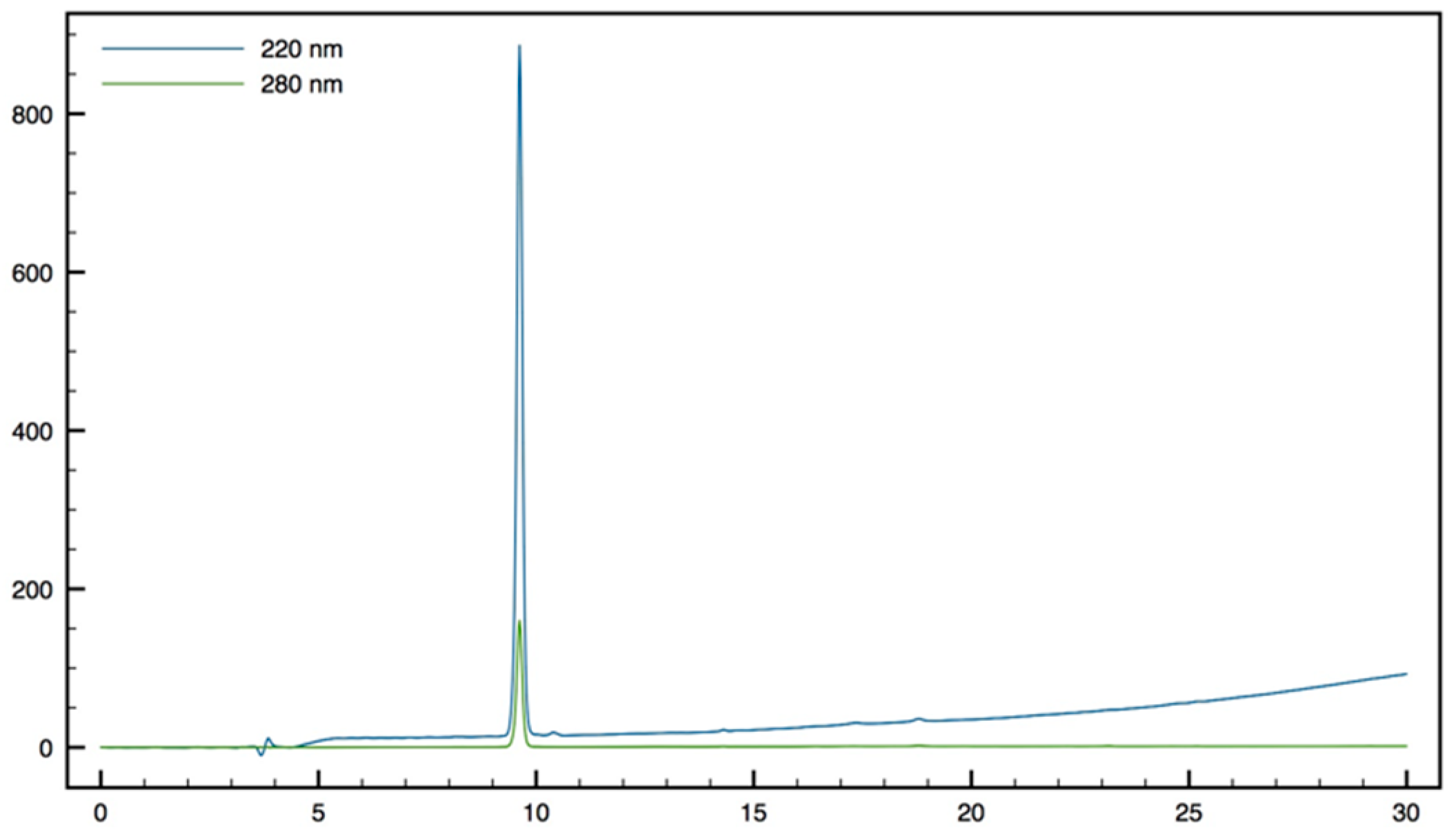
NMR spectra were recorded on Bruker ARX 250 (Bruker, France SAS, Wissembourg, France) or Brucker Avance III 300 spectrometers (Bruker, France SAS, Wissembourg, France) unless otherwise noted. Proton chemical shifts values (δ) are reported in parts per million (ppm) downfield from tetramethylsilane (TMS) unless noted otherwise. Coupling constants (J) are reported in Hertz (Hz). Carbon chemical shifts values (δ) are reported in parts permillion (ppm) with reference to internal solvent CDCl3 (77.00 ppm) or CD3OD (49.00 ppm). Multiplicities are abbreviated as follows: Singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad singlet (bs). Signal assignments were made using COSY and HSQC experiments, and for peptides NOESY (250 ms mixing time), TOCSY (80 ms mixing time), and DQF-COSY spectra. High-resolution mass spectra (HRMS) were obtained on a Finnigan MAT 95 instrument and are given as experimental (found) and theoretical (calcd). Analytical RP-HPLC were performed on either a Waters system connected to a Breeze software or a Dionex system connected to a Chromeleon software. Waters system consisted of a binary pump (Waters 1525) and a dual wavelength Uv/visible Absorbance detector (Waters 2487, Saint-Quentin-en-Yveline, France). Dionex system consisted in an analytical automated LC system (Ultimate 3000) equipped with an auto sampler, a pump block composed of two ternary gradient pumps, and a dual wavelength detector. The analyses were performed on C18 analytical columns (from AIT (Paris, France) or Higgins (San Diego, CA, USA)) using as eluent A, H2O containing 0.1% of TFA and as eluent B, CH3CN containing 0.1% of TFA, at a flow rate of 1 mL/min. UV detection was done at 220 and 280 nm. Peptides were characterized by MALDI-TOF MS (DE-Pro, PerSeptive Biosystems, Framingham, MA, USA) in positive ion reflector mode using the matrix α-cyano-4-hydroxy-cinnamic acid (CHCA). Peptide molecular weights were determined for the free amine and not for the TFA salts.
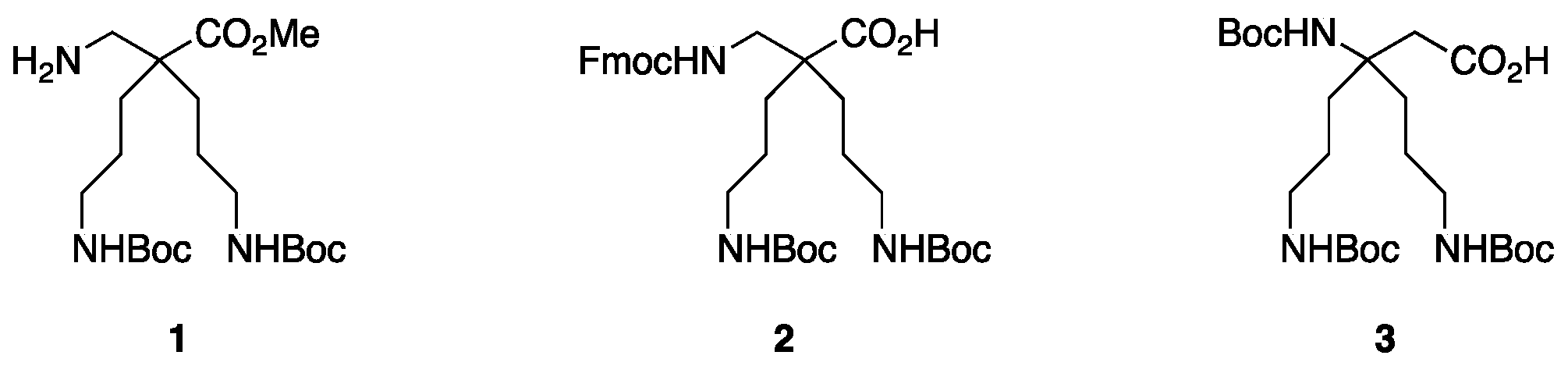
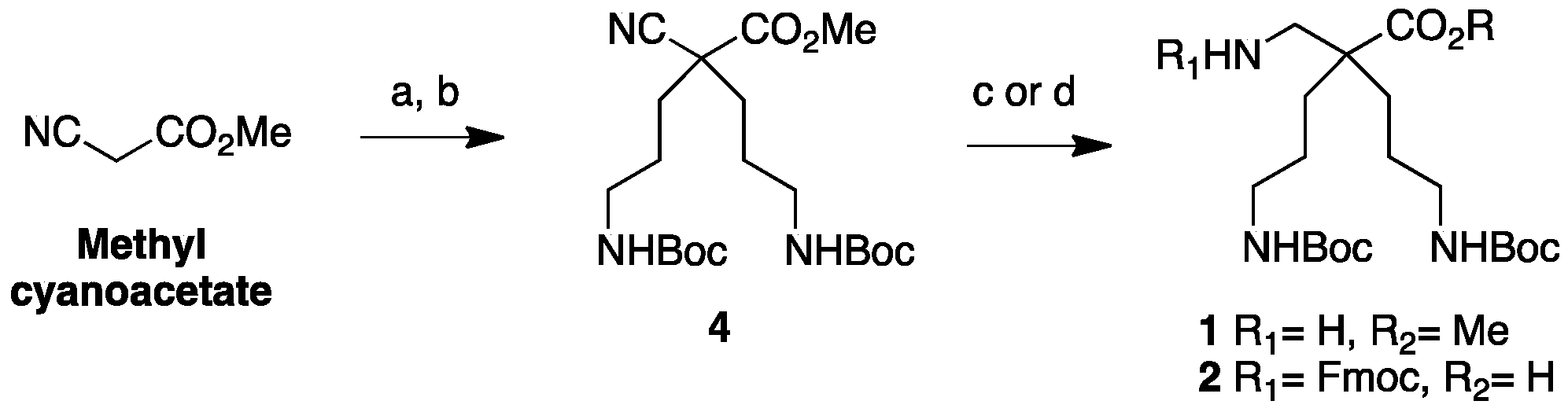
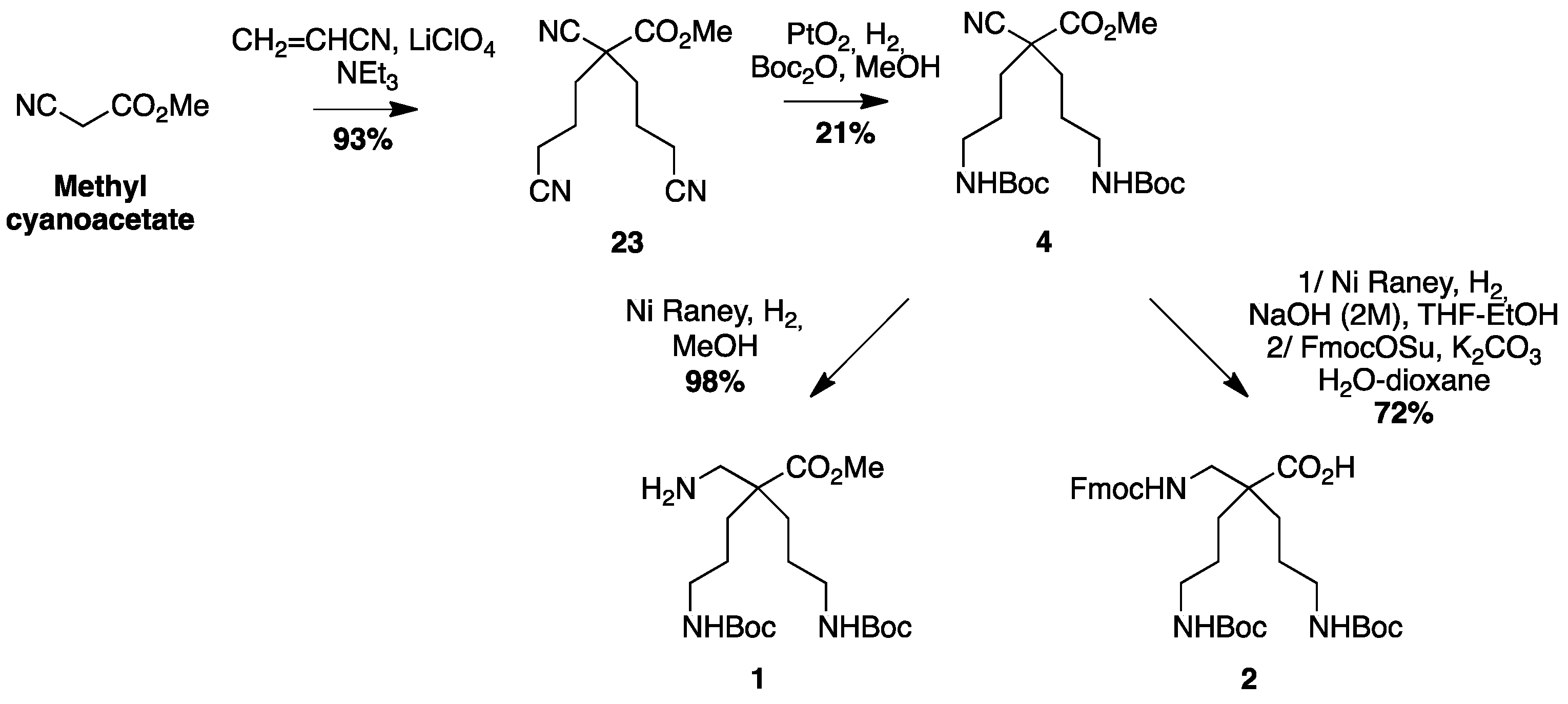
4.3.1. Synthesis of H-β2,2 hbis-Orn(Boc)2OMe 1 and Fmoc β2,2 hbis-Orn(Boc)2OH 2 (Scheme 5)
Methyl 2,4-dicyano-2-(2-cyanoethyl)butanoate23: Methyl 2-cyanoacetate (10 g, 100 mmol) was mixed with acrylonitrile (11.7 g, 220 mmol) in a three-necked round bottom flask equipped with a condenser and an addition funnel. Triethylamine (6.8 mL, 50 mmol) was added dropwise at 0 °C through the addition funnel. The reaction was stirred continuously and allowed to react overnight at rt. After confirming completion of the reaction by TLC, AcOEt was added. The organic layer was washed with 5% citric acid solution and brine, dried over MgSO4, filtered, and evaporated. The product precipitated overnight. The solid was washed with AcOEt and obtained as a pale yellow powder (19.27 g, 93% yield); Rf (Cy/AcOEt, 1:1) = 0.47; 1H NMR (250 MHz, CDCl3) δ 3.87 (s, 3H, CO2CH3), 2.37–2.62 (m, 4H, CH2β), 2.31 (ddd, J = 15.5 Hz, 8.6 Hz, 6.8 Hz, 2H, CH2γ), 2.14 (ddd, J = 14.2 Hz, 8.6 Hz, 6.1 Hz, 2H, CH2γ); 13C NMR (75 MHz, CDCl3): δ 166.6 (C, C=O), 117.2 (2C, C≡Nγ), 116.1 (C, C≡N α), 54.30 (CH3, CO2CH3), 47.6 (C, Cα), 32.1 (2CH2, CH2β), 13.6 (2CH2, CH2γ); MS-ESI+: calcd for C10H11N3O2 205.09, calcd for C10H11N3O2Na 228.08, found 228.07 [M + Na]+.
Methyl 2-cyano-4-(Boc)amine-2-(3-(Boc)amine propyl)pentanoate4: Compound 23 (10 g, 49 mmol) was dissolved in methanol (25 mL). Boc2O (23.5 g, 108 mmol) and PtO2 (2.2 g, 9.8 mmol) were added and the reaction mixture was stirred at rt for 3 days under 5 bars of H2 pressure. The reaction mixture was filtered through a celite pad and evaporated to dryness. The crude compound was purified by flash chromatography (Cy/AcOEt 100:0 → 70:30) to afford yellowish oil (5 g, 21% yield); Rf (Cy/AcOEt, 1:1) = 0.56; 1H NMR (300 MHz, CDCl3): δ (ppm) 4.69 (bs, 2H, NH), 3.76 (s, 3H, CO2CH3), 3.08–3.20 (m, 4H, CH2δ), 1.44–2 (m, 8H, CH2βανδCH2γ), 1.37–1.50 (m, 18H, C(CH3)3).; 13C NMR (62.5 MHz, CDCl3) δ 169.1 (C, C=O ester), 155.8 (2C, C=O carbamate), 118.7 (C, C≡N), 79.3 (2C, C(CH3)3), 53.3 (CH3, CO2CH3), 49.1 (C, Cα), 39.7 (2CH2, CH2δ), 34.4 (2CH2, CH2β), 28.2 (6CH3, C(CH3)3), 26.1 (2CH2, CH2γ).; MS-ESI+: calcd for C20H35N3O6 413.25, calcd for C20H35N3O6Na 436.24, found 436.24 [M + Na]+.
H-β2,2 hbis-Orn(Boc)2OMe1: Compound 4 (2.35 g, 4.9 mmol) was dissolved in methanol (100 mL). Raney nickel was added, and the mixture was stirred under 5 bars of H2 pressure at rt for 3 days. The reaction mixture was filtered through a celite pad and evaporated to dryness. The product was used in peptide synthesis without further purification. (2.0 g, 98% yield); Rf (Cy/AcOEt, 1:1) = 0.56; 1H NMR (300 MHz, MeOD) δ 3.68 (s, 3H, CO2CH3), 3.01 (t, J = 6.7 Hz, 4H, CH2δ), 2.77 (s, 2H, CH2βε), 1.55–1.60 (m, 4H, CH2β), 1.32–1.43 (m, 22H, CH2γ and C(CH3)3); 13C NMR (75 MHz, MeOD) δ 177.9 (C, C=O ester), 158.6 (2C, C=O carbamate), 79.9 (2C, C(CH3)3), 52.4 (CH3, CO2CH3), 51.5 (C, Cα), 45.9 (CH2, CH2βε), 41.6 (2CH2, CH2δ), 31.2 (2CH2, CH2β), 28.8 (6CH3, C(CH3)3), 25.4 (2CH2, CH2γ); HRMS-ESI+: calcd for C20H39N3O6 417.2839, found 418.2915 [M + H]+.
Fmoc β2,2 hbis-Orn(Boc)2OH2: Compound 4 (2.3 g, 5.6 mmol) was dissolved in methanol (125 mL). An aqueous solution of sodium hydroxide (2 M) (12.5 mL, 25 mmol) and Raney nickel were added. The mixture was stirred under 5 bars of H2 pressure at rt for 7 days. The reaction mixture was filtered through a celite pad and evaporated to dryness. The crude compound was dissolved in a 1:1 mixture of THF and water (150 mL). FmocOSu (2.3 g, 6.8 mmol) and K2CO3 (1.7 g, 12.2 mmol) were added. The solution was allowed to react at rt overnight. After confirming the completion of the reaction by TLC, THF was evaporated. The resulting aqueous solution was acidified to pH = 2 by dropwise addition of 1M hydrochloric acid at 0 °C. The product was extracted with AcOEt, dried over MgSO4, filtered, and concentrated in vacuo. The crude compound was purified by flash chromatography (Cy/AcOEt/AcOH 100:0:1 → 75:25:1) to afford a white powder (2.5 g, 72% yield); Rf (Cy/AcOEt/AcOH, 7:3:0.1) = 0.27; 1H NMR (250 MHz, CDCl3) δ 7.75 (d, J = 7.2 Hz, 2H, CH Ar), 7.58 (d, J = 7.2 Hz, 2H, CH Ar), 7.19–7.39 (m, 4H, CH Ar), 5.54 (bs, 1H, NH Fmoc), 4.95 (bs, 2H, NH Boc), 4.40 (d, J = 6.5 Hz, 2H, CH2 Fmoc), 4.20 (t, J = 6.5 Hz, 1H, CH Fmoc), 3.38–3.41 (m, 2H, CH2βε), 3.07 (m, 4H, CH2δ), 1.20–1.67 (m, 26H, CH2β, CH2γ and C(CH3)3); 13C NMR (62.5 MHz, CDCl3) δ 176.5 (C, C=O acid), 157.20, 156.4 (3C, C=O carbamate), 143.9, 141.3 (4C, C Ar), 129.1, 128.2, 127.7, 127.1, 125.3, 125.1, 120.0 (8CH, CH Ar), 79.3 (2C, C(CH3)3), 67.0 (CH2, CH2 Fmoc), 49.8 (CH2, CH2βε), 47.2 (CH, CH Fmoc), 40.7 (2CH2, CH2δ), 40.6 (C, Cα), 30.6 (2CH2, CH2β), 28.4 (6CH3, C(CH3)3), 24.3 (2CH2, CH2γ); HRMS-ESI+: calcd for C34H47N3O8 625.3255, calcd for C34H47N3O8Na 648,3153, found 648.3261 [M + Na]+.
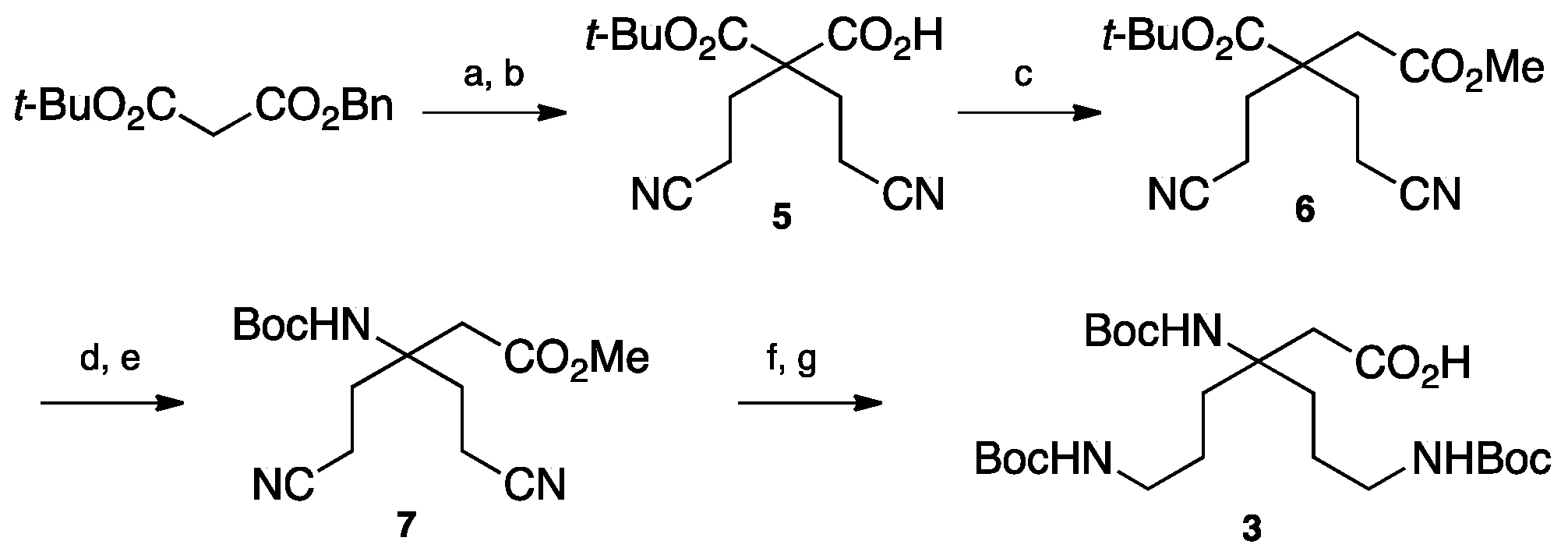
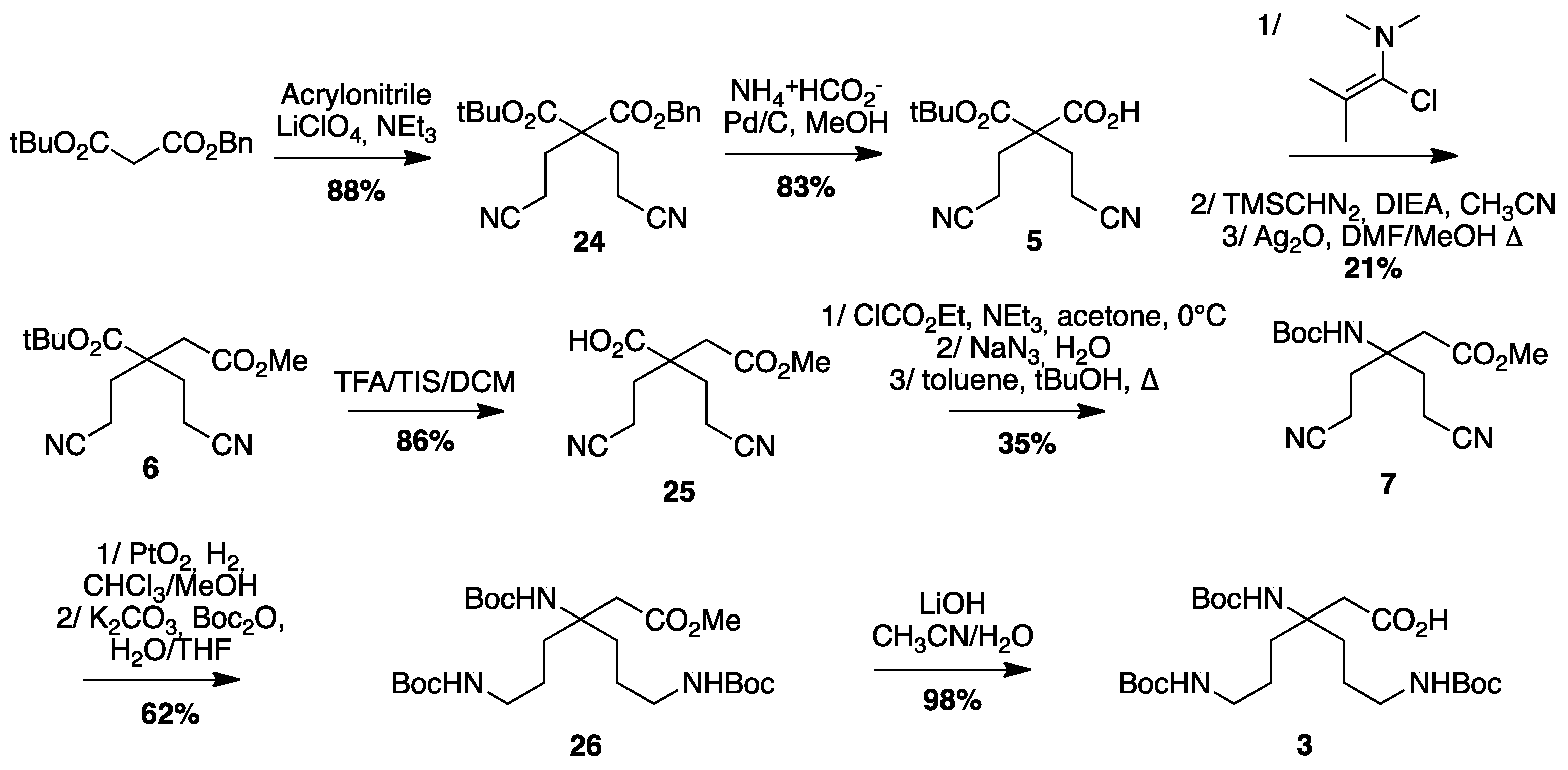
4.3.2. Synthesis of Boc-β3,3 hbis-Orn(Boc)2OH 3 (Scheme 6)
1-Benzyl 3-tert-butyl 2,2-bis(2-cyanoethyl)malonate24: Benzyl tert-butylmalonate (25 g, 96.4 mmol) was mixed with acrylonitrile (14 mL, 210 mmol). Triethylamine (5.3 mL, 40 mmol) was added dropwise, followed by lithium perchlorate (5.4 g, 50 mmol). The reaction was stirred continuously and allowed to react overnight. After confirming completion of the reaction by TLC, AcOEt was added to the reaction mixture. The organic layer was washed with 5% citric acid solution and brine, dried over MgSO4, filtered, and evaporated. The crude compound was purified by flash chromatography (Cy/AcOEt, 100:0 to 8:2) to afford a yellow oil (30.2 g, 88% yield); Rf (Cy/AcOEt, 8:2) = 0.37; 1H NMR (250 MHz, CDCl3) δ 7.37 (m, 5H, CH Ar), 5.20 (s, 2H, CH2Ph), 2.17–2.43 (m, 8H, CH2βανδCH2γ), 1.35 (s, 9H, C(CH3)3); 13C NMR (62.5 MHz, CDCl3) δ 169.2, 167.9 (2C, C=O), 134.5 (C, C Ar), 128.9, 128.8, 128.5, 127, 126.2 (5CH, CH Ar), 118.5 (2C, C≡N), 84 (C, C(CH3)3), 67.9 (CH2, CH2Ph), 56.2 (C, Cα), 29.5 (2CH2, CH2β), 27.6 (3CH3, C(CH3)3), 13 (2CH2, CH2γ); HRMS-ESI+: calcd for C20H24N2O4 356.1736, calcd for C20H24N2O4Na 379,1634, found 379.1628 [M + Na]+.
2-(Tert-butoxycarbonyl)-4-cyano-2-(2-cyanoethyl)butanoic acid5: Compound 24 (18 g, 51 mmol) was dissolved in MeOH (500 mL). Ammonium formate (16.7 g, 265 mmol) and Pd/C (5.1 g, 100 mg/mmol) were added and the reaction mixture was stirred for 3 h. Afterward, the reaction mixture was filtered through a celite pad to remove the Pd/C before evaporation to dryness. The product was diluted with dichloromethane. The organic layer was washed with 10% citric acid solution and brine, dried over MgSO4, filtered, and evaporated to afford an oil. The product was used in the following step without further purification. (11.34 g, 83% yield); Rf (Cy/AcOEt/AcOH, 8:2:0.1) = 0.1; 1H NMR (250 MHz, CDCl3) δ 2.41–2.54 (m, 4H, CH2β), 2.20 (t, J = 7.5 Hz, 4H, CH2γ), 1.51 (s, 9H, C(CH3)3); 13C NMR (62.5 MHz, CDCl3) δ 173.1 (C, C=O acid), 168.3 (C, C=O ester), 118.5 (2C, C≡N), 84.8 (C, C(CH3)3), 56.2 (C, Cα), 30.0 (2CH2, CH2β), 27.8 (3CH3, C(CH3)3), 13.1 (2CH2, CH2γ); HRMS-ESI+: calcd for C13H18N2O4 266.1267, calcd for C13H18N2O4Na 289,1165, found 289.1159 [M + Na]+.
1-Tert-butyl 4-methyl 2,2-bis(2-cyanoethyl)succinate6: Compound 5 (9.0 g, 34 mmol) was dissolved in DCM under Argon. 1-Chloro-N,N-2-trimethylpropenylamine (9.0 mL, 68 mmol) was added. The solution was stirred for 2 h then concentrated in vacuo. The residue was dissolved in dry acetonitrile (170 mL) and cooled to 0 °C. DIEA (11.9 mL, 68 mmol) and a 2M solution of trimethylsilyldiazomethane in Et2O (34 mL, 68 mmol) was added. The reaction mixture was stirred at 0 °C for 16 h. The organic solvents were evaporated in vacuo. The residue was dissolved in AcOEt and washed with 10% citric acid, saturated NaHCO3 and brine. Finally, the organic layer was dried over MgSO4, filtered, and evaporated to dryness. The crude compound was dissolved in DMF (180 mL) and MeOH (90 mL) then Ag2O (39.4 g, 170 mmol) was added. The reaction mixture was refluxed for 10 min. After evaporation of MeOH, diethyl ether and a saturated solution of NH4Cl were added slowly and the mixture was filtered through a celite pad. The organic layer was separated and washed with a saturated solution of NH4Cl, dried over MgSO4, filtered, and evaporated. The crude compound was purified by flash chromatography (Cy/AcOEt, 100:0 to 60:40) to afford a yellow oil (2.1 g, 21% yield); Rf (Cy/AcOEt, 1:1) = 0.6; 1H NMR (300 MHz, CDCl3) δ 3.71 (s, 3H, CO2CH3), 2.61 (s, 2H, CH2α), 2.27–2.37 (m, 4H, CH2γ), 1.91–2.07 (m, 4H, CH2δ), 1.48 (s, 9H, C(CH3)3); 13C NMR (75 MHz, CDCl3) δ 171.4, 170.1 (2C, C=O), 118.8 (2C, C≡N), 82.5 (C, C(CH3)3), 51.6 (CH3, CO2CH3), 46.6 (CH2, CH2α), 37.4 (C, Cβ), 30.6 (2CH2, CH2γ), 27.5 (3CH3, C(CH3)3), 12.3 (2CH2, CH2δ); HRMS-ESI+: calcd for C15H22N2O4 294.1580, calcd for C15H22N2O4Na 317,1478, found 317.4718 [M + Na]+.
2,2-Bis(2-cyanoethyl)-4-methoxy-4-oxobutanoic acid25: Compound 6 (1.3 g, 4.4 mmol) was dissolved in DCM (40 mL). Triisopropylsilane (900 µL, 4.4 mmol) and TFA (40 mL) were added. The reaction mixture was stirred for 1 hour before evaporation to dryness. The crude compound was purified by flash chromatography (Cy/AcOEt/AcOH, 100:0:1 to 50:50:1) to afford a colorless oil (900 mg, 86% yield); Rf (Cy/AcOEt/AcOH, 5:5:0.1) = 0.34; 1H NMR (300 MHz, CDCl3) δ 3.66 (s, 3H, CO2CH3), 2.65 (s, 2H, CH2α), 2.30–2.49 (m, 4H, CH2γ), 1.98–2.16 (m, 4H, CH2δ); 13C NMR (75 MHz, CDCl3) δ 177.9 (C, C=O acid), 170.5 (C, C=O ester), 118.7 (2C, C≡N), 52.4 (CH3, CO2CH3), 46.3 (C, C β), 37.0 (CH2, CH2α), 30.8 (2CH2, CH2γ), 12.8 (2CH2, CH2δ); HRMS-ESI+: calcd for C11H14N2O4 238.0954, calcd for C11H14N2O4Na 261,0852, found 261.0845 [M + Na]+.
Methyl 3-((tert-butoxycarbonyl)amino)-5-cyano-3-(2-cyanoethyl)pentanoate7: Compound 25 (450 mg, 1.9 mmol) was dissolved in dry acetone (15 mL) and cooled to 0° C. NEt3 (300 µL, 2.3 mmol) and ClCO2Et (200 µL, 2.1 mmol) were added. The reaction mixture was stirred for 1.5 h. A solution of NaN3 (309 mg, 4.75 mmol) in H2O (8.5 mL) was added and the mixture was stirred at 0 °C for 2 additional hours. Acetone was evaporated and the compound was extracted with toluene. The organic layer was dried over MgSO4 and filtered. The volume was reduced by evaporation to 20 mL. tert-BuOH (15 mL) was added and the reaction was refluxed for 16 h. The solvent was evaporated and the crude compound purified by flash chromatography (Cy/AcOEt, 100:0 to 7:3) to afford a white powder (200 mg, 35% yield); Rf (Cy/AcOEt, 1:1) = 0.44; 1H NMR (300 MHz, CDCl3) δ 5.17 (bs, 1H, NHBoc), 3.71 (s, 3H, CO2CH3), 2.60 (s, 2H, CH2α), 2.22–2.45 (m, 6H, CH2γ and CH2δ1), 1.99–2.12 (m, 2H, CH2δ2), 1.40 (s, 9H, C(CH3)3); 13C NMR (75 MHz, CDCl3) δ 170.4 (C, C=O ester), 154.3 (C, C=O carbamate), 119.2 (2C, C≡N), 80.5 and 80.4 (2C, C(CH3)3), 55.3 (C, Cβ), 52.4 (CH3, CO2CH3), 39.5 (CH2, CH2α), 31.8 (2CH2, CH2γ), 28.3 (3CH3, C(CH3)3), 12.0 (2CH2, CH2δ); HRMS-ESI+: calcd for C15H23N3O4 309.1689, calcd for C15H23N3O4Na 332,1587, found 332.1581 [M + Na]+.
Methyl-3,6-bis((tert-butoxycarbonyl)amino)-3-(3-((tert-butoxycarbonyl)amino)propyl) hexanoate26: Compound 7 (145 mg, 0.47 mmol) was dissolved in a 9:1 mixture of methanol and chloroform. PtO2 (16 mg, 0.07 mmol) was added and the reaction mixture was stirred under 5 bars of H2 pressure at rt for 3 days. The reaction mixture was filtered through a celite pad and evaporated to dryness. The crude product was dissolved in a 1:1 mixture of THF/H2O and Boc2O was added. After stirring overnight, THF was evaporated and the product was extracted with DCM. The organic layer was washed with brine, dried over MgSO4, filtered, and evaporated in vacuo. The crude compound was purified by flash chromatography (Cy/AcOEt, 7:3) to afford a colorless oil (150 mg, 62% yield); Rf (Cy/AcOEt, 1:1) = 0.68; 1H NMR (300 MHz, CDCl3) δ 4.85 (bs, 1H, NH Boc), 4.72 (bs, 2H, NH Boc), 3.64 (s, 3H, CO2CH3), 3.02–3.11 (m, 4H, CH2ε), 2.61 (s, 2H, CH2α), 1.56–1.77 (m, 4H, CH2γ), 1.34–1.48 (m, 31H, CH2δ and C(CH3)3); 13C NMR (75 MHz, CDCl3) δ 171.7 (C, C=O ester), 156.0 (2C, C=O carbamate), 154.5 (C, C=O carbamate), 79.1 (3C, C(CH3)3), 56.1 (C, Cβ), 51.6 (CH3, CO2CH3), 40.5 (2CH2, CH2ε), 40.3 (CH2, CH2α), 33.3 (2CH2, CH2β), 28.4 (9CH3, C(CH3)3), 23.8 (2CH2, CH2γ); HRMS-ESI+: calcd for C25H47N3O8 517.3363, calcd for C25H47N3O8Na 540,3261, found 540.3255 [M + Na]+.
Boc-β3,3 hbis-Orn(Boc)2OH3: Compound26 (0.130 g, 0.25 mmol) was dissolved in a 1:1 mixture of THF/H2O. LiOH (12 mg, 0.5 mmol) was added and the reaction mixture was stirred at rt for 5 days. THF was evaporated and the resulting aqueous solution was acidified to pH = 2 by dropwise addition of 1 M hydrochloric acid at 0 °C. The product was extracted with DCM and the organic layer was washed with brine, dried over MgSO4, filtered, and evaporated to afford a white powder (120 mg, 98% yield). The product was used in following step without any further purification; Rf (Cy/AcOEt, 1:1) = 0.20; 1H NMR (300 MHz, MeOD) δ 3.01 (t, J = 6.6 Hz, 4H, CH2ε), 2.62 (s, 2H, CH2α), 1.73 (m, 4H, CH2γ), 1.42 (m, 31H, CH2δ and C(CH3)3); 13C NMR (75 MHz, MeOD) δ 158.6 (3C, C=O carbamate), 80.0 (3C, C(CH3)3), 57.2 (C, Cβ), 41.7 (3CH2, CH2α and CH2ε), 34.3 (2CH2, CH2γ), 29.0 (9CH3, C(CH3)3), 25.0 (2CH2, CH2δ); HRMS-ESI+: calcd for C24H45N3O8 503.3207, calcd for C24H45N3O8Na 526,3105, found 526.3099 [M + Na]+; IR (ATR) υ (cm−1): 3346.9 (-OH acid), 2962.3, 2975.8, 2872.9, 2495.0, 1686.5 (C=O acid), 1514.6, 1479.5, 1453.5, 1392.0, 1365.3, 1273.7, 1248.7, 1162.3, 1092.6, 985.2, 866.5, 778.8.
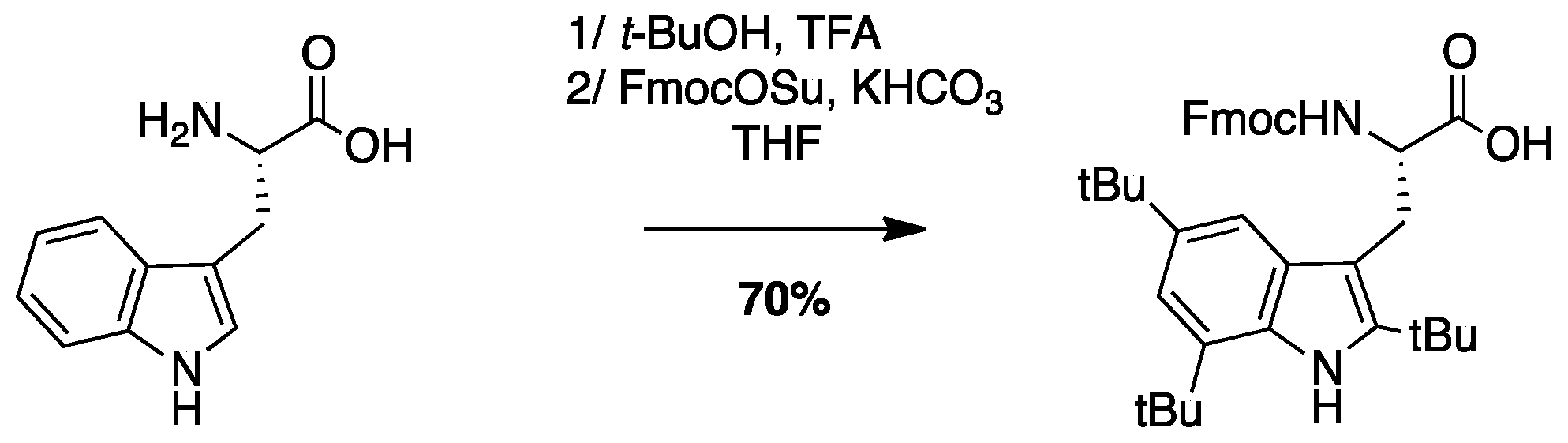
4.3.3. Synthesis of Fmoc-Tbt-OH (Scheme 7)
(S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(2,4,6-tri-tert-butyl-1H-indol-3-yl) propanoic Acid: A mixture of H-Trp-OH (3 g, 14.6 mmol) and tert-BuOH (31 mL, 323 mmol) in TFA (90 mL) was stirred at rt for 20 days. The resulting dark solution was evaporated to dryness to give a black oil, and water (50 mL) was added. To the resulting suspension was added KHCO3 until pH = 8–9. THF (50 mL) and FmocOSu (5.4 g, 16.0 mmol) were added and the mixture was stirred for 16 h. THF was evaporated and the solution was acidified to pH = 2. The compound was extracted with AcOEt, dried over MgSO4, filtered, and concentrated in vacuo. The crude compound was purified by flash chromatography (Cy/AcOEt/AcOH 100:0:1 to 50:50:1) to afford a white powder (6 g, 70% yield); Rf (Cy/AcOEt/AcOH, 5:5:0.1) = 0.66; 1H NMR (300 MHz, CDCl3) δ 7.12–8.08 (m, 10H, CH Ar), 4.65–4.88 (m, 1H, CH Fmoc), 4.22–4.43 (m, 2H, CH2 Fmoc), 4.17 (t, J = 6.8 Hz, 1H, CHα), 3.56-3.74 (m, 1H, CH2β1), 3.42 (dd, J = 14.8 Hz, 9.1 Hz, 1H, CH2β2), 1.57 (s, 18H, C(CH3)3), 1.45 (s, 9H, C(CH3)3); 13C NMR (75 MHz, CDCl3) δ 177.7 (C, C=O acid), 156.1 (C, C=O carbamate), 143.8, 143.7, 142.9, 142.7, 141.2, 132.0, 130.2, 129.8 (9C, C Ar), 127.6, 127.0, 125.2, 125.1, 119.8, 116.9, 111.6 (10C, CH Ar), 103.9 (C, C Ar), 67.2 (CH2, CH2 Fmoc), 55.3 (CH, CHα), 47.0 (CH, CH Fmoc), 34.8 (2C, C(CH3)3), 33.1 (C, C(CH3)3), 32.0, 30.9, 30.6 (9CH3, C(CH3)3), 27.6 (CH2, CH2β); HRMS-ESI+: calcd for C38H46N2O4 594.3458, calcd for C38H46N2O4Na 617.3356, found 617.3350 [M + Na]+.
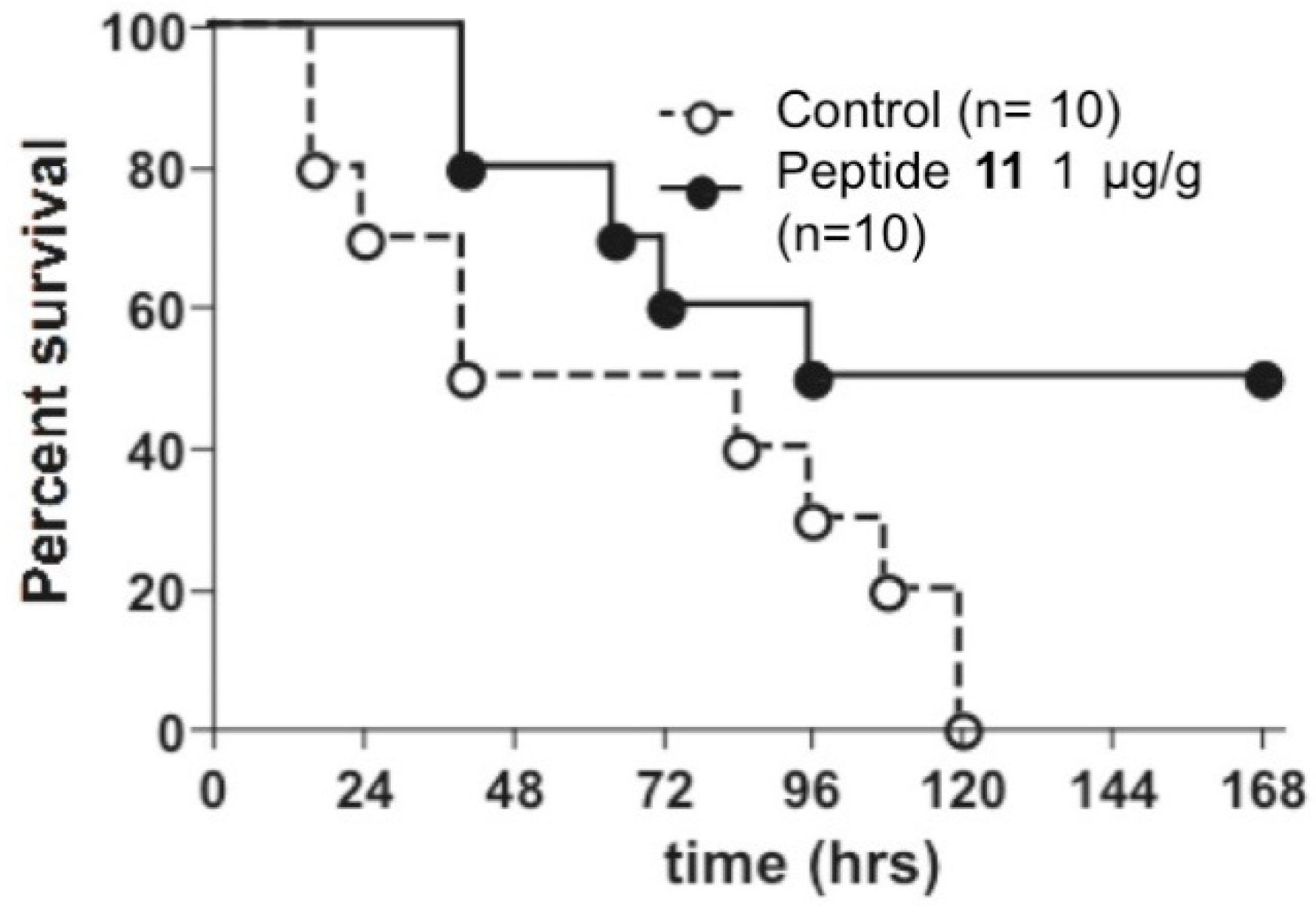
4.3.4. Synthesis of Peptide A: Arg-Tbt-Arg-NH2
![Molecules 24 01702 i001]()
Fmoc Rink Amide resin loaded at 0.43 mmol/g (162 mg, 0.07 mmol) was washed with DMF and allowed to swell in DMF for 15 min. Fmoc deprotection was achieved through treatment of the resin with a solution of 20% piperidine (v:v) in DMF (5 min, 3 times), followed by washing with NMP. Fmoc-Arg(Pbf)-OH (4 eq, 0.28 mmol, 182 mg) was dissolved in dry NMP and HATU (3.6 eq, 0.25 mmol, 95 mg) and DIEA (10 eq, 0.7 mmol, 130 µL) were added. The resulting solution was added to the resin and the mixture was stirred for 2 h then filtrated and washed with NMP. Removal of the Fmoc protecting group was achieved by treatment of the resin with 20% (v:v) piperidine in DMF (3 times for 5 min). The resin was washed with NMP. Fmoc-Tbt-OH (4 eq, 0.28 mmol, 166 mg) was dissolved in NMP (1.5 mL). HATU (3.6 eq, 0.25 mmol, 95 mg) and DIEA (10 eq, 0.7 mmol, 130 µL) were added. The solution was added to the resin and the coupling reaction was allowed to proceed for 1.5 h at room temperature. The solution was removed by filtration and the resin was washed with DMF. After removal of the Fmoc protective group (20% piperidine in DMF, 5 min, 3 times) and washing of the resin with NMP, a solution of Fmoc-Arg(Pbf)-OH (4 eq, 0.28 mmol, 182 mg), HATU (3.6 eq, 0.25 mmol, 95 mg), and DIEA (10 eq, 0.7 mmol, 130 µL) in NMP (2 mL) was added and the reaction mixture was stirred for 2 h then filtrated and washed with NMP. Simultaneous final deprotection and cleavage from the resin was achieved by treating the resin with a TFA/TIS/H2O cocktail (95:2.5:2.5, 3 mL) for 4 h. The crude peptide was precipitated through addition of cold diethyl ether. Purification by preparative RP-HPLC using a gradient of 15% to 90% MeCN in 30 min gives after lyophilisation peptide A as a white powder with a purity of >95%. MALDI-TOF: calcd for C36H62N10O3 683, found 684.4 [M + H]+, 706.4 [M + Na]+, 722.4 [M + K]+; HPLC (Water/ACN (0.1% TFA); 15% to 100% ACN in 30 min): tr = 10.19 min.
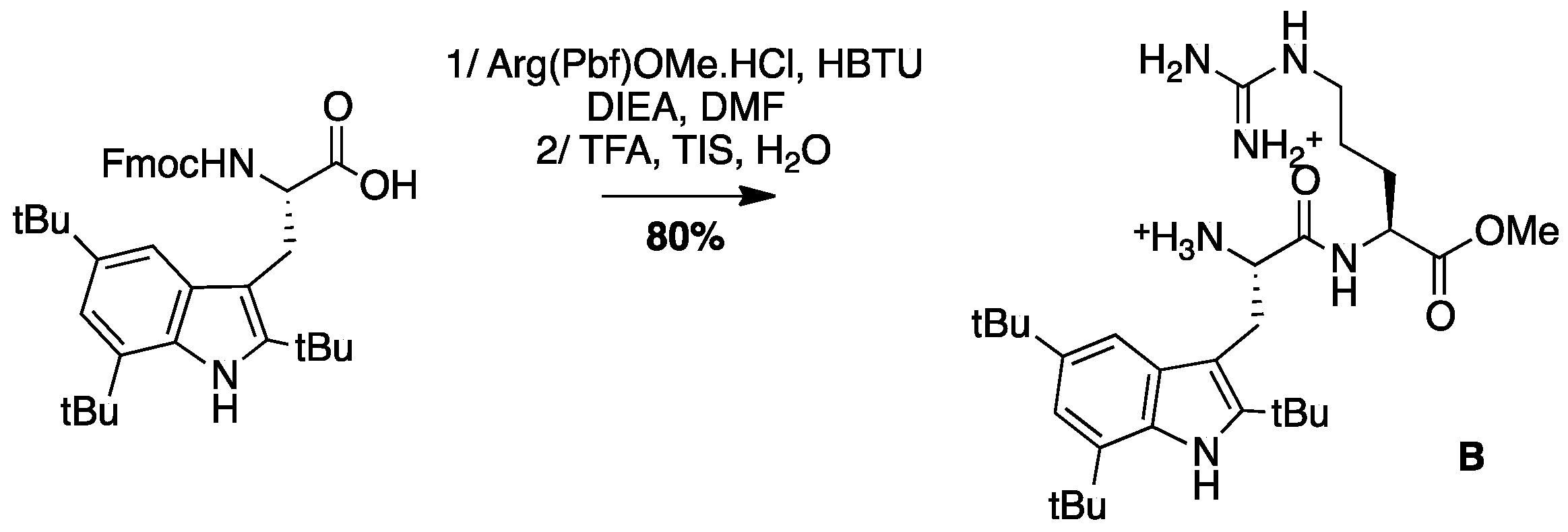
4.3.5. Synthesis of Peptide B, Tbt-Arg-OMe B (Scheme 8)
Boc-Tbt-OH (50 mg, 0.11 mmol) was dissolved in DMF. HBTU (42 mg, 0.11 mmol) and DIEA (40 µL, 0.22 mmol) were added and the mixture was stirred for 5 min before addition of H-Aργ(Pbf)OMe (52 mg, 0.11 mmol). The reaction mixture was stirred at room temperature for 5 h, then diluted with Et2O and washed with an aqueous saturated solution of NH4Cl. The organic layer was dried over MgSO4, filtered, and evaporated to dryness. The crude compound was purified by flash chromatography (Cy/AcOEt, 100:0 to 50:50) to afford the pure protected dipeptide as a white powder (80 mg, 80% yield). Treatment of this compound with a cocktail of TFA/TIS/H2O (95:2.5:2.5) for 4 h, followed by evaporation to dryness lead to peptide B, which was purified by preparative RP-HPLC using a gradient of 30% to 50% MeCN in 30 min. After lyophilisation, peptide B was obtained as white powder with purity >98%; 1H NMR (300 MHz, MeOD) δ 7.24 (s, 1H, CH indole), 7.11 (s, 1H, CH indole), 4.25 (t, J = 6, 1H, CHα Arg), 4.08 (t, J = 8.1, 1H, CHα Tbt), 3.42 (d, J = 8.1, 1H, CH2β Tbt), 3.39 (s, 3H, COOCH3), 3.09–3.15 (m, 2H, CH2δ Arg), 1.70–1.74 (m, 1H, CH2γ1 Arg), 1.44–1.57 (m, 3H, CH2γ2 and CH2β Arg), 1.54 (s, 9H, C(CH3)3), 1.50 (s, 9H, C(CH3)3), 1.37 (s, 9H, C(CH3)3); MALDI-TOF: calcd for C30H50N6O3 542.4, found 543.2 [M + H]+, 565.2 [M + Na]+; HPLC (Water/ACN (0.1% TFA); 5% to 100% ACN in 30 min: tr = 15.15 min.
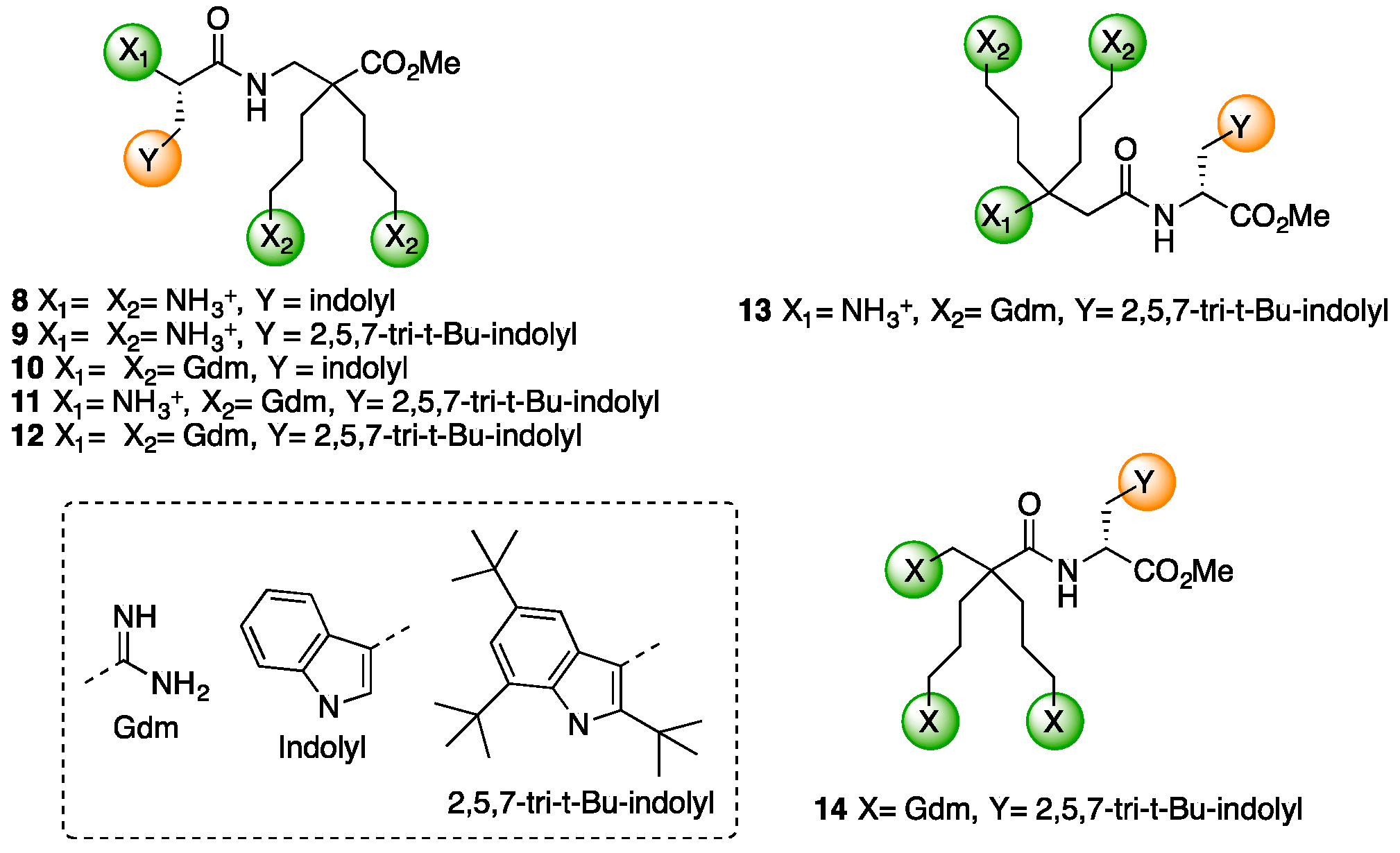
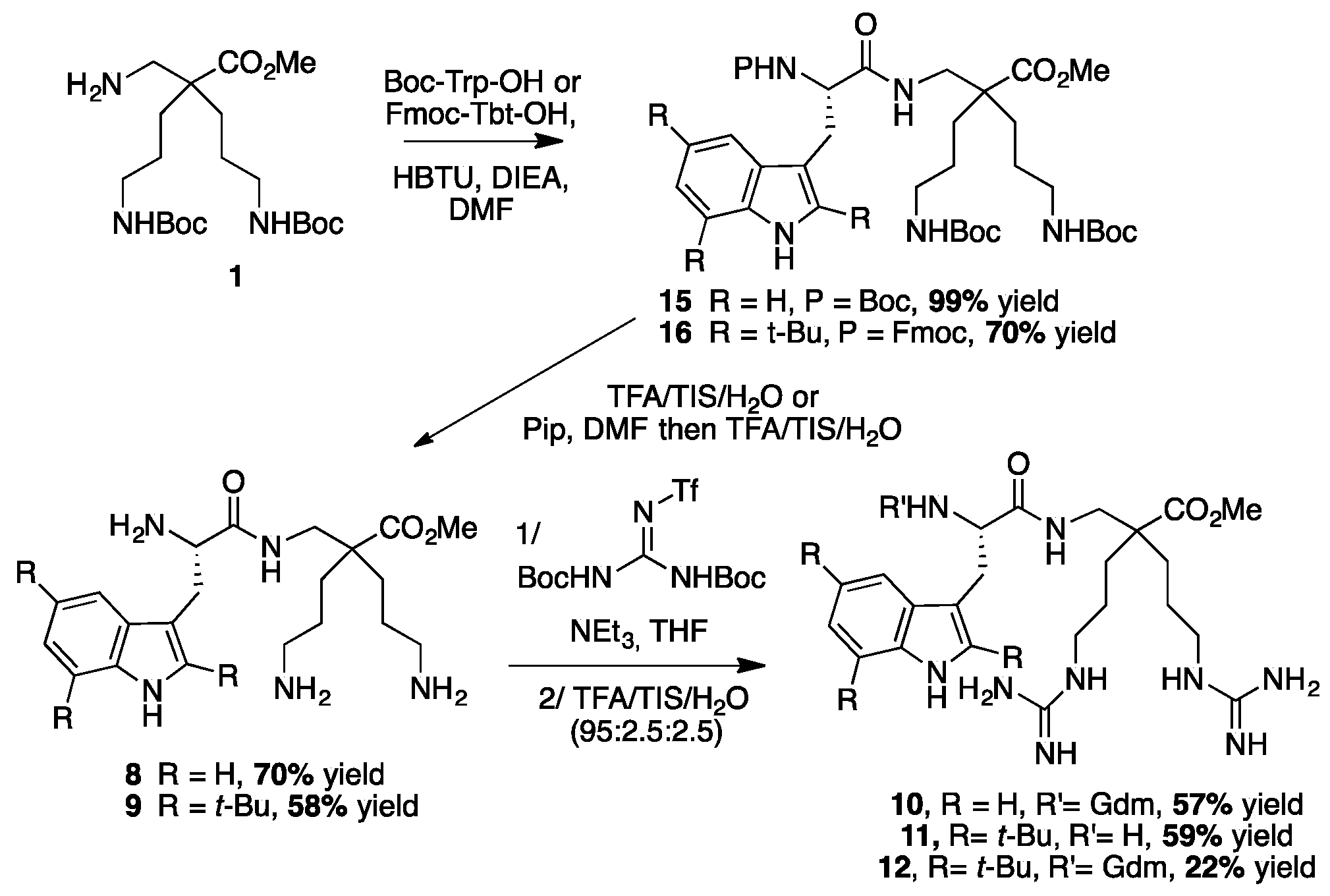
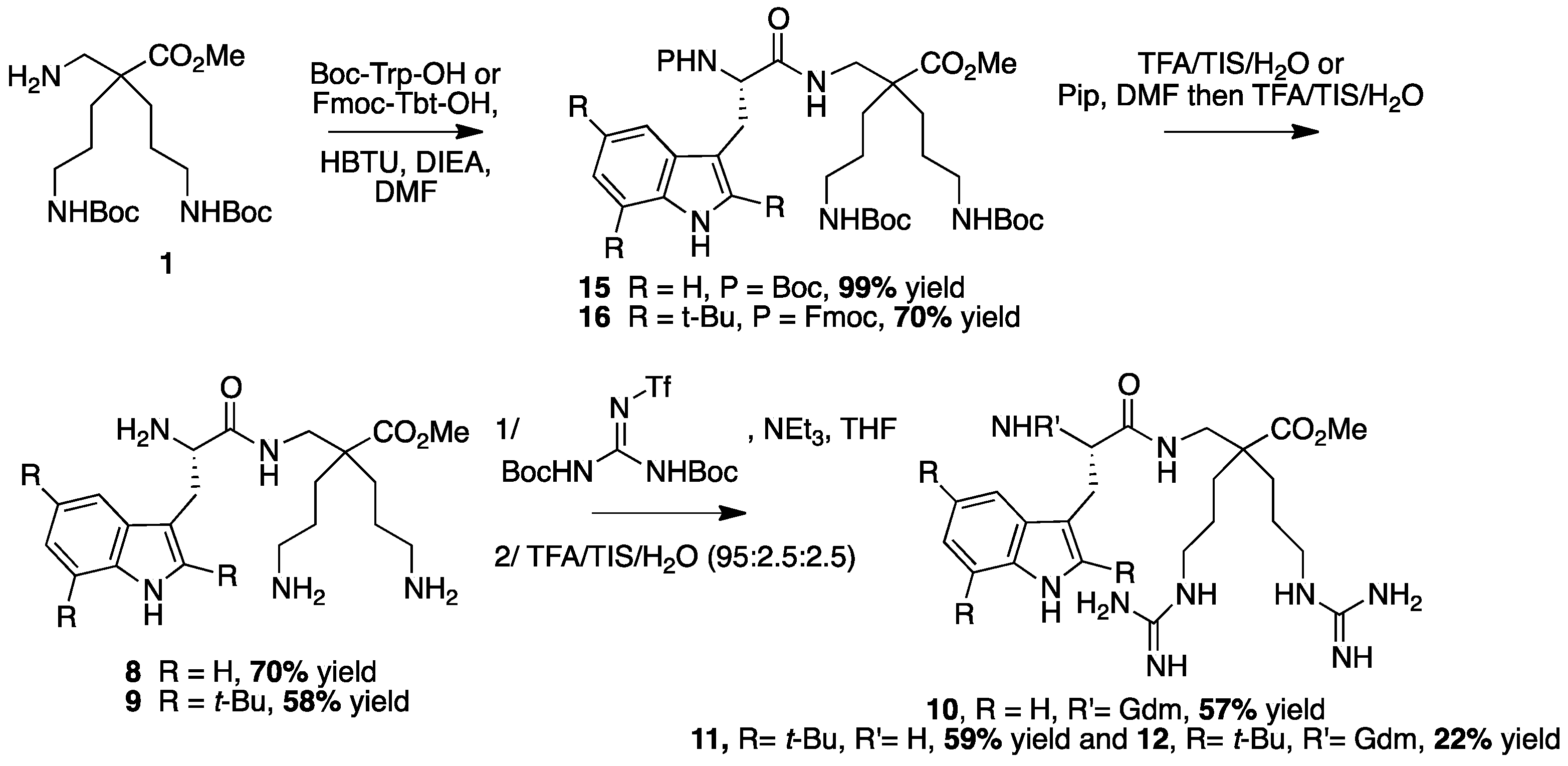
4.3.6. Synthesis of Peptides 8–12 by LPPS (Scheme 9)
Synthesis of Trp-β2,2 hbis-Orn-OMe 8 and Gdm-Trp-β2,2 hbis-Arg-OMe 10
Boc Trp-β2,2 hbis-Orn(Boc)2-OMe15: Boc-Tbt-OH (60 mg, 0.2 mmol) was dissolved in DMF (6 mL). HBTU (76 mg, 0.2 mmol) and DIEA (80 µL, 0.4 mmol) were added and the mixture was stirred for 5 min before addition of H-β2,2 hbis-Orn(Boc)2OMe 1 (84 mg, 0.2 mmol). The reaction mixture was stirred at room temperature overnight, then diluted with Et2O and washed with an aqueous saturated solution of NH4Cl. The organic layer was dried over MgSO4, filtered, and evaporated to dryness. The crude compound was purified by flash chromatography (Cy/AcOEt, 70:30) to afford 15 as a white powder (140 mg, 99% yield). 1H NMR (300 MHz, MeOD) δ 7.71 (d, J = 7.8, 1H, CH Ar), 7.38 (d, J = 7.2, 1H, CH Ar), 7.19 (td, J = 7.2, 1.1, 1H, CH Ar), 7.13 (td, J = 7.8, 1.1, 1H, CH Ar), 7.06 (d, J = 2.1, 1H, CH Ar), 5.91 (br, 1H, NH Boc), 5.36 (br, 1H, NH Boc), 4.78 (br, 1H, CHα Trp), 4.72 (br, 1H, CH2β1 Trp), 4.52 (br, 1H, CH2β2 Trp), 3.66 (s, 3H, CO2CH3), 3.30–3.35 (m, 2H, CH2βε1β2,2hbis-Orn), 3.09–3.23 (m, 2H, CH2βε2β2,2hbis-Orn), 2.95–3.04 (m, 4H, CH2δβ2,2hbis-Orn), 1.52 (s, 27H, C(CH3) 3), 1.42–1.15 (m, 8H, CH2βανδCH2γβ2,2hbis-Orn).
Trp-β2,2 h bis-Orn-OMe8: Compound
15 (70 mg, 0.1 mmol) was dissolved in DCM (∼0.4 M) and an equivalent volume of TFA/TIS/H
2O (95:2.5:2.5). The mixture was stirred at rt for 1 h then evaporated to dryness. The crude product was purified by preparative RP-HPLC using a gradient of 10% to 50% MeCN in 30 min. After lyophilisation compound
8 was obtained as white powder with purity >95% (30 mg, 70% yield);
1H NMR (500 MHz, D
2O) δ 7.68 (d,
J = 8, 1H, C
H Ar), 7.55 (d,
J = 12.8, 1H, C
H Ar), 7.33 (s, 1H, C
H Ar), 7.30 (t,
J = 8, 1H, C
H Ar), 7.22 (t,
J = 7.5, 1H, C
H Ar), 4.42 (dd,
J = 9.5, 6, 1H, C
Hα Trp), 3.66 (s, 3H, CO
2C
H3), 3.51 (d,
J = 14.5, 1H, C
H2βε
1 β
2,2 h bis-Arg), 3.41 (dd,
J = 14.2, 6, 1H, C
H2β
1 Trp), 3.35 (dd,
J = 14.2, 9.5, 1H, C
H2β
2 Trp), 3.15 (d,
J = 14.5, 1H, C
H2βε
2 β
2,2 hbis-Arg), 2.81 (t,
J = 7.8, 1H, C
H2δ
1 β
2,2 h bis-Arg), 2.71 (t,
J = 7.8, 1H, C
H2δ
2 β
2,2 h bis-Arg), 1.49–1.53 (m, 1H, C
H2γ
1 β
2,2 h bis-Arg), 1.35–1.39 (m, 3H, C
H2γ
1’ and C
H2γ
2 β
2,2 h bis-Arg), 1.24 (td,
J = 13.2, 3.7, 1H, C
H2β
1 β
2,2 h bis-Arg), 1.05–1.12 (m, 2H, C
H2β
1′ and C
H2β
2β
2,2 hbis-Arg), 0.88–0.95 (m, 1H, C
H2β
2′β
2,2 hbis-Arg); MALDI-TOF: calcd for C
21H
33N
5O
3 403.3, calcd for C
21H
33N
5O
3Na 426.3, found 404.5 [M + H]
+, 426.5 [M + Na]
+, 442.5 [M + K]
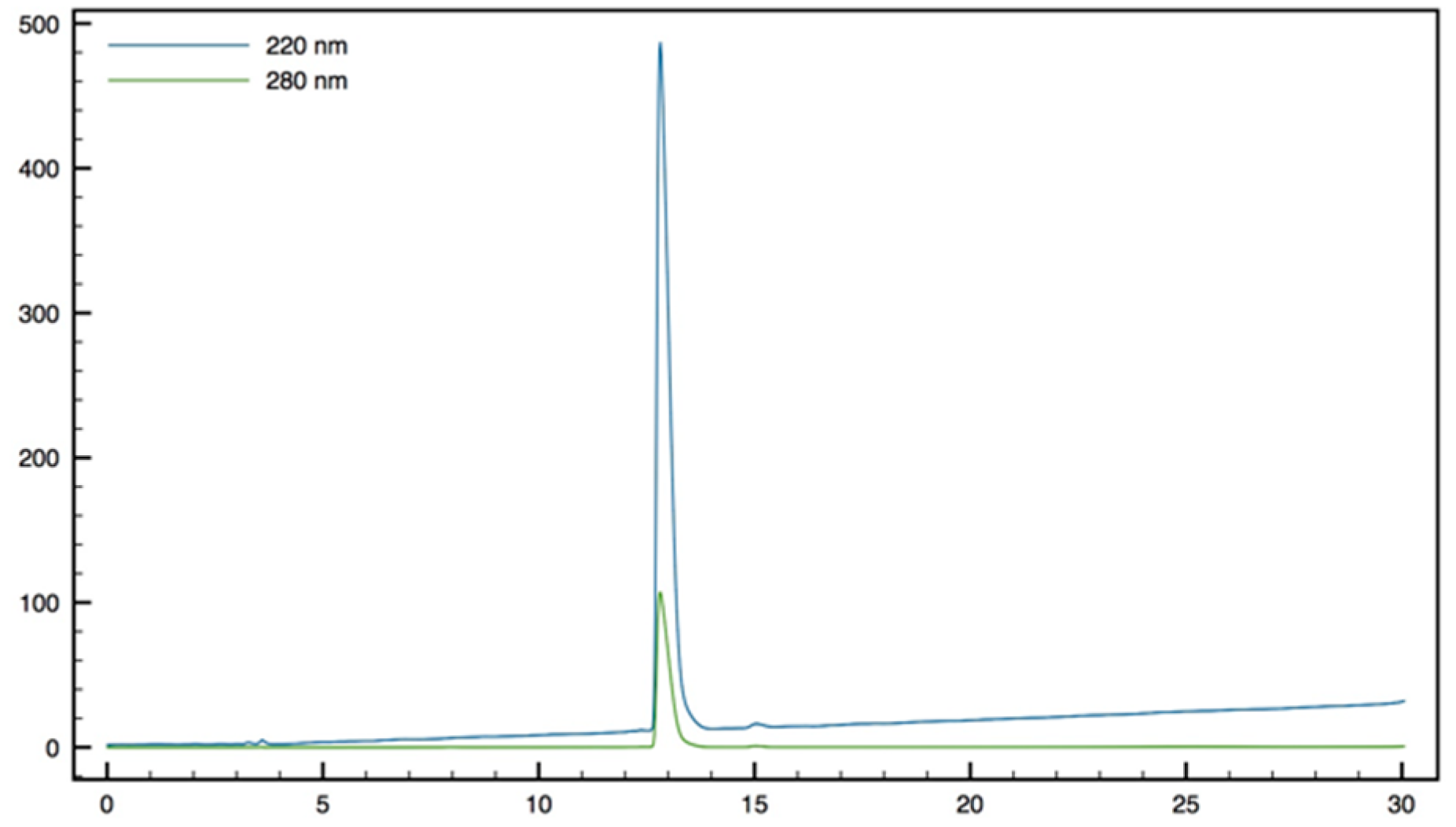
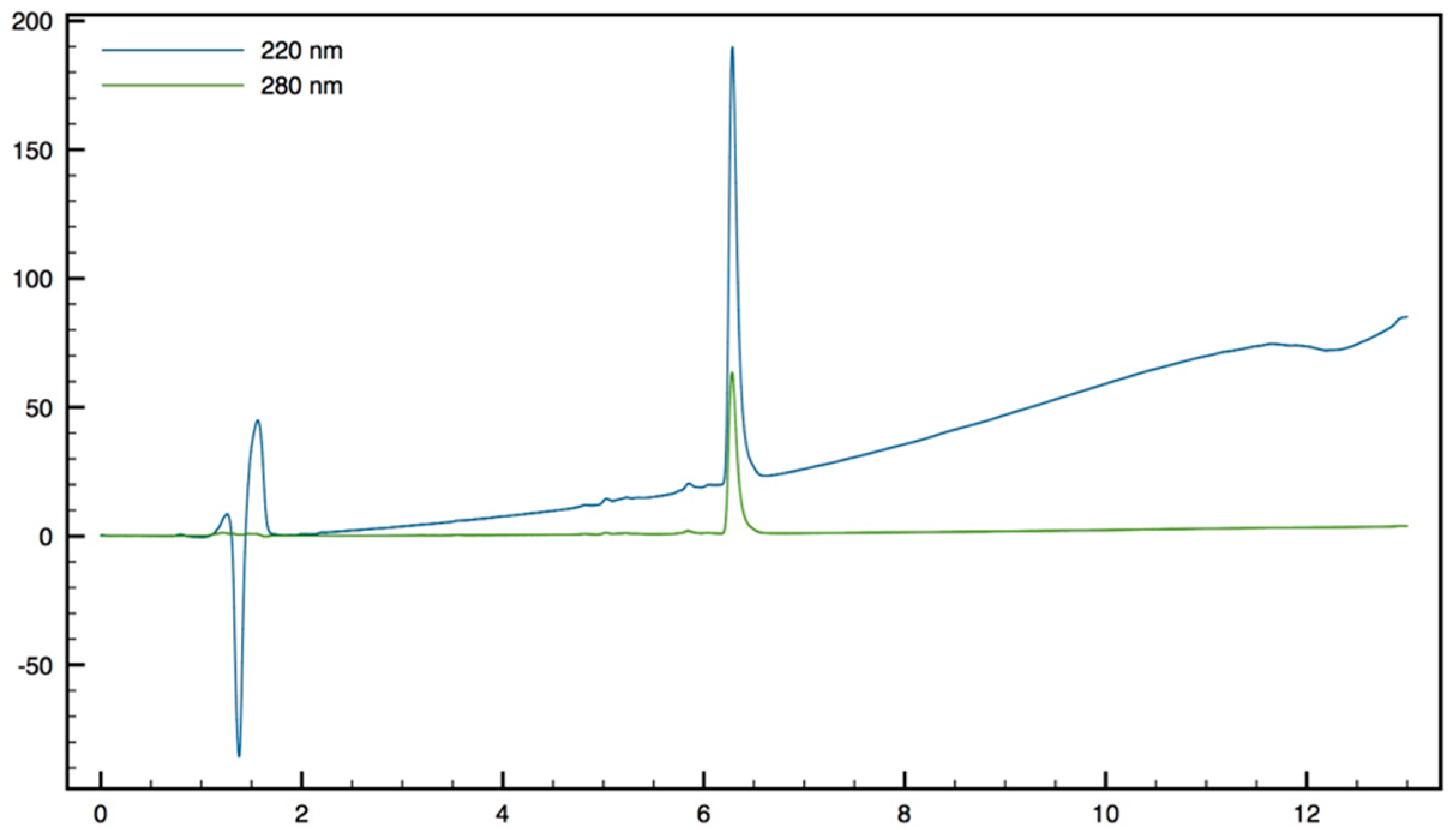
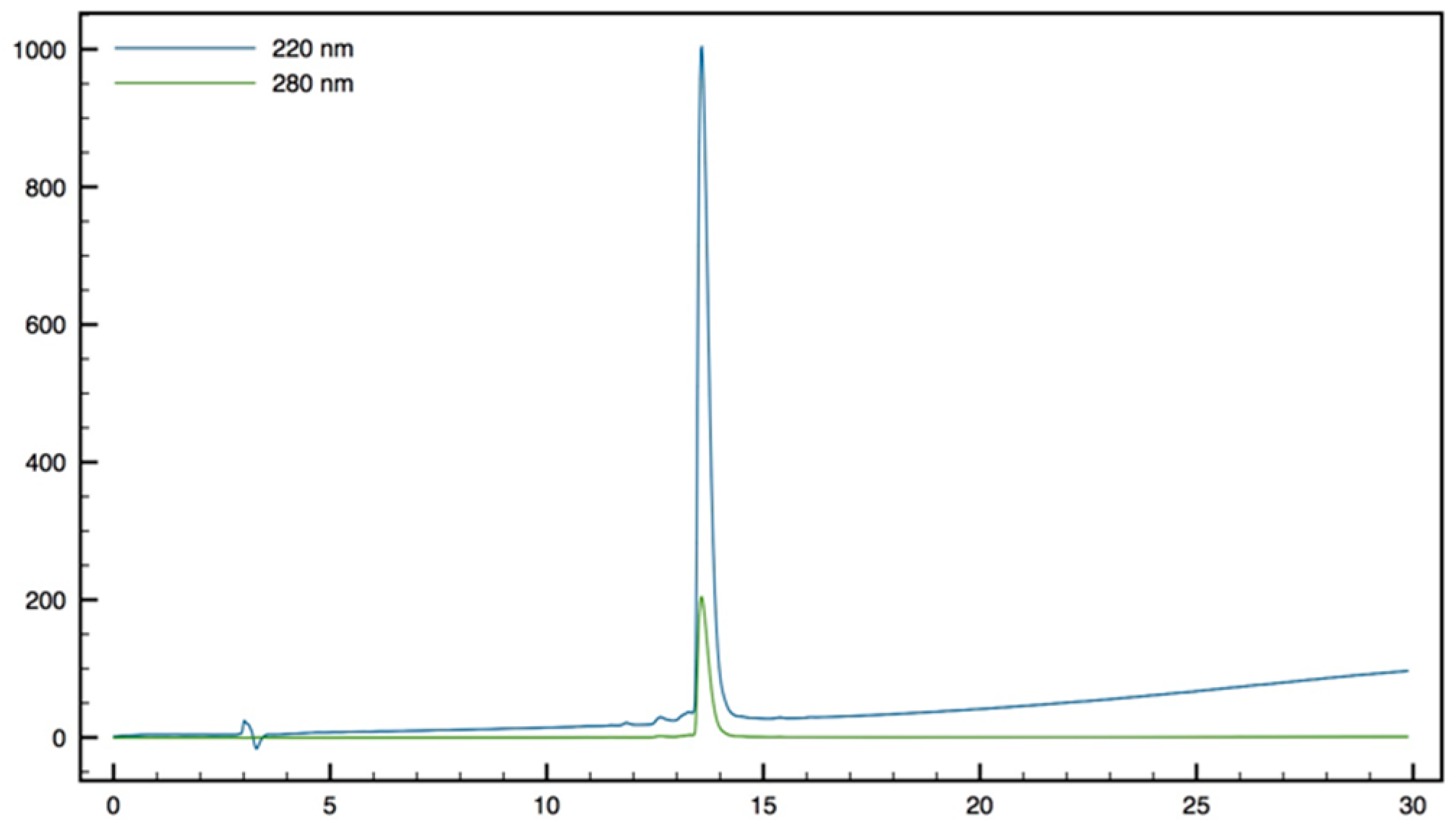
+; HPLC (Water/ACN (0.1% TFA); 5% to 100% ACN in 30 min): tr = 7.18 min (
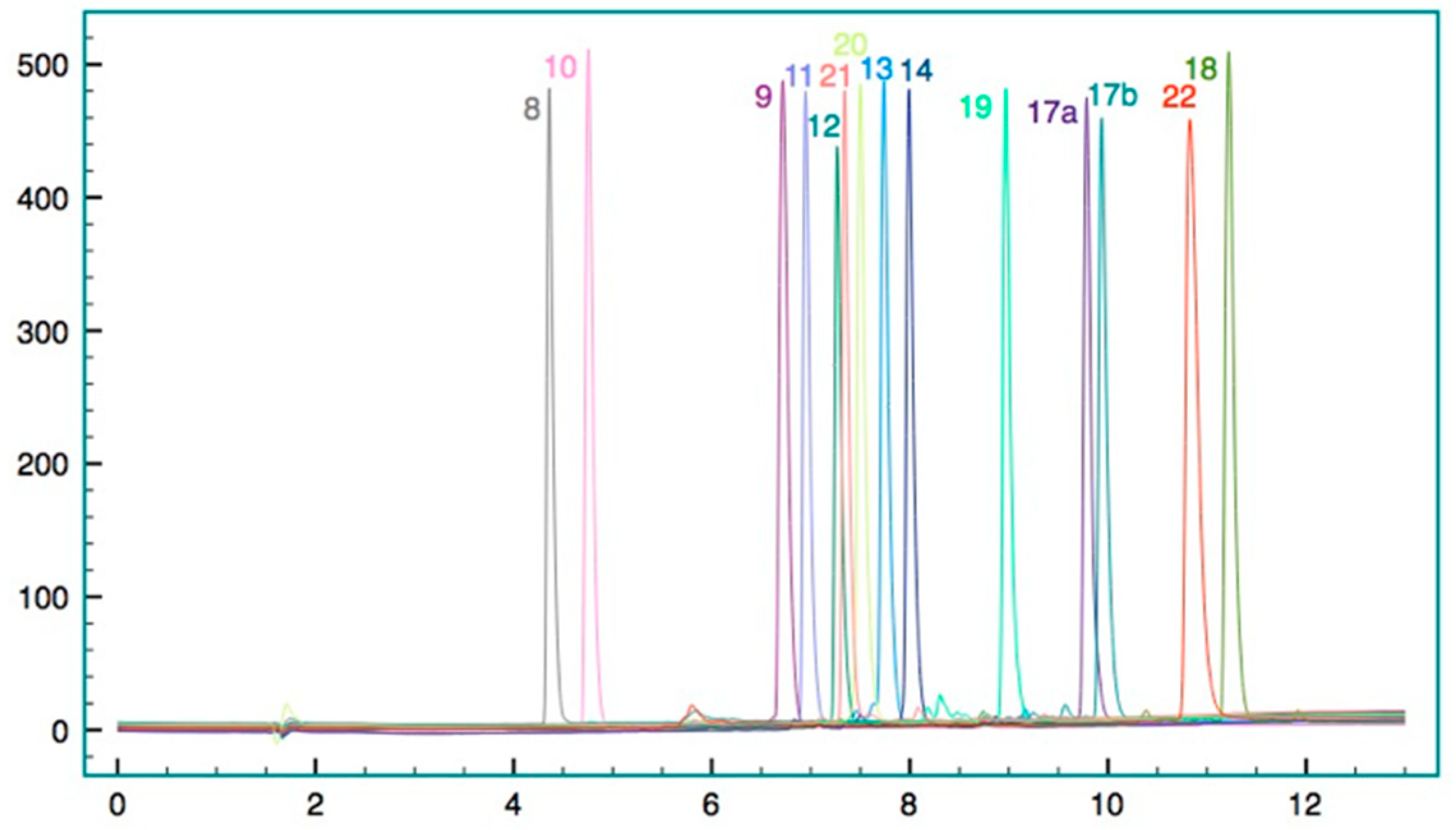
Figure 11).
Gdm-Trp-β2,2 hbis
-Arg-OMe10: Compound
15 (70 mg, 0.1 mmol) was dissolved in DCM (∼0.4 M) and an equivalent volume of TFA/TIS/H
2O (95:2.5:2.5). The mixture was stirred at rt for 1.5 h then evaporated to dryness. The crude compound was dissolved in 6 mL of THF 1,3-Di-Boc-2-(trifluoromethylsulfonyl)guanidine (137 mg, 0.35 mmol) and NEt
3 (60 µL, 0.4 mmol) were added and the reaction mixture was stirred at rt overnight. After evaporation of THF, a solution of TFA/TIS/H
2O (95:2.5:2.5) was added and the mixture was stirred at rt for 2 h. The crude product was purified by preparative RP-HPLC using a gradient of 10% to 50% MeCN in 30 min. After lyophilisation, compound
10 was obtained as white powder with purity >98% (31 mg, 57% yield);
1H NMR (300 MHz, D
2O) δ 7.69 (d,
J = 7.5, 1H, C
H Ar), 7.36 (d,
J = 8.1, 1H, C
H Ar), 7.31 (s, 1H, C
H Ar), 7.26 (td,
J = 7.5, 0.9, 1H, C
H Ar), 7.22 (td,
J = 7.2, 0.9, 1H, C
H Ar), 4.62 (t,
J = 7.5, 1H, C
Hα Trp), 3.69 (s, 3H, CO
2C
H3), 3.47 (d,
J = 14.1, 1H, C
H2βε
1 β
2,2 hbis-Arg), 3.35 (d,
J = 7.5, 2H, C
H2β Trp), 3.21 (d,
J = 14.4, 1H, C
H2βε
2 β
2,2 hbis-Arg), 3.05 (t,
J = 6.6, 1H, C
H2δ
1 β
2,2 h bis-Arg), 2.98 (dd,
J = 11.7, 6.6, 1H, C
H2δ
2 β
2,2 h bis-Arg), 1.29–1.43 (m, 4H, C
H2γ β
2,2 hbis-Arg), 1.09-1.28 (m, 4H, C
H2β β
2,2 hbis-Arg); MALDI-TOF: calcd for C
32H
57N
5O
3 529.3, calcd for C
32H
57N
5O
3Na 552.3, found 530.6 [M + H]
+, 552.6 [M + Na]
+, 513.6 [M + H − NH
3]
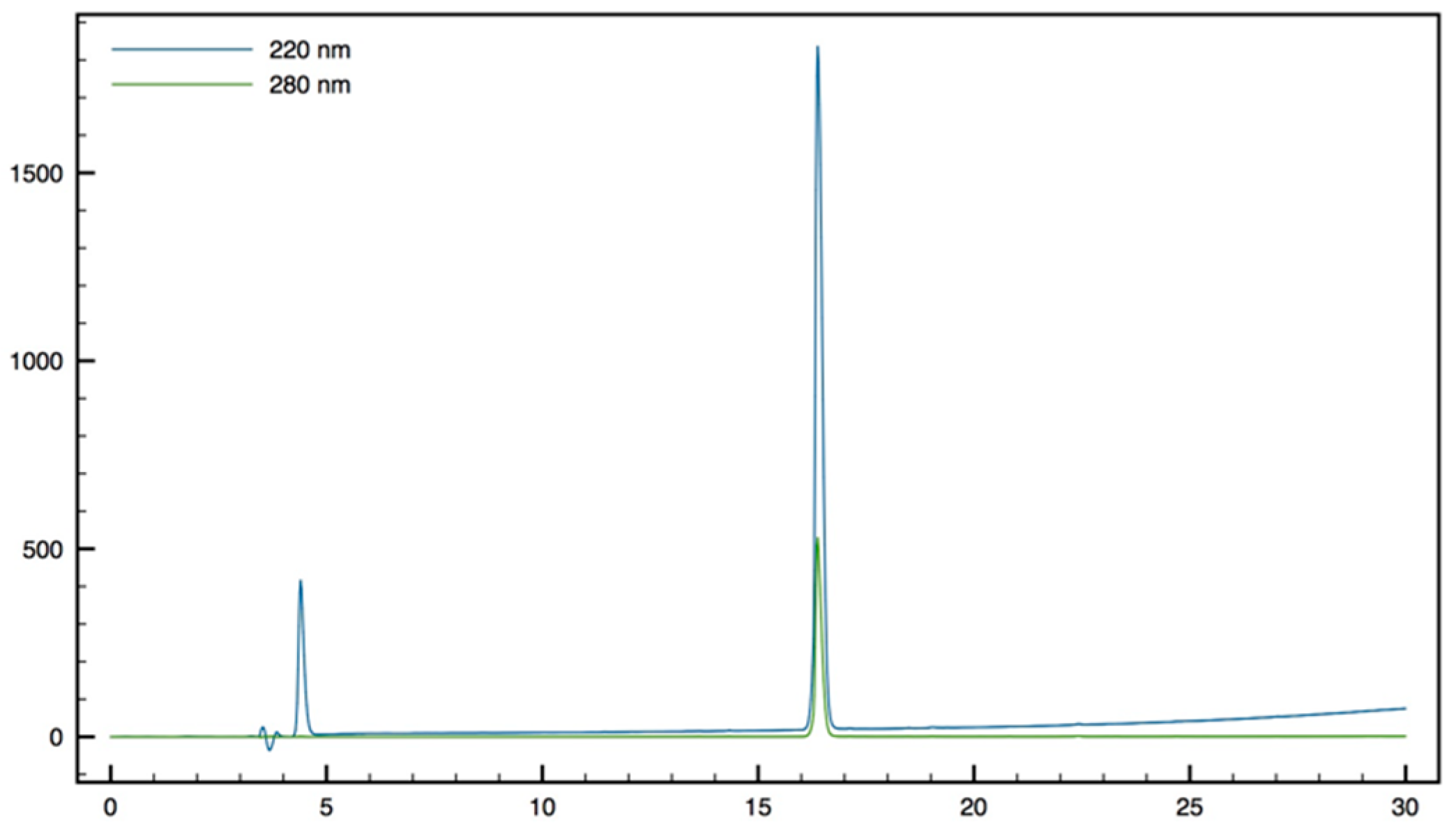
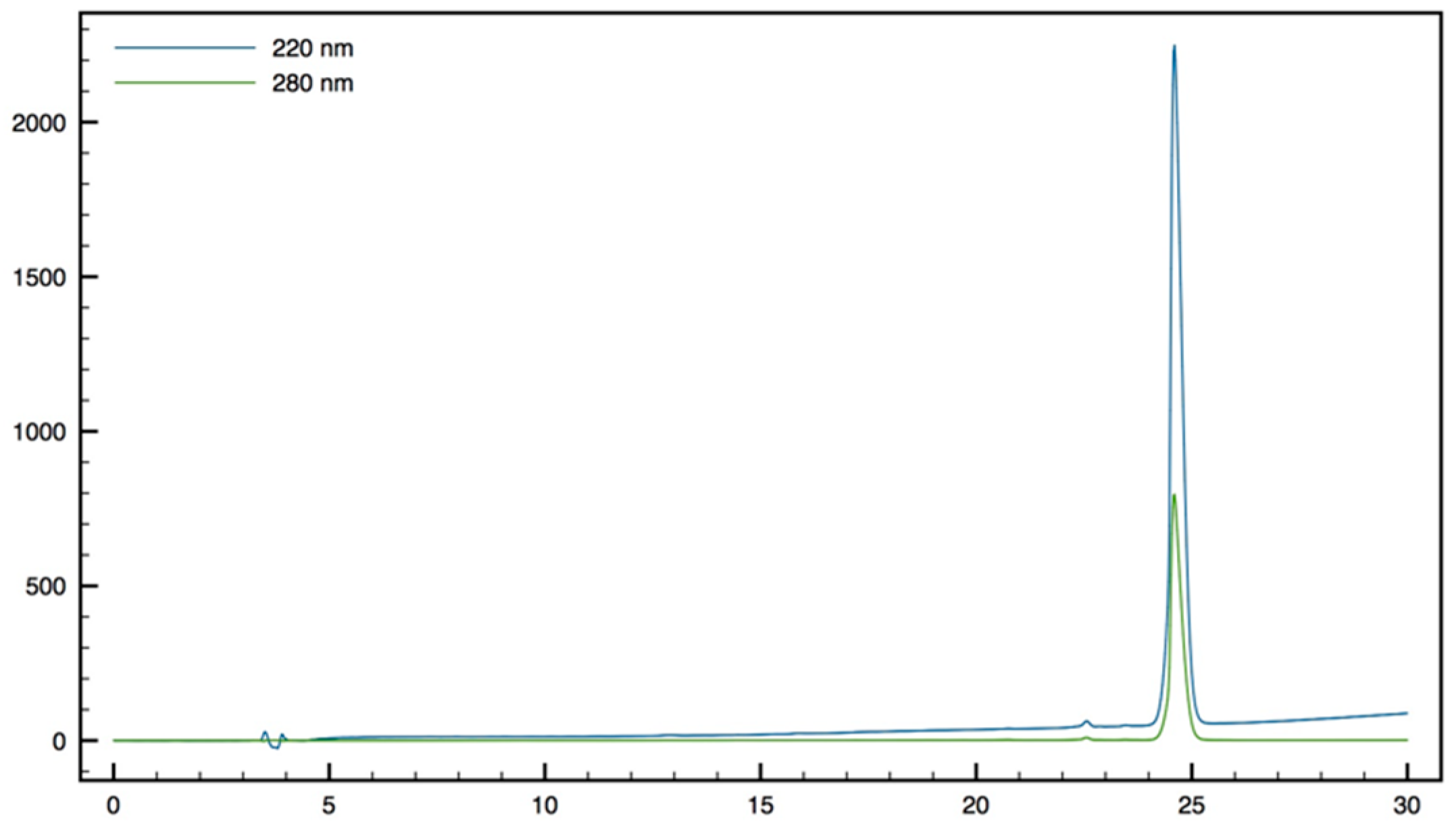
+; HPLC (Water/ACN (0.1% TFA); 5% to 100% ACN in 30 min): tr = 9.62 min (
Figure 12).
Synthesis of Tbt-β2,2 hbis-Orn-OMe 9, Tbt-β2,2 h bis-Arg-OMe 11 and Gua-Tbt-β2,2 h bis-Arg-OMe 12
Fmoc-Tbt-β2,2 hbis-Orn(Boc)2OMe16: Fmoc-Tbt-OH (400 mg, 0.64 mmol) was dissolved in DMF (24 mL). HBTU (244 mg, 0.64 mmol) and DIEA (240 µL, 1.28 mmol) were added and the mixture was stirred for 3 h before addition of H-β2,2 h bis-Orn(Boc)2OMe 1 (268 mg, 0.64 mmol). The reaction mixture was stirred at room temperature overnight, then diluted with Et2O and washed with an aqueous saturated solution of NH4Cl. The organic layer was dried over MgSO4, filtered, and evaporated to dryness. The crude compound was purified by flash chromatography (Cy/AcOEt, 100:0 to 70:30) to afford the pure protected dipeptide as a white powder (450 mg, 70% yield). 1H NMR (300 MHz, MeOD) δ 8.22 (s, 1H, NH indole), 7.75 (d, J = 7.2, 1H, CH Ar Fmoc), 7.55 (d, J = 7.2, 1H, CH Ar Fmoc), 7.41 (s, 1H, CH Ar indole), 7.35 (t, J = 7.2, 1H, CH Ar Fmoc), 7.24 (dt, J = 11.7 and 7.2, 1H, CH Ar Fmoc), 7.12 (s, 1H, CH indole), 4.27–4.33 (m, 3H, CHα Tbt and CH2 Fmoc), 4.12 (t, J = 6.9, 1H, CH Fmoc), 3.55 (s, 3H, CO2CH3), 3.43 (dd, J = 14.1 9.3, 1H, CH2β1 Tbt), 3.39 (d, J = 14.1, 1H, CH2βε1 β2,2 h bis-Orn), 3.23 (dd, J = 14.4, 6.3, 1H, CH2β2 Tbt), 2.9 (m, 4H, CH2δ β2,2 h bis-Orn), 2.79 (d, J = 14.1, 1H, CH2βε2 β2,2 hbis-Orn), 1.52 (s, 9H, C(CH3)3 indole), 1.47 (s, 9H, C(CH3)3 indole), 1.36–1.44 (m, 35H, C(CH3)3 indole, C(CH3)3 Boc, CH2β β2,2 hbis-Orn and CH2γ β2,2 hbis-Orn); 13C NMR (75 MHz, MeOD) δ 177.1 (C, C=O amide), 174.9 (C, C=O ester), 158.4 (C=O Boc), 157.9 (C=O Fmoc), 145.2, 145.1, 143.6, 142.9, 142.5, 133.1, 131.8, 131.3 (8C, C Ar), 128.7, 128.2, 126.2, 120.9 (4CH, CH Ar Fmoc), 117.3, 113.4 (2CH, CH Ar indole), 106.2 (C, C Ar), 79.8 (C, C(CH3)3 Boc), 68.1 (CH2, CH2 Fmoc), 58.5 (CH, CHα Tbt), 52.3 (CH3, CO2CH3), 50.7 (C, Cα β2,2 hbis-Orn), 48.3 (CH, CH Fmoc), 42.9 (CH2, CH2βε β2,2 hbis-Orn), 41.6 (CH2, CH2δ β2,2 hbis-Orn), 35.7, 35.5 and 34.3 (3C, C(CH3)3 indole), 32.7 (CH3, C(CH3)3 indole), 32.4 and 32.1 (CH2, CH2γ β2,2 h bis-Orn), 31.3 (CH3, C(CH3)3 indole), 30.9 (CH3, C(CH3)3 indole), 29 (CH2, CH2β Tbt), 28.8 (6CH3, C(CH3)3 Boc), 25.5 and 25.3 (CH2, CH2β β2,2 h bis-Orn).
H-Tbt-
β2,2 h bis-OrnOMe9: Compound
16 (60 mg, 0.06 mmol) was dissolved in a 20% solution of piperidine in DCM and allowed to react for 1 h before evaporation to dryness. A solution of TFA/TIS/H
2O (95:2.5:2.5) was added and the mixture was stirred at rt for 1 h. The crude product was purified by preparative RP-HPLC using a gradient of 30% to 50% MeCN in 30 min. After lyophilisation
9 was obtained as white powder with a purity of 98% (20 mg, 58% yield);
1H NMR (300 MHz, D
2O) δ 8.54 (s, 1H, N
H indole), 7.26 (s, 1H, C
H Ar), 7.22 (s, 1H, C
H Ar), 4.03 (dd,
J = 9.3, 6, 1H, C
Hα Tbt), 3.50 (s, 3H, CO
2C
H3), 3.37 (d,
J = 14.2, 1H, C
H2βε
1 β
2,2 h bis-Orn), 3.34 (d,
J = 14.1, 2H, C
H2β Tbt), 2.80 (m, 4H, C
H2δ β
2,2 h bis-Orn), 2.33 (d,
J = 14.2, 1H, C
H2βε
2 β
2,2 h bis-Orn), 1.31–1.44 (m, 35H, C
H2β β
2,2 h bis-Orn, C
H2γ β
2,2 h bis-Orn and C(C
H3)
3); MALDI-TOF: calcd for C
32H
57N
5O
3 571.5, found 572.6 [M + H]
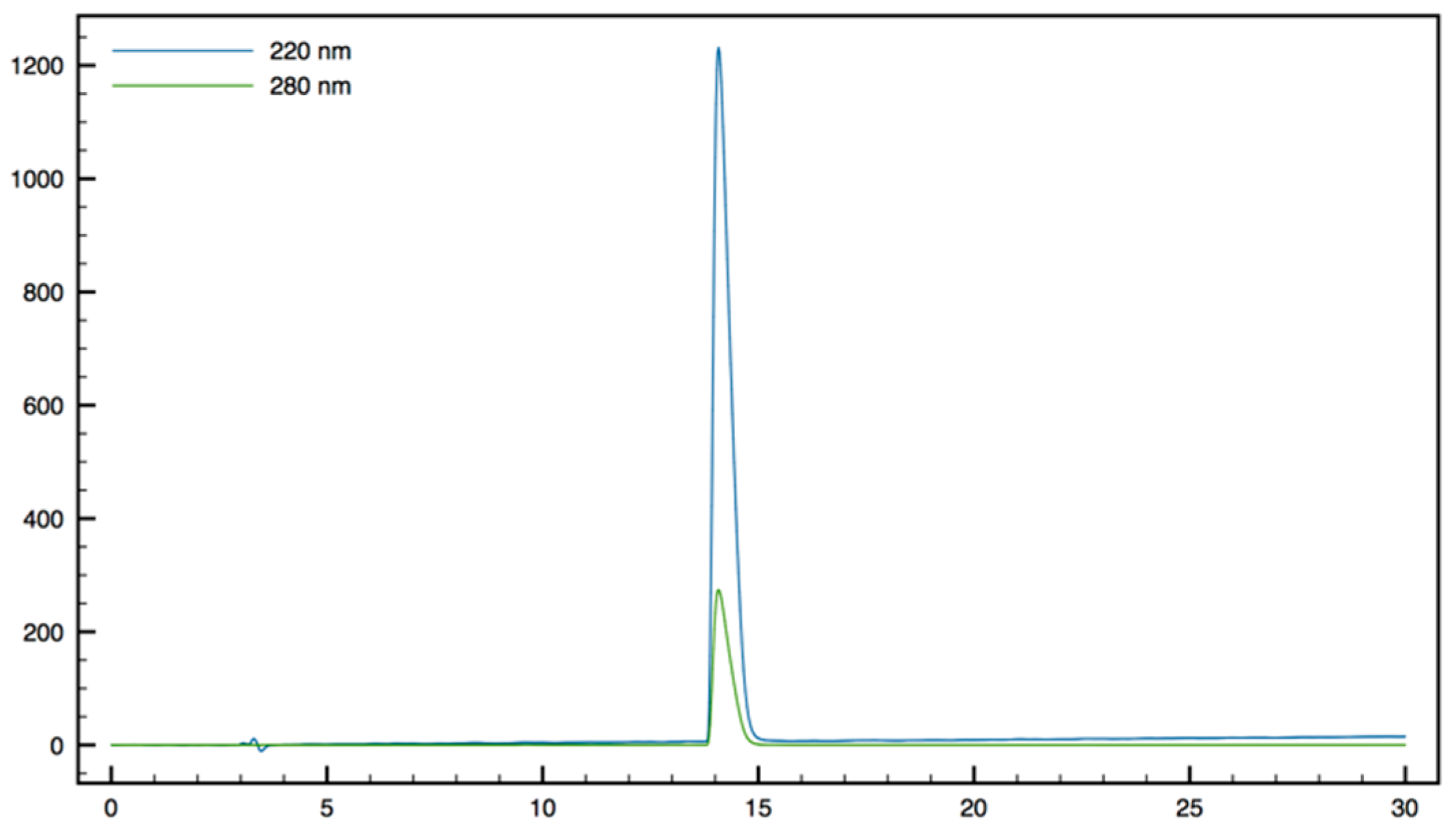
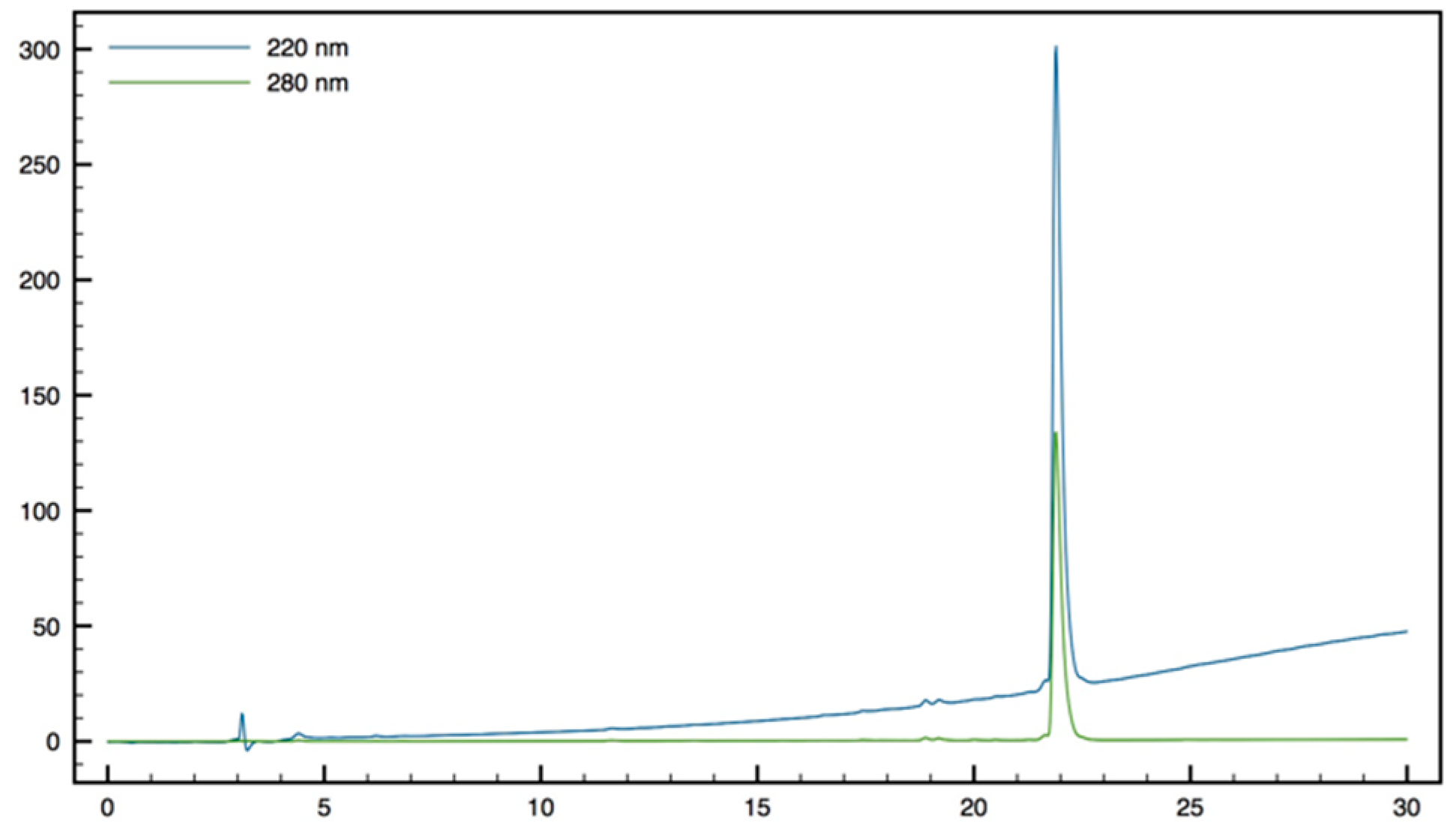
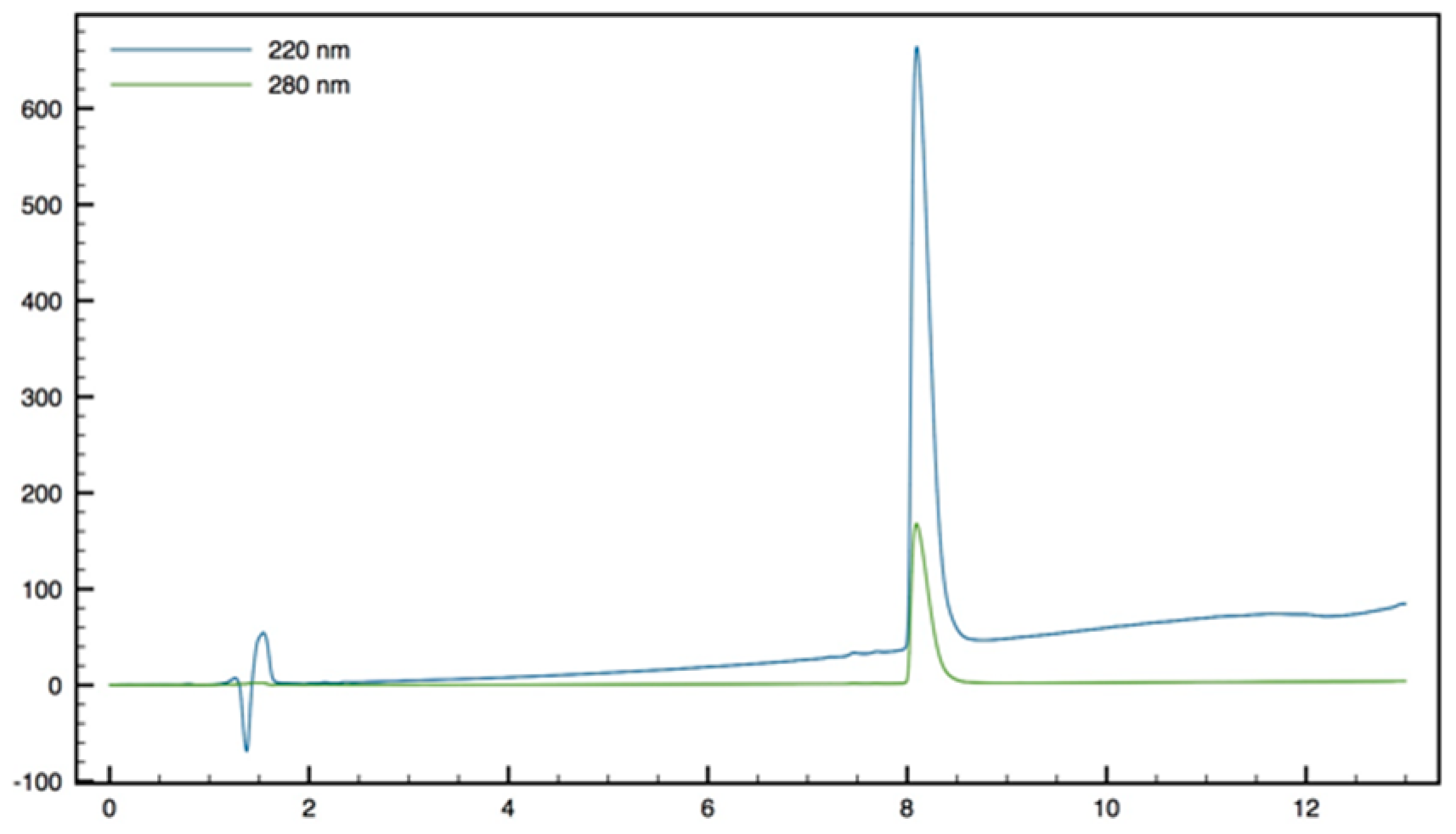
+; HPLC (Water/ACN (0.1% TFA); 30% to 50% ACN in 30 min): tr = 12.73 min (
Figure 13).
H-Tbt-β2,2 h bis-Arg-OMe 11 and Gua-Tbt-β2,2 h bis-Arg-OMe12: Compound 9 (10 mg, 0.013 mmol) was dissolved in 1 mL of THF. 1,3-Di-Boc-2-(trifluoromethylsulfonyl) guanidine (30 mg, 0.08 mmol) and DIEA (27 µL, 0.156 mmol) were added and the reaction mixture was stirred at rt for 2 h. After evaporation of THF, a solution of TFA/TIS/H2O (95:2.5:2.5) was added and the mixture was stirred at rt for 2 h. The crude product was evaporated in vacuo and purified by preparative RP-HPLC using a gradient of 30% to 50% MeCN in 30 min. Two pics were collected separately at 14 and 18 min corresponding, respectively, to compounds 11 and 12. After lyophilisation, the two compounds 11 (5 mg, 59% yield) and 12 (2 mg, 22% yield) were obtained as white powders with purity >99%.
H-Tbt-
β2,2 h bis-Arg-OMe11:
1H NMR (500 MHz, D
2O) δ 7.29 (s, 1H, C
H indole), 7.27 (s, 1H, C
H indole), 4.14 (dd,
J = 11.2, 5.5, 1H,
CHα Tbt), 3.61 (dd,
J = 14.5, 5, 1H, C
H2β
1 Tbt), 3.57 (s, 3H, CO
2C
H3), 3.44 (dd,
J = 13.5, 12, 2H, C
H2β
2 Tbt), 3.21 (d,
J = 14.5, 1H, C
H2βε
1 β
2,2 h bis-Arg), 3.04 (m,
J = 4H, C
H2δ β
2,2 h bis-Arg), 1.83 (d,
J = 14.5, 1H, C
H2βε
2 β
2,2 h bis-Arg), 1.55 (s, 9H, C(C
H3)
3), 1.49 (s, 9H, C(C
H3)
3), 1.39 (s, 9H, C(C
H3)
3), 1.1-1.34 (m, 8H, C
H2β β
2,2 h bis-Arg and C
H2γ β
2,2 h bis-Arg); MALDI-TOF: calcd for C
35H
61N
5O
3 655.5, calcd for C
35H
61N
5O
3Na 678.5, found 656.4 [M + H]
+, 678.4 [M + Na]
+, 694.4 [M + K]
+, 639.4 [M + H − NH
3]
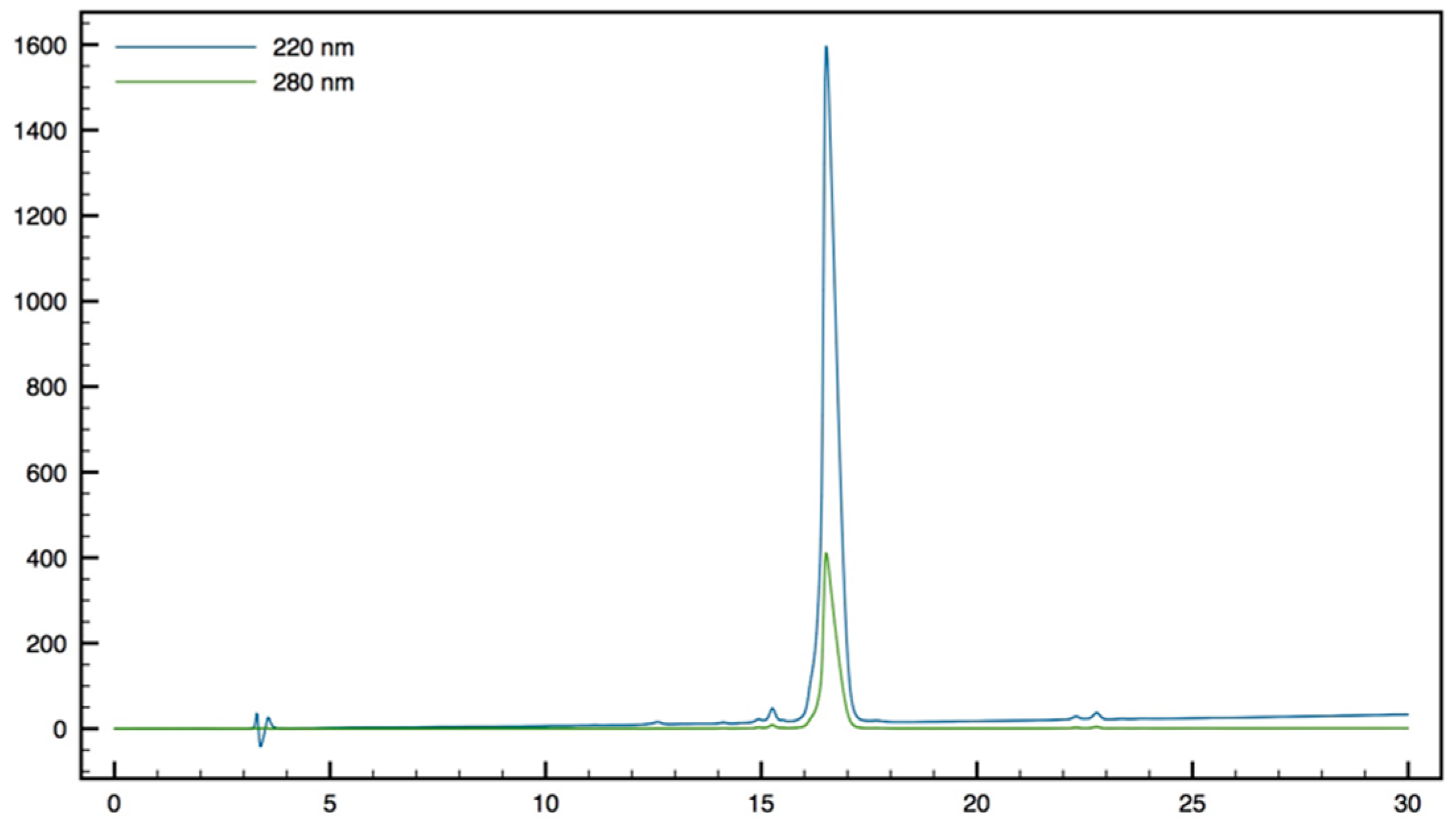
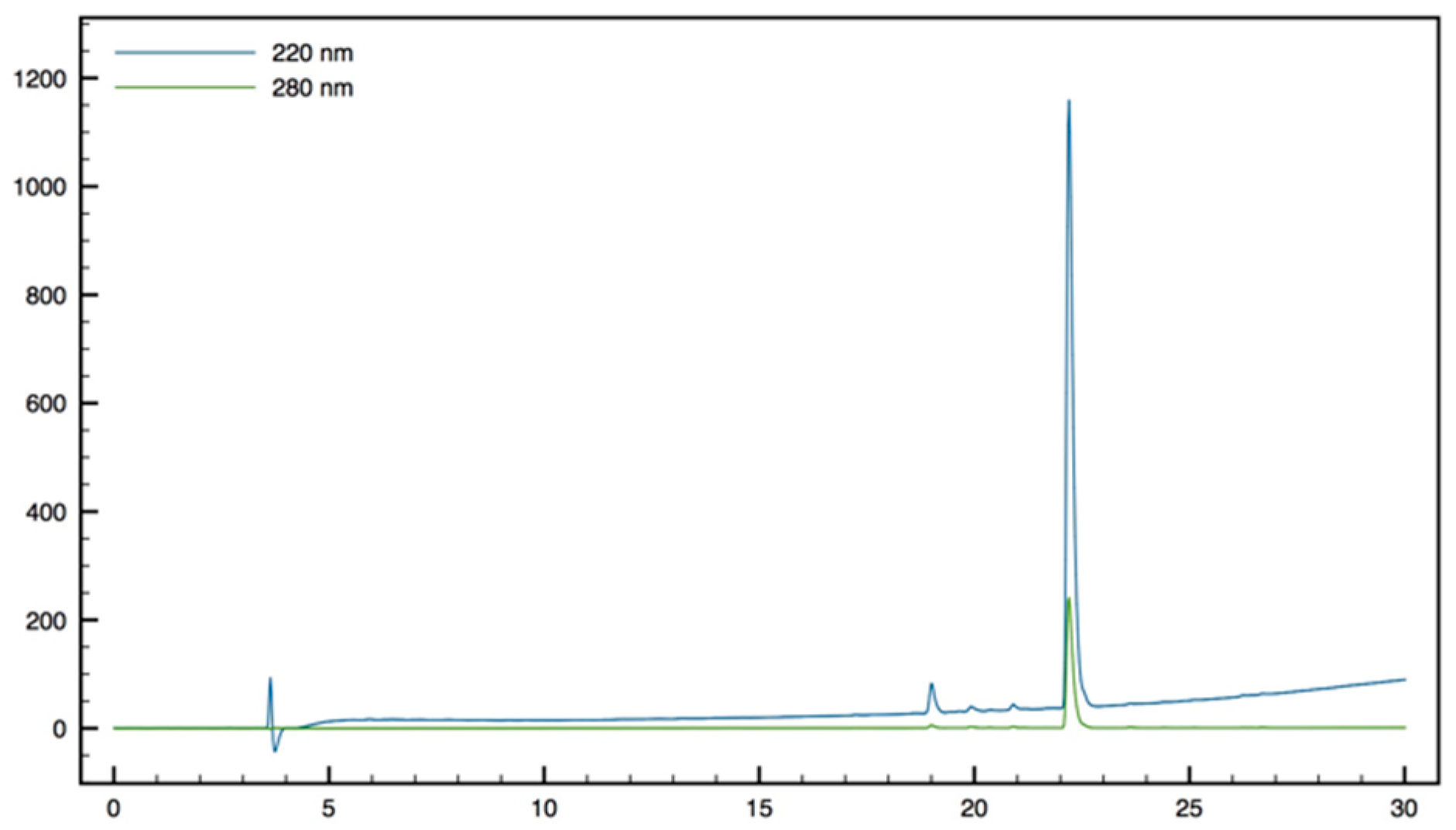
+; HPLC (Water/ACN (0.1% TFA); 30% to 50% ACN in 30 min: tr = 16.11 min (
Figure 14).
Gua-Tbt-
β2,2 h bis-Arg-OMe12:
1H NMR (300 MHz, MeOD) δ 8.35 (s, 1H, N
H indole), 7.31 (d,
J = 1.5, 1H, C
H Ar), 7.14 (d,
J = 1.5, 1H, C
H Ar), 4.39 (t,
J = 7.2, 1H, C
Hα Tbt), 3.65 (s, 3H, CO
2C
H3), 3.52 (d,
J = 14.2, 1H, C
H2βε
1 β
2,2 h bis-Arg), 3.47 (dd,
J = 11.1, 7.2, 2H, C
H2β Tbt), 3.01–3.13 (m, 4H, C
H2δ β
2,2 h bis-Arg), 2.82 (d,
J = 14.2, 1H, C
H2βε
2 β
2,2 h bis-Arg), 1.26–1.64 (m, 35H, C
H2β β
2,2 h bis-Arg, C
H2γ β
2,2 h bis-Arg and C(C
H3)
3); MALDI-TOF: calcd for C
36H
63N
11O
3 697.5, calcd for C
36H
63N
11O
3Na 720.5, found 698.4 [M + H]
+, 720.4 [M + Na]
+, 736.3 [M + K]
+, 681.3 [M + H − NH
3]
+; HPLC (Water/ACN (0.1% TFA); 30% to 70% ACN in 30 min): tr = 14.08 min (
Figure 15).
4.3.7. Synthesis of Peptides 13 and 14 by SPPS (Scheme 10)
H-β3,3-h-bis-Arg-Tbt-OMe13: HMBA-AM resin (108 mg, 0.1 mmol) was washed five times with DMF, DCM, and DMF, then allowed to swell in DMF for 30 min. Fmoc-Tbt-OH (4 eq, 0.4 mmol, 238 mg) was dissolved in dry DCM. The solution was cooled to 0 °C, DIC (4 eq, 0.4 mmol, 60 µL) was added. The reaction was stirred for 1.5 h and the solvent was then removed
in vacuo. The resulting anhydride was dissolved in DMF and added to the resin. A solution of DMAP (0.1 eq, 0.04 mmol, 5 mg) in DMF was added and the resin was shaken for 1 h before washing with DMF, DCM, and DMF (resin loading = 0.84 mmol/g). Removal of the Fmoc protecting group was achieved by treatment of the resin with 20% (
v:
v) piperidine in DMF 3 times for 5 min. The resin was washed five times with DMF. Boc-β
3,3h bis-Orn(Boc)
2OH
3 (2 eq, 0.18 mmol, 90 mg) was dissolved in DMF (1.2 mL). HATU (1.8 eq, 0.17 mmol, 65 mg) and DIEA (2 eq, 0.18 mmol, 23 µL) were added. The solution was added to the resin (0.09 mmol, 108 mg) and the coupling reaction was allowed to proceed for 2 h at room temperature. The solution was removed by filtration and the resin was washed with DMF five times. Reaction completion was monitored by Kaiser test. Boc removal was performed by treating the resin with a TFA/TIS/H
2O cocktail (95:2.5:2.5) for 5 h. The free amines were then reacted with 1,3-Di-Boc-2-(trifluoromethylsulfonyl)guanidine (10 eq, 1.8 mmol, 700 mg) and NEt
3 (10 eq, 1.8 mmol, 240 µL) in DMF overnight. The resin was filtrated and the Boc groups were removed with a TFA/TIS/H
2O cocktail (95:2.5:2.5) at rt for 3 h. After filtration, peptide
14 was cleaved from the resin using a mixture of MeOH/DMF/DIEA (5:5:1) for 16 h at 50 °C. The solution was filtrated, and solvents were evaporated. The crude product was purified by preparative RP-HPLC using a gradient of 30% to 70% MeCN in 30 min. After lyophilisation peptide
13 was obtained as a white powder with a purity of >95% (47 mg, 75% yield);
1H NMR (500 MHz, D
2O) δ 7.37 (d,
J = 1.7, 1H, C
H Ar indole), 7.28 (d,
J = 1.7, 1H, C
H Ar indole), 4.69 (t,
J = 7.5, 1H, C
Hα Tbt), 3.48–3.53 (m, 1H, C
H2β
1 Tbt), 3.68 (dd,
J = 10.5, 1, 1H, C
H2β
2 Tbt), 3.25 (t,
J = 6.5, 1H, C
H2ε
1 β
2,2 h bis-Arg), 3.17 (t,
J = 6.5, 1H C
H2ε
2 β
2,2 h bis-Arg), 2.69 (d,
J = 16, 1H, C
H2α
1 β
2,2 h bis-Arg), 2.55 (d,
J = 16, 1H, C
H2α
2 β
2,2 h bis-Arg), 1.70–1.75 (m, 4H, C
H2γ β
2,2 h bis-Arg), 1.64-1.68 (m, 2H, C
H2δ
1 β
2,2 h bis-Arg), 1.55–1.61 (m, 2H, C
H2δ
2 β
2,2 h bis-Arg), 1.42 (s, 9H, C(C
H3)
3), 1.40 (s, 9H, C(C
H3)
3), 1.29 (s, 9H, C(C
H3)
3); MALDI-TOF: calcd for C
35H
61N
9O
2 655.5, calcd for C
35H
61N
9O
2Na 678.5, found 656.5 [M + H]
+, 678.5 [M + Na]
+, 694.5 [M + K]
+; HPLC (Water/ACN (0.1% TFA); 5% to 100% ACN in 30 min): tr = 19.29 min (
Figure 16).
Gdm-β2,2-h-bis-Arg-Tbt-OMe14: HMBA-AM resin (185 mg, 0.2 mmol) was washed five times with DMF, DCM. and DMF, then allowed to swell in DMF for 30 min. Fmoc-Tbt-OH (4 eq, 0.4 mmol, 238 mg) was dissolved in dry DCM. The solution was cooled to 0 °C, and DIC (4 eq, 0.4 mmol, 60 µL) was added. The reaction was stirred for 30 min and the solvent was then removed
in vacuo. The resulting anhydride was dissolved in DMF and added to the resin. A solution of DMAP (0.1 eq, 0.04 mmol, 5 mg) in DMF was added and the resin was shaken for 1 h before washing with DMF, DCM, and DMF (resin loading = 0.4 mmol/g). Removal of the Fmoc protecting group was achieved by treatment of the resin with 20% (
v:
v) piperidine in DMF 3 times for 5 min. The resin was washed five times with DMF. Fmoc-β
2,2 h bis-Orn(Boc)
2OH
2 (3 eq, 0.22 mmol, 141 mg) was dissolved in DMF (1.5 mL). HATU (1.8 eq, 0.21 mmol, 80 mg) and DIEA (4 eq, 0.88 mmol, 150 µL) were added. The solution was added to the resin (0.07 mmol, 185 mg) and the reaction was allowed to proceed at 50 °C for 16 h. The solution was removed by filtration and the resin was washed with DMF five times. The reaction being incomplete as revealed by a Kaiser test, the same coupling procedure was repeated a second time. The resin was then treated with a 20% solution of piperidine in DMF for 5 min 3 times. Boc removal was performed by treatment with a TFA/TIS/H
2O cocktail (95:2.5:2.5) for 1 h. The free amines were then reacted with 1,3-Di-Boc-2-(trifluoromethylsulfonyl)guanidine (5 eq, 0.35 mmol, 137 mg) and NEt
3 (10 eq, 0.7 mmol, 90 µL) in DMF overnight. The resin was filtrated and the Boc groups were removed with a TFA/TIS/H
2O cocktail (95:2.5:2.5) at rt for 3 h. After filtration, peptide
14 was cleaved from the resin using a mixture of MeOH/DMF/DIEA (5:5:1) for 16 h at 50 °C. The solution was filtrated, and solvents were evaporated. The crude product was purified by preparative RP-HPLC using a gradient of 40% to 90% MeCN in 30 min. After lyophilisation, peptide
14 was obtained as white powder with a purity of >95% (34 mg, 72% yield);
1H NMR (300 MHz, D
2O) δ 7.36 (s, 1H, C
H indole), 7.19 (s, 1H, C
H indole), 4.7 (m, 1H, C
Hα Tbt), 3.61 (s, 3H, CO
2C
H3), 3.53 (dd,
J = 15.3, 6.6, 1H, C
H2β
1 Tbt), 3.31–3.36 (m, 2H, C
H2β
2 Tbt and C
H2βε
1 β
2,2 h bis-Arg), 2.88 (t,
J = 7, 1H, C
H2δ
1 β
2,2 h bis-Arg), 2.85 (t,
J = 7, 1H, C
H2δ
2 β
2,2 h bis-Arg), 2.83 (d,
J = 14.5, 1H, C
H2βε
2 β
2,2 h bis-Arg), 1.42 (s, 9H, C(C
H3)
3), 1.39 (s, 9H, C(C
H3)
3), 1.27 (s, 9H, C(C
H3)
3), 1.14–1.33 (m, 8H, C
H2β and C
H2γ β
2,2 h bis-Arg); MALDI-TOF: calcd for C
36H
63N
11O
3 697.5, calcd for C
36H
63N
11O
3Na 720.5, found 698.5 [M + H]
+, 720.4 [M + Na]
+, 736.4 [M + K]
+; HPLC (Water/ACN (0.1% TFA); 30% to 70% ACN in 30 min): tr = 16.5 min (
Figure 17).
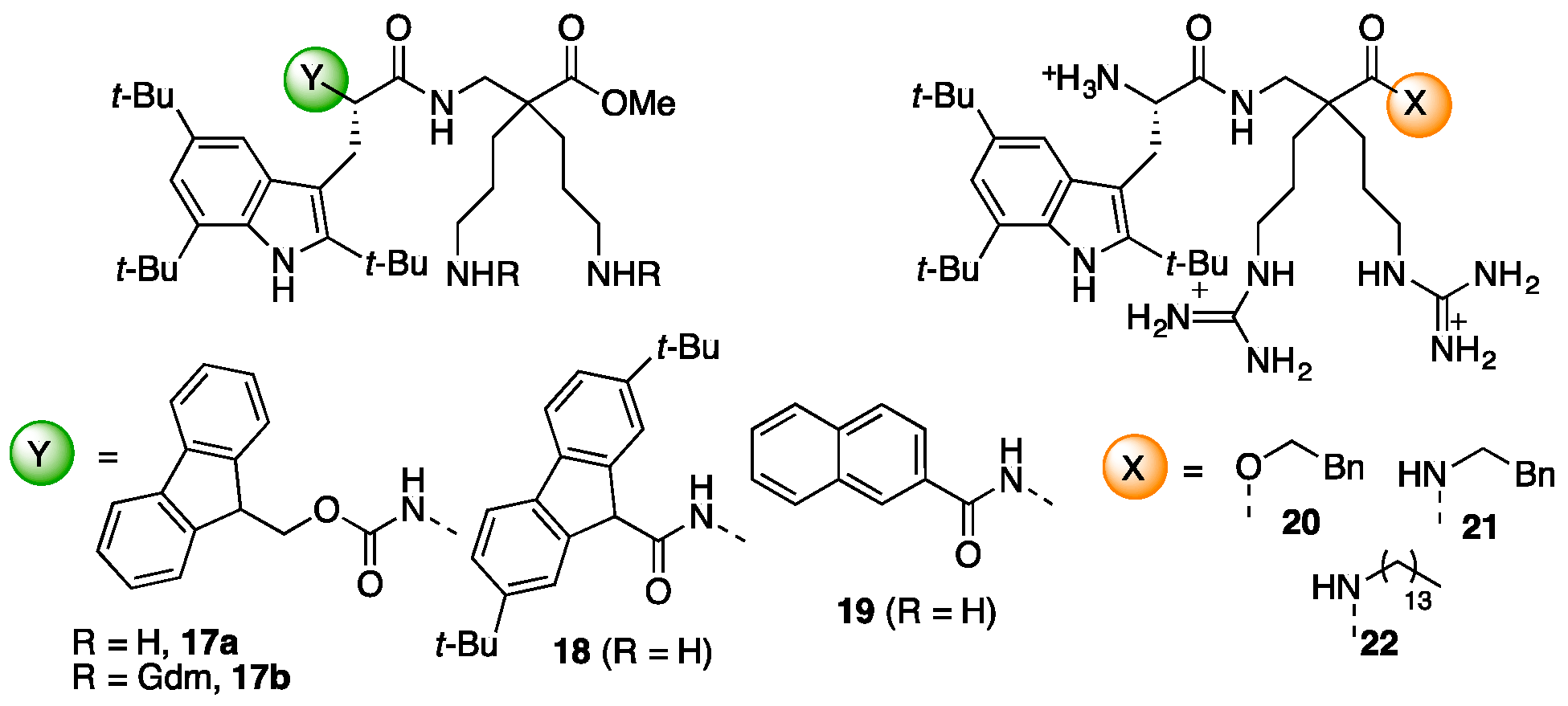
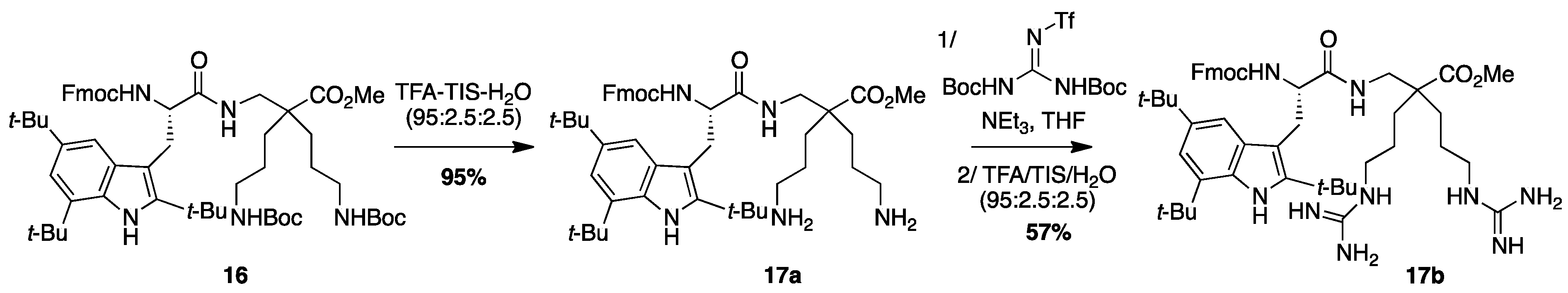
4.3.8. Synthesis of Fmoc-Tbt-β2,2-h-bis-Orn-OMe 17a and Fmoc-Tbt-β2,2-h-bis-Arg-OMe 17b (Scheme 11)
Fmoc-Tbt-
β2,2-h-bis-Orn-OMe17a: Compound
16 (42 mg, 0.042 mmol) was treated with a mixture of TFA/TIS/H
2O (95:2.5:2.5, V = 1mL) at rt for 3 h and then evaporated to dryness. The crude product was purified by preparative RP-HPLC using a gradient of 50% to 100% MeCN in 30 min. After lyophilisation compound
17a was obtained as white powder with purity >99% (27 mg, 95% yield);
1H NMR (300 MHz, CD
3OD) δ 7.80 (d,
J = 7.5 Hz, 2H, C
H Fmoc), 7.60 (t,
J = 8.8 Hz, 2H, C
H Fmoc), 7.37–7.42 (m, 5H, C
H indole and C
H Fmoc), 7.13 (d,
J = 1.4 Hz, 1H, C
H indole), 4.44 (dt,
J = 9.8 Hz, 7.7 Hz, 1H, C
H Fmoc), 4.17–4.24 (m, 3H, C
H2 Fmoc and C
Hα Tbt), 3.59 (d,
J = 14.2 Hz, 1H, C
H2βε
1 β
2,2 h bis-Arg), 3.41 (dd,
J = 14.6 Hz, 9 Hz, 1H, C
H2β
1 Tbt), 3.26 (dd,
J = 14.2 Hz, 5.8 Hz, 1H, C
H2β
2 Tbt), 2.88 (d,
J = 14.2 Hz, 1H, C
H2βε
2 β
2,2 h bis-Arg), 2.79–2.82 (m, 4H, C
H2δ β
2,2 h bis-Arg), 1.22–1.69 (m, 35H, C(C
H3)
3, C
H2β β
2,2 h bis-Arg and C
H2γ β
2,2 h bis-Arg);
MALDI-TOF: calcd for C
48H
67N
5O
5 793.5, found 794.5 [M + H]
+, 816.4 [M + Na]
+, 832.4 [M + K]
+;
HPLC (Water/ACN (0.1% TFA); 50% to 100% ACN in 10 min: tr = 6.29 min (
Figure 18).
Synthesis of Fmoc-Tbt-β2,2-h-bis-Arg-OMe 17b
Compound
17a (50 mg, 0.05 mmol) was dissolved 1 mL of THF. 1,3-Di-Boc-2-(trifluoromethylsulfonyl)guanidine (51 mg, 0.13 mmol) and NEt
3 (35 µL, 0.26 mmol) were added and the reaction mixture was stirred at rt for 24 h. After evaporation of THF, a solution of TFA/TIS/H
2O (95:2.5:2.5, 2 mL) was added and the mixture was stirred at rt for 45 min. The crude product was evaporated
in vacuo and purified by preparative RP-HPLC using a gradient of 30% to 100% MeCN in 30 min. After lyophilisation compound
17b was obtained as white powders with purity >99% (25 mg, 57% yield);
1H NMR (300 MHz, CD
3OD) δ 7.80 (d,
J = 7.5 Hz, 2H, C
H arom Fmoc), 7.60 (d,
J = 7.5 Hz, 2H, C
H arom Fmoc), 7.39 (t,
J = 7.5 Hz, 2H, C
H arom Fmoc), 7.33 (s,
1H, C
H arom indole), 7.27 (t,
J = 7.5 Hz, 2H, C
H arom Fmoc), 7.13 (s, 1H, C
H indole), 4.27–4.38 (m, 2H, C
H2 Fmoc), 4.18–4.22 (m, 2H, C
H Fmoc and C
Hα Tbt), 3.56 (d,
J = 11.8 Hz, 1H, C
H2βε
1 β
2,2 h bis-Arg), 3.40 (dd,
J = 14.6 Hz, 9.1 Hz, 1H, C
H2β
1 Tbt), 3.25 (dd,
J = 14.6 Hz, 5.5 Hz, 1H, C
H2β
2 Tbt), 3–3.07 (m,
4H, C
H2δ β
2,2 h bis-Arg), 2.72 (d,
J = 14.3 Hz, 2H, C
H2βε
2 β
2,2 h bis-Arg), 1.34–1.53 (m, 35H, C(C
H3)
3, C
H2β β
2,2 h bis-Arg and C
H2γ β
2,2 h bis-Arg); MALDI-TOF: calcd for C
50H
71N
9O
5 877.6, found 878.4 [M + H]
+, 916.4 [M + K]
+; HPLC (Water/ACN (0.1% TFA); 5% to 100% ACN in 30 min: tr = 24.59 min (
Figure 19).
4.3.9. Synthesis of Fluo-Tbt-β2,2 h bis-Orn-OMe 18 (Scheme 12)
Compound
16 (43 mg, 0.043 mmol) was dissolved in THF (1 mL) and treated with DBU (0.2 µL, 0.0013 mmol) and octanethiol (75 µL, 0.43 mmol) at rt for 10 min and then evaporated to dryness. The crude compound was purified by flash chromatography (DCM/MeOH/NEt
3, 100:0:0 to 80:20:1) affording a white powder (17 mg, 99% yield). The product was dissolved in 2 mL of THF. 2,7-di-
tert-butylfluorène-9-carboxylic acid (17 mg, 0.05 mmol), HBTU (16 mg, 0.043 mmol), and DIEA (75 µL, 0.43 mmol) were added and the reaction mixture was stirred at rt overnight. After evaporation of THF, a solution of TFA/TIS/H
2O (95:2.5:2.5, 2 mL) was added and the mixture was stirred at rt for 45 min and then evaporated. The crude product was purified by preparative RP-HPLC using a gradient of 50% to 100% MeCN in 30 min. After lyophilisation compound
18 was obtained as white powder with purity >99% (24 mg, 65% yield);
1H NMR (300 MHz, CD
3OD) δ 7.79 (s, C
H arom fluorenyl), 7.71 (d,
J = 8 Hz, 2H, C
H arom fluorenyl), 7.59 (s, C
H arom fluorenyl), 7.49 (d,
J = 8 Hz, 2H, C
H arom fluorenyl), 7.36 (s, 1H, C
H indole), 7.14 (s, 1H, C
H indole), 4.38 (dd,
J = 10 Hz, 4 Hz, 1H, C
Hα Tbt), 3.65 (d,
J = 14 Hz, 1H, C
H2βε
1 β
2,2 h bis-Arg), 3.56 (dd,
J = 14.8 Hz, 10.5 Hz, 1H, C
H2β
1 Tbt), 3.39 (dd,
J = 14.8 Hz, 4 Hz, 1H, C
H2β
2 Tbt), 2.65 (d,
J = 14.3 Hz, 2H, C
H2βε
2 β
2,2 h bis-Arg), 2.52–2.66 (m,
2H, C
H2δ
1 β
2,2 h bis-Arg), 2.38–2.45 (m,
2H, C
H2δ β
2,2 h bis-Arg), 1.30–1.53 (m, 35H, C(C
H3)
3, C
H2β β
2,2 h bis-Arg and C
H2γ β
2,2 h bis-Arg); MALDI-TOF: calcd for C
55H
81N
5O
4 875.6, found 876.6 [M + H]
+, 898.6 [M + Na]
+, 914.6 [M + K]
+; HPLC (Water/ACN (0.1% TFA); 45% to 100% ACN in 30 min: tr = 21.9 min (
Figure 20).
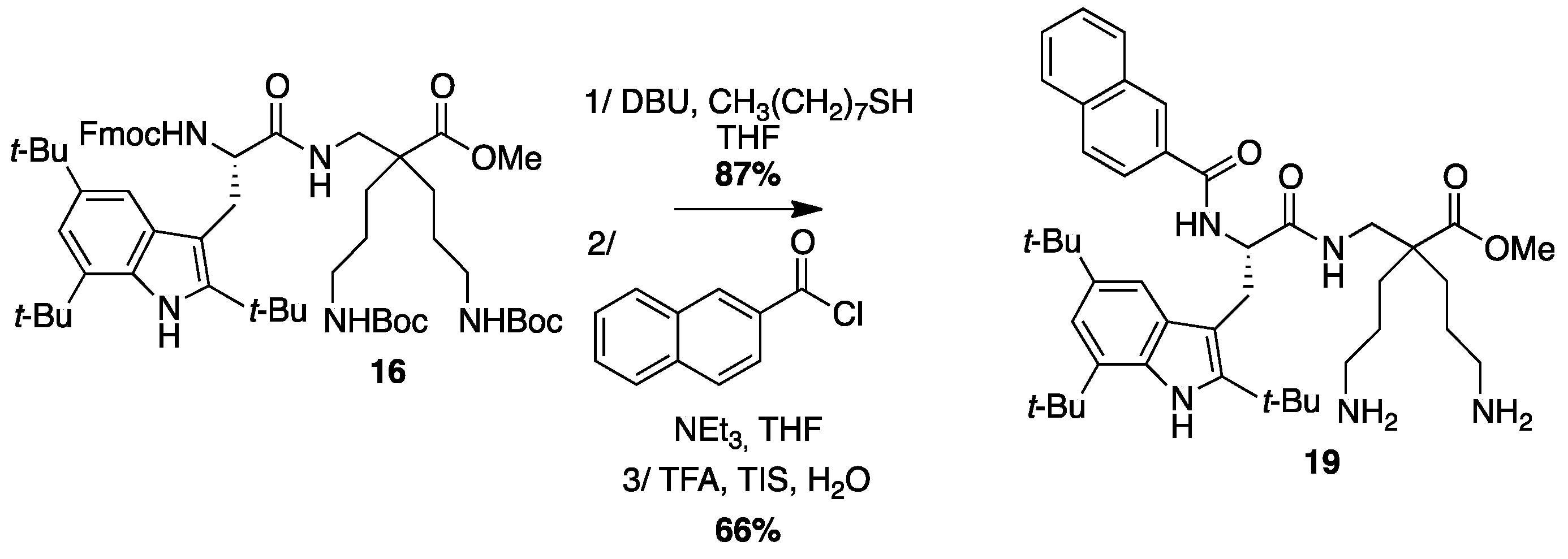
4.3.10. Synthesis of Np-Tbt-β2,2 h bis-Orn-OMe 19 (Scheme 13)
Compound
16 (50 mg, 0.05 mmol) was dissolved in THF (1 mL) and treated with DBU (0.3 µL, 0.002 mmol) and octanethiol (90 µL, 0.5 mmol) at rt for 10 min and then evaporated to dryness. The crude compound was purified by flash chromatography (DCM/MeOH/NEt
3, 100:0:0 to 80:20:1) affording a white powder (34 mg, 87% yield). This compound was dissolved in 4 mL of THF. 2-Naphtoyl chloride (9.5 mg, 0.05 mmol) and NEt
3 (14 µL, 0.1 mmol) were added and the reaction mixture was stirred at rt overnight. After evaporation of THF, a solution of TFA/TIS/H
2O (95:2.5:2.5, 2 mL) was added and the mixture was stirred at rt for 30 min and then evaporated. The crude product was purified by preparative RP-HPLC using a gradient of 30% to 100% MeCN in 30 min. After lyophilisation compound
19 was obtained as white powder with 96% purity (22 mg, 60% yield);
1H NMR (300 MHz, CD
3OD) δ 8.06 (s, 1H, C
H Np), 7.91 (d,
J = 8.4 Hz, 2H, C
H Np), 7.86 (d,
J = 7.8 Hz, 1H, C
H Np), 7.75 (d,
J = 8.4 Hz, 1H, C
H Np), 7.60 (t,
J = 6.4 Hz, 1H, C
H Np), 7.56 (t,
J = 6.4 Hz, 1H, C
H Np), 7.41 (s, 1H, C
H indole), 7.18 (s, 1H, C
H indole), 4.57 (t,
J = 7.5 Hz, 1H,
CHα Tbt), 3.69 (d,
J = 14.2, 2H, C
H2βε
1 β
2,2 h bis-Arg), 3.63 (dd,
J = 14.7 Hz, 8.3 Hz, 1H, C
H2β
1 Tbt), 3.48 (dd,
J = 14.7 Hz, 6.6 Hz, 1H, C
H2β
2 Tbt), 2.99 (d,
J = 14.2, 2H, C
H2βε
1 β
2,2 h bis-Arg), 2.78–2.88 (m, 4H, C
H2δ β
2,2 h bis-Arg), 1.18–1.37 (m, 35H, C(C
H3)
3, C
H2β β
2,2 h bis-Arg and C
H2γ β
2,2 h bis-Arg); MALDI-TOF: calcd for C
44H
65N
5O
5 725.5, found 726.4 [M + H]
+, 748.4 [M + K]
+, 764.4 [M + K]
+; HPLC (Water/ACN (0.1% TFA); 5% to 100% ACN in 30 min: tr = 21.9 min (
Figure 21).
4.3.11. Synthesis of Tbt-β2,2 h bis-Arg-OBn 20 (Scheme 14)
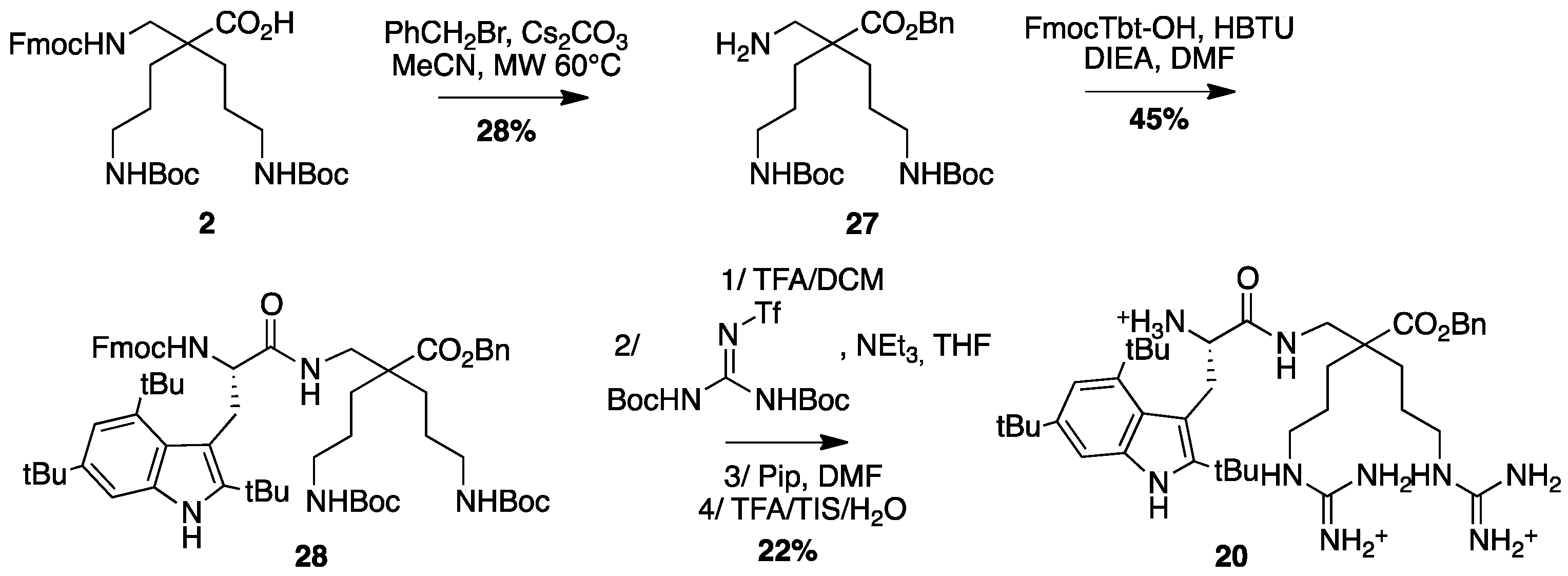
β2,2-h-bis-Orn(Boc)2OBn27: Fmoc β2,2-h-bis-Orn(Boc)2OH 2 (300 mg, 0.48 mmol) was dissolved in MeCN (1.7 mL). After addition of Cs2CO3 (188 mg, 0.58 mmol) and benzyl bromide (63 µL, 0.53 mmol), the reaction mixture was heated at 60 °C under microwave (150W) for 10 min. The solution was filtered and evaporated to dryness. The crude compound was dissolved in AcOEt and washed with an aqueous solution of NaHCO3 5% followed by a solution of citric acid 5%, then dried over MgSO4, filtered, and concentrated in vacuo. The crude compound was purified by flash chromatography (DCM/MeOH/NEt3 100:0:0.1 to 95:5:0.1) to afford a white powder (80 mg, 28% yield); 1H NMR (300 MHz, CD3OD) δ 7.34–7.43 (m, 5H, CH Ar), 5.17 (s, 2H, CH2Ph), 3.01 (t, J = 6.8 Hz, 4H, CH2δ), 2.84 (s, 2H, CH2βε), 1.62 (dd, J = 9.3 Hz, 5.3 Hz, 4H, CH2β), 1.43 (s, 18H, C(CH3)3), 1.24–1.41 (m, 4H, CH2γ); 13C NMR (75 MHz, CD3OD) δ 176.9 (C, C=O ester), 158.5 (2C, C=O carbamate), 137.5 (C, C Ar), 129.6, 129.4, 129.3 (3CH, CH Ar), 79.9 (2C, C(CH3)3), 67.5 (CH2, CH2Ph), 51.3 (CH2, CH2βε), 45.5 (C, Cα), 41.5 (2CH2, CH2δ), 31.1 (2CH2, CH2β), 28.8 (6CH3, C(CH3)3), 25.3 (2CH2, CH2γ); HRMS-ESI+: calcd for C26H43N3O6 493.3152, found 494.3225 [M + H]+.
Fmoc-Tbt-β2,2-h-bis-Orn(Boc)2OBn28: Fmoc-Tbt-OH (83 mg, 0.14 mmol) was dissolved in DMF (6 mL). HBTU (53 mg, 0.14 mmol) and DIEA (24 µL, 0.14 mmol) were added and the mixture was stirred for 5 min before addition of H-β2,2-h-bis-Orn(Boc)2OBn 28 (70 mg, 0.14 mmol). The reaction mixture was stirred at room temperature overnight, then diluted with Et2O and washed with an aqueous saturated solution of NH4Cl. The organic layer was dried over MgSO4, filtered, and evaporated to dryness. The crude compound was purified by flash chromatography (Cy/AcOEt, 100:0 to 70:30) to afford the pure protected dipeptide as a white powder (67 mg, 45% yield). 1H NMR (300 MHz, MeOD) δ 7.91 (s, 1H, NH indole), 7.66 (d, J = 7.5, 1H, CH Ar Fmoc), 7.45 (d, J = 7.2, 1H, CH Ar Fmoc), 7.37 (s, 1H, CH Ar indole), 7.3 (t, J = 7.5, 1H, CH Ar Fmoc), 7.18 (dt, J = 14.7 and 6.9, 1H, CH Ar Fmoc), 7.09 (s, 1H, CH indole), 5.89 (bs, 2H, NH Boc), 5.71 (bs, 1H, NH Fmoc), 4.87 (s, 2H, CH2Ph), 4.20–4.34 (m, 3H, CHα Tbt and CH2 Fmoc), 4.09 (t, J = 7.2, 1H, CH Fmoc), 3.37 (d, J = 14.1, 1H, CH2βε1 β2,2 h bis-Orn), 3.31 (d, J = 7.5 Hz, 2H, CH2β Tbt), 2.86 (m, 4H, CH2δ β2,2 h bis-Orn), 2.55 (d, J = 14.1, 1H, CH2βε2 β2,2 h bis-Orn), 1.52 (s, 9H, C(CH3)3 indole), 1.47 (s, 9H, C(CH3)3 indole), 1.36–1.44 (m, 35H, C(CH3)3 indole, C(CH3)3 Boc, CH2β β2,2 h bis-Orn and CH2γ β2,2 h bis-Orn).
H-Tbt-β2,2 h bis-Arg-OBn20: Compound 28 (68 mg, 0.064 mmol) was dissolved in DCM (∼0.4 M) and an equivalent volume of TFA. The mixture was stirred at rt for 1.5 h then evaporated to dryness. The crude compound was dissolved in 4 mL of THF.
1,3-Di-Boc-2-(trifluoromethylsulfonyl)guanidine (50 mg, 0.128 mmol) and NEt
3 (500 µL, 3.2 mmol) were added and the reaction mixture was stirred at rt for 16 h. After evaporation of THF, the crude mixture was dissolved in a 20% solution of piperidine in DCM and allowed to react for 2 h before evaporation to dryness. A solution of TFA/TIS (95:5) was added and the mixture was stirred at rt for 1.5 h. After evaporation, the crude product was purified by preparative RP-HPLC using a gradient of 20% to 90% MeCN in 30 min. After lyophilisation compound
20 was obtained as white powder with purity >99% (10 mg, 22% yield);
1H NMR (300 MHz, CD
3OD) δ 8.36 (s, 1H, N
H indole), 7.20–7.32 (m, 5H, C
H Ph), 7.28 (s, 1H, C
H indole), 7.15 (s, 1H, C
H indole), 5.03 (d,
J = 12 Hz, 1H, C
H2 Ph), 5.01 (d,
J = 12 Hz, 1H, C
H2 Ph), 4.03 (t,
J = 6.1 Hz, 1H,
CHα Tbt), 3.60 (d,
J = 14.2, 1H, C
H2βε
1 β
2,2 h bis-Arg), 3.42 (d,
J = 6.6 Hz, 2H, C
H2β Tbt), 2.97–3 (m, 4H, C
H2δ β
2,2 h bis-Arg), 2.33 (d,
J = 1.24 Hz, 1H, C
H2βε
2 β
2,2 h bis-Arg), 1.20–1.59 (m, 35H, C(C
H3)
3, C
H2β β
2,2 h bis-Arg and C
H2γ β
2,2 h bis-Arg); MALDI-TOF: calcd for C
48H
67N
5O
5 731.5, found 732.4 [M + H]
+, 770.3 [M + K]
+, 716.3 [M + H − NH
3]
+; HPLC (Water/ACN (0.1% TFA); 40% to 90% ACN in 10 min: tr = 7.51 min (
Figure 22).
4.3.12. Synthesis of Tbt-β2,2 h bis-Arg-NHBn 21 (Scheme 15)
β2,2-h-bis-Orn(Boc)2NHBn29: Fmoc β2,2-h-bis-Orn(Boc)2OH 2 (300 mg, 0.48 mmol) was dissolved in DCM (20 mL). DCC (100 mg, 0.48 mmol), HOBt (64 mg, 0.48 mmol), DMAP (5 mg, 0.05 mmol), and benzyl amine (56 mg, 0.53 mmol) were added. The reaction mixture was stirred at rt overnight. The solution was washed with an aqueous saturated solution of NaCl, dried over MgSO4, filtered, and concentrated in vacuo. The crude compound was purified by flash chromatography (Cy/AcOEt 100:0 to 50:50) to afford a colorless oil (251 mg, 73% yield); Rf (Cy/AcOEt, 7:3) = 0.5; 1H NMR (300 MHz, CDCl3) δ 7.76 (d, J = 7.5 Hz, 2H, CH Ar), 7.58 (d, J = 7.5 Hz, 2H, CH Ar), 7.40 (t, J = 7.5 Hz, 2H, CH Ar), 7.25–7.33 (m, 7H, CH Ar), 6.48 (bs, 1H, NH amide), 5.47 (bs, 1H, NH Fmoc), 4.78 (bs, 2H, NH Boc), 4.36–4.42 (m, 4H, CH2Ph, CH2 Fmoc), 4.15–4.20 (m, 1H, CH Fmoc), 3.36–3.38 (m, 2H, CH2βε), 2.98–3.04 (m, 4H, CH2δ), 1.31–1.69 (m, 26H, CH2β, C(CH3)3, CH2γ); 13C NMR (75 MHz, CD3OD) δ 175.3 (C, C=O amide), 157.3 (C, C=O carbamate), 156.3 (C, C=O carbamate), 143.9, 141.4, 138.3 (3C, C Ar), 128.8, 127.8, 127.6, 127.1, 125.1, 120.1 (6CH, CH Ar), 79.2 (C, C(CH3)3), 66.9 (CH2, CH2 Fmoc), 49.8 (C, Cα), 47.3 (CH, CH Fmoc), 44.6 (CH2, CH2βε), 43.8 (CH2, CH2Ph), 40.7 (CH2, CH2δ), 30.8 (CH2, CH2β), 29.0 (CH3, C(CH3)3), 24.2 (CH2, CH2γ); Fmoc β2,2-h-bis-Orn(Boc)2NHBn (251 mg, 0.35 mmol) was dissolved in THF (6 mL). Octanethiol (600 µL, 3.5 mmol) and DBU (1.5 µL, 0.01 mmol) were added. The reaction mixture was stirred for 15 min then concentrated in vacuo. The crude compound was purified by flash chromatography (DCM/MeOH/NEt3 100:0:0 to 80:20:0.1) to afford 29 as a colorless oil (150 mg, 87% yield); 1H NMR (300 MHz, CDCl3) δ 8.94 (bs, 2H, NH2), 7.15–7.24 (m, 5H, CH Ar), 4.75 (bs, 2H, NH Boc), 4.33–4.35 (m, 2H, CH2Ph), 2.98–3.11 (m, 4H, CH2δ), 2.80 (s, 2H, CH2βε), 1.01–1.73 (m, 26H, CH2β, C(CH3)3, CH2γ); 13C NMR (75 MHz, CD3OD) δ 176.3 (C, C=O amide), 156.1 (C, C=O carbamate), 139 (C, C Ar), 128.5, 127.3, 127 (3CH, CH Ar), 78.9 (C, C(CH3)3), 47.3 (C, Cα), 45.3 (CH2, CH2βε), 42.9 (CH2, CH2Ph), 40.8 (CH2, CH2δ), 31.6 (CH2, CH2β), 28.4 (CH3, C(CH3)3), 22.4 (CH2, CH2γ); HRMS-ESI+: calcd for C26H44N4O5 492.3312, found 515.3199 [M + Na]+.
Fmoc-Tbt-β2,2 h bis-Orn(Boc)2NHBn30: Fmoc-Tbt-OH (90 mg, 0.15 mmol) was dissolved in DMF (6 mL). HBTU (57 mg, 0.15 mmol) and DIEA (30 µL, 0.15 mmol) were added and the mixture was stirred for 5 min before addition of H-β2,2h bis-Orn(Boc)2NHBn 29 (75 mg, 0.15 mmol). The reaction mixture was stirred at room temperature overnight, then diluted with Et2O and washed with an aqueous saturated solution of NH4Cl. The organic layer was dried over MgSO4, filtered, and evaporated to dryness. The crude compound was purified by flash chromatography (Cy/AcOEt, 100:0 to 60:40) to afford the pure protected dipeptide 30 as a white powder (64 mg, 40% yield). 1H NMR (300 MHz, MeOD) δ 7.91 (s, 1H, NH indole), 7.65 (d, J = 7.5, 2H, CH Ar Fmoc), 7.40 (d, J = 7.4, 2H, CH Ar Fmoc), 7.35 (s, 1H, CH Ar indole), 7.28 (t, J = 7.4, 2H, CH Ar Fmoc), 7.03–7.20 (m, 8H, CH Ar Fmoc, CH indole, CH benzyl), 6.35 and 6.19 (2 bs, 1H, NH Amide), 5.61 (bs, 1H, NH Fmoc), 4.69 and 4.76 (2bs, 2H, NH Boc), 4.15–4.27 (m, 5H, CHα Tbt, CH2 Fmoc, CH2Ph), 4.08 (t, J = 76.8, 1H, CH Fmoc), 3.24–3.39 (m, 3H, CH2β1 Tbt, CH2βε β2,2 h bis-Orn), 2.84–2.94 (m, 4H, CH2δ β2,2 h bis-Orn), 2.67–2.74 (m, 1H, CH2β2 Tbt), 1.30–1.34 (m, 53H, C(CH3)3 indole, C(CH3)3 Boc, CH2β β2,2 h bis-Orn and CH2γ β2,2 h bis-Orn) 13C NMR (75 MHz, CD3OD) δ 175.1 and 172.4 (C, C=O amide), 156.2 (C, C=O carbamate), 143.9, 143.6, 142.8, 142.6, 141.3, 141.2, 138.3, 132.1, 130.2, 129.8 (10C, C Ar), 128.7, 127.7, 127.4, 127.1, 125.3, 125.2, 120, 116.8, 112.1 (9CH, CH Ar), 104.5 (C, C indolyl), 79.1 (C, C(CH3)3), 67.2 (CH2, CH2 Fmoc), 56.7 (CH, CHα Tβτ), 49.2 (C, Cα β2,2 h bis-Orn), 47.1 (CH, CH Fμοχ), 43.6 (CH2, CH2Ph), 42.9 (CH2, CH2β Tbt), 40.7 (2CH2, CH2δ β2,2 h bis-Orn), 34.9, 34.7, 33.1 (3CH2, CH2β ανδ CH2β’ β2,2 h bis-Orn), 32.2, 30.9, 30.8, 30.7 (4CH3, C(CH3)3), 24.2, 24.3 (2CH2, CH2γ).
H-Tbt-
β2,2 h bis-Arg-NHBn21: Compound
30 (64 mg, 0.06 mmol) was dissolved in DCM (∼0.4 M) and an equivalent volume of TFA containing 5% of TIS. The mixture was stirred at rt for 2 h then evaporated to dryness. The crude compound was dissolved in 4 mL of THF. 1,3-Di-Boc-2-(trifluoromethylsulfonyl)guanidine (117 mg, 0.3 mmol) and NEt
3 (80 µL, 0.6 mmol) were added and the reaction mixture was stirred at rt for 48 h. After evaporation of THF, the crude mixture was dissolved in a 20% solution of piperidine in DCM and allowed to react for 2 h before evaporation to dryness. A solution of TFA/TIS (95:5) in DCM was added and the mixture was stirred at rt for 1.5 h. After evaporation, the crude product was purified by preparative RP-HPLC using a gradient of 40% to 100% MeCN in 30 min. After lyophilisation, compound
21 was obtained as white powder with purity >99% (20 mg, 45% yield);
1H NMR (300 MHz, CD
3OD) δ 7.32 (s, 1H, C
H indole), 7.25–7.32 (m, 5H, C
H arom), 7.17 (s, 1H, C
H indole), 4.28 (s, 2H, C
H2Ph), 4.06 (dd,
J = 9.4 Hz, 6.1 Hz, 1H,
CHα Tbt), 3.44–3.56 (m, 3H, C
H2βε
1 β
2,2 h bis-Arg and C
H2β Tbt), 3.11–3.17 (m, 4H, C
H2δ β
2,2 h bis-Arg), 2.39 (d,
J = 14 Hz, 1H, C
H2βε
2 β
2,2 h bis-Arg), 1.22–1.54 (m, 35H, C(C
H3)
3, C
H2β β
2,2 h bis-Arg and C
H2γ β
2,2 h bis-Arg); MALDI-TOF: calcd for C
41H
66N
10O
2 730.5, found 731.6 [M + H]
+; HPLC (Water/ACN (0.1% TFA); 40% to 100% ACN in 30 min: tr = 13.57 min (
Figure 23).
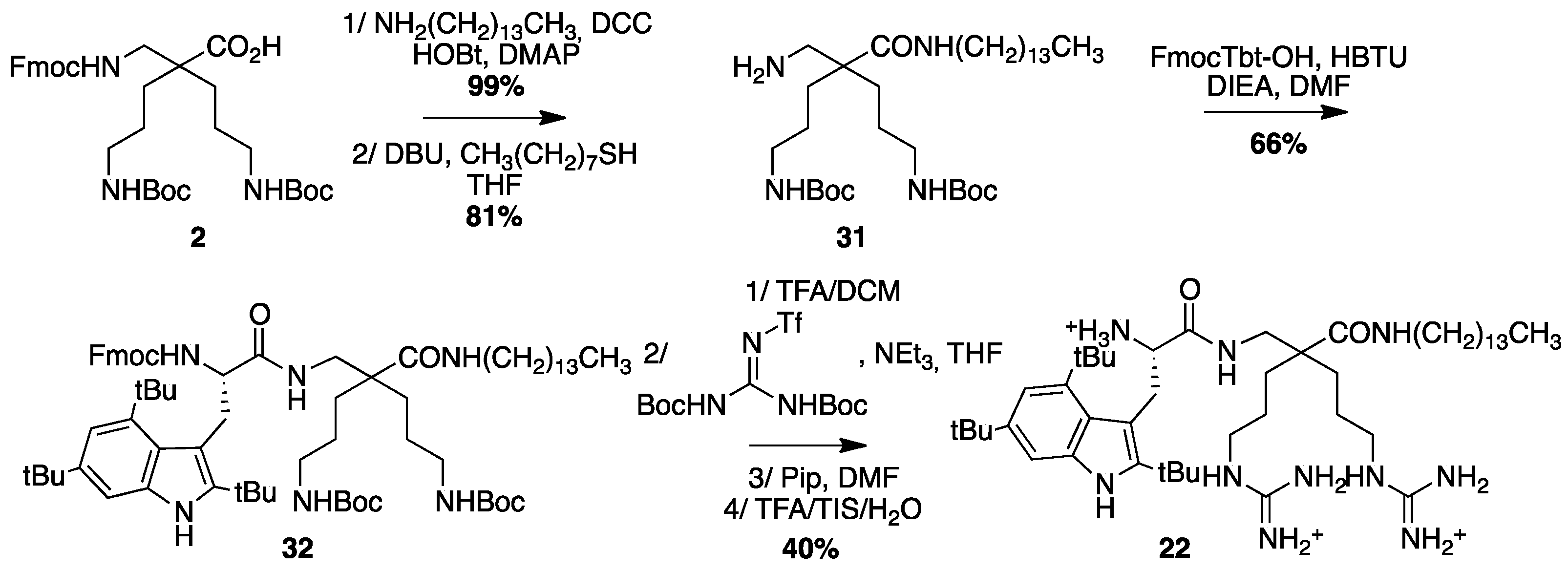
4.3.13. Synthesis of Tbt-β2,2 h bis-Arg-NH(CH2)13CH3 22 (Scheme 16)
β2,2-h-bis-Orn(Boc)2NH(CH2)13CH331: Fmoc β2,2h bis-Orn(Boc)2OH 2 (300 mg, 0.48 mmol) was dissolved in DCM (25 mL). DCC (109 mg, 0.53 mmol), HOBt (72 mg, 0.53 mmol), DMAP (5 mg, 0.05 mmol), and tetradecyl amine (113 mg, 0.53 mmol) were added. The reaction mixture was stirred at for 4 h. The solution was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The crude compound was purified by flash chromatography (Cy/AcOEt 100:0 to 50:50) to afford a colorless oil (390 mg, 99% yield); Rf (Cy/AcOEt, 1:1) = 0.76; 1H NMR (300 MHz, CDCl3) δ 7.67 (d, J = 7.3 Hz, 2H, CH Ar), 7.51 (d, J = 7.3 Hz, 2H, CH Ar), 7.31 (t, J = 7.3 Hz, 2H, CH Ar), 7.22 (t, J = 7.3 Hz, 2H, CH Ar), 6.05 (bs, 1H, NH amide), 5.65 and 5.47 (2bs, 1H, NH Fmoc), 4.80 (bs, 2H, NH Boc), 4.30 (d, J = 6.2 Hz, 2H, CH2 Fmoc), 4.11 (t, J = 6.9 Hz, 1H, CH Fmoc), 3.28 (d, J = 6.1 Hz, 1H, CH2βε β2,2 h bis-Orn), 3.13 (m, 2H, CH2C13H27), 3 (m, 4H, CH2δ β2,2 h bis-Orn), 1.41 (m, 4H, CH2γ β2,2 h bis-Orn), 1.17-1.35 (m, 46H, C(CH3)3 Boc, (CH2)12CH3 and CH2β β2,2 h bis-Orn), 0.8 (t, 3H, (CH2)12CH3); 13C NMR (75 MHz, CD3OD) δ 175.2 (C, C=O amide), 157.3 and 156.3 (2C, C=O carbamate), 143.9 and 141.3 (C, C arom Fmoc), 127.7, 127.1, 125.1, 120 (5CH, CH arom Fmoc), 79.1 (C, C(CH3)3), 66.9 (CH2, CH2 Fmoc), 49.6 (C, Cα β2,2 h bis-Orn), 47.2 (C, CH Fmoc), 44.6 (CH2, CH2βε β2,2 h bis-Orn), 40.7 (CH2, CH2C13H27), 39.8 (CH2, CH2δ β2,2 h bis-Orn), 33.9 (CH2, CH2C12H25), 31.9 (CH2, CH2β β2,2 h bis-Orn), 30.8 (CH2, CH2C11H23), 29.72, 29.68, 29.62, 29.59, 29.39, 29.33 (CH2, CH2C7H17), 28.4 (CH3, C(CH3)3), 27.1, 27.0, 24.2 (CH2, (CH2)3CH3), 22.7 (CH2, CH2γ β2,2 h bis-Orn), 14.2 (CH3, (CH2)13CH3); HRMS-ESI+: calcd for C48H76N4O7 820.5714, found 843.5606 [M + Na]+. The obtained Fmoc β2,2-h-bis-Orn(Boc)22NH(CH2)13CH3 (130 mg, 0.12 mmol) was dissolved in THF (2 mL). Octanethiol (210 µL, 1.2 mmol) and DBU (0.5 µL, 0.0036 mmol) were added. The reaction mixture was stirred for 20 min then concentrated in vacuo. The crude compound was purified by flash chromatography (DCM/MeOH/NEt3 100:0:0.1 to 80:20:0.1) to afford a colorless oil (58 mg, 81% yield); 1H NMR (300 MHz, CD3OD) δ 3.21 (t, J = 7.2 Hz, 2H, CH2C12H27), 33.04 (t, J = 6.9 Hz, 4H, CH2δ β2,2 h bis-Orn), δ 2.78 (s, 2H, CH2β’ β2,2 h bis-Orn), 1.32-1.58 (m, 48H, C(CH3)3 Boc, CH2(CH2)11CH3, CH2β β2,2 h bis-Orn and CH2γ β2,2 h bis-Orn), 0.93 (t, 3H, J = 6.6 Hz, (CH2)13CH3); 13C NMR (75 MHz, CD3OD) δ 178.2 (C, C=O amide), 159.4 (C, C=O carbamate), 79.8 (C, C(CH3)3), 50.2 (C, Cα), 45.8 (CH2, CH2β’), 41.7 (CH2, CH2δ), 40.4 (CH2, CH2(CH2)12CH3), 33.1, 31.6, 30.8, 30.5 (4CH2), 28.8, (6CH3, C(CH3)3), 28.2, 25.3, 23.7 (CH2), 14.5 (CH3, CH2CH3); HRMS-ESI+: calcd for C33H66N4O5 598.5033, found 599.5112 [M + H]+.
Fmoc-Tbt-β2,2-h-bis-Orn(Boc)2NH(CH2)13CH332: Fmoc-Tbt-OH (54 mg, 0.09 mmol) was dissolved in DMF (5 mL). HBTU (34 mg, 0.09 mmol) and DIEA (20 µL, 0.09 mmol) were added and the mixture was stirred for 5 min before addition of β2,2-h-bis-Orn(Boc)2NH(CH2)13CH3 31 (55 mg, 0.09 mmol). The reaction mixture was stirred at room temperature for 4 h, then diluted with Et2O and washed with an aqueous saturated solution of NH4Cl. The organic layer was dried over MgSO4, filtered, and evaporated to dryness. The crude compound was purified by flash chromatography (Cy/AcOEt, 100:0 to 50:50) to afford the pure protected dipeptide as a white powder (70 mg, 66% yield). 1H NMR (300 MHz, CDCl3) δ 7.99 (s, 1H, NH indole), 7.23 (d, J = 7.5, 2H, CH Ar Fmoc), 7.50 (d, J = 7.2, 2H, CH Ar Fmoc), 7.43 (s, 1H, CH Ar indole), 7.38 (t, J = 7.5, 2H, CH Ar Fmoc), 7.24 (t, J = 7.5, 2H, CH Ar Fmoc), 7.17 (s, 1H, CH indole), 6.23 (bs, 1H, NH amide), 5.92 and 5.66 (2bs, 1H, NH Fmoc), 4.78 and 4.86 (2bs, 2H, NH Boc), 4.25–4.37 (m, 3H, CHα Tbt and CH2 Fmoc), 4.13–4.17 (m, 1H, CH Fmoc), 3.33–3.44 (m, 3H, CH2βε β2,2 h bis-Orn and CH2β1 Tbt), 3.02 (m, 6H, CH2δ β2,2 h bis-Orn and CH2(CH2)12CH3), 2.75–2.80 (m, 1H, CH2β2 Tβτ), 1.30–1.47 (m, 77H, 3C(CH3)3 indole, CH2(CH2)12CH3, 2C(CH3)3 Boc, 2CH2β β2,2 h bis-Orn and 2CH2γβ2,2h bis-Orn), 0.86 (t, J = 6.9, 3H, (CH2)13CH3) 13C NMR (75 MHz, CDCl3) δ 174.8 and 172.3 (C, C=O amide), 156.1 and 156.2 (C, C=O carbamate), 143.9, 143.6, 142.8, 142.6, 141.3, 141.2, 132.1, 130.2, 129.8 (9C, C Ar), 127.7, 127.1, 125.3, 125.2, 120, 116.8, 112.1 (7CH, CH Ar), 104.5 (C, C indolyl), 79.1 (C, C(CH3)3), 67.2 (CH2, CH2 Fmoc), 56.7 (CH, CHα Tβτ), 49.2 (C, Cα β2,2 h bis-Orn), 47.1 (CH, CH Fμοχ), 43 (CH2, CH2β Tbt), 40.7 (CH2, CH2C13H29, 39.7 (2CH2, CH2δ β2,2 h bis-Orn), 34.9 (CH2, CH2C12H27), 34.8 (CH2, CH2C11H25), 33.1 (CH2, CH2C10H23), 32.1 (CH3, C(CH3)3), 32 (CH2, CH2C9H21), 30.6 and 30.9 (2CH3, C(CH3)3), 29.8, 29.7, 29.6, 29.5, 29.4, 29.3 (7CH2, (CH2)8CH3), 28.5 (2CH3, C(CH3)3 Boc), 27 and 26.9 (2CH2, CH2β β2,2 h bis-Orn), 24.2 (CH2, CH2βε β2,2 h bis-Orn), 22.7 (2CH2, CH2γ), 14.2 (CH3, (CH2)13CH3).
H-Tbt-
β2,2h bis-Arg-NH(CH2)13CH322: Compound
32 (52 mg, 0.045 mmol) was dissolved in DCM (3 mL) and a mixture of TFA/TIS/H
2O (3 mL/150 µL/150 µL) as added. The solution was stirred at rt for 4 h then evaporated to dryness. The crude compound was dissolved in 4 mL of THF. 1,3-Di-Boc-2-(trifluoromethylsulfonyl)guanidine (53 mg, 0.135 mmol) and NEt
3 (40 µL, 0.27 mmol) were added and the reaction mixture was stirred at rt overnight. After evaporation of THF, the crude mixture was dissolved in a 20% solution of piperidine in DCM and allowed to react for 2 h before evaporation to dryness. A solution of TFA/TIS/H
2O (95:2.5:2.5) in DCM was added and the mixture was stirred at rt for 3 h. After evaporation, the crude product was purified by preparative RP-HPLC using a gradient of 40% to 90% MeCN in 30 min. After lyophilisation, compound
22 was obtained as white powder with purity >99% (12 mg, 40%);
1H NMR (300 MHz, CD
3OD) δ 8.33 (s, 1H, N
H indole), 7.60 (t,
J = 5.6 Hz, 1H, N
H amide), 7.30 (d,
J = 1.5 Hz, 1H, C
H indole), 7.15 (d,
J = 1.5 Hz, 1H, C
H indole), 4.06 (dd,
J = 10.1 Hz, 5.6 Hz,
CHα Tbt), 3.34–3.55 (m, 3H, C
H2βε
1 β
2,2 h bis-Arg and C
H2β Tbt), 3.11-3.17 (m, 2H, C
H2C
13H
29), 3.01–3.09 (m, 4H, C
H2δ β
2,2 h bis-Arg), 2.37 (d,
J = 14.1 Hz, 1H, C
H2βε
2 β
2,2 h bis-Arg), 1.68–1.84 (m, 2H, C
H2C
12H
27), 1.16–1.62 (m, 57H, 3C(C
H3)
3, (C
H2)
11CH
3, C
H2β β
2,2 h bis-Arg and C
H2γ β
2,2 h bis-Arg), 0.90 (t,
J = 6.7, 3H, (CH
2)
13C
H3); MALDI-TOF: calcd for C
48H
88N
10O
2 836.7, found 837.6 [M + H]
+, 859.6 [M + Na]
+; HPLC (Water/ACN (0.1% TFA); 50% to 100% ACN in 10 min: tr = 8.09 min (
Figure 24).


 ,
, 










































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}