Protein Kinase B and Extracellular Signal-Regulated Kinase Inactivation is Associated with Regorafenib-Induced Inhibition of Osteosarcoma Progression In Vitro and In Vivo



Abstract
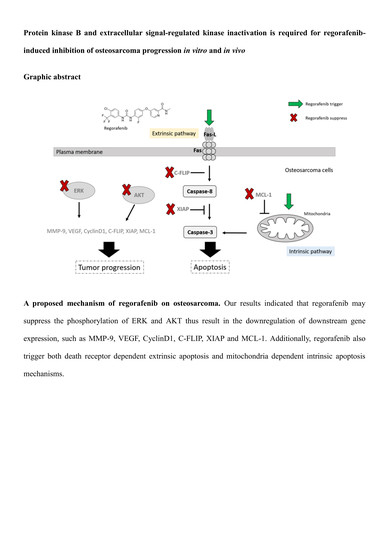
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Establishment of Stable U-2 OS/luc2 Cells
2.4. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide MTT Assay
2.5. Cell Cycle Analysis
2.6. Annexin-V/PI Double Staining for Flow Cytometry Assay
2.7. Caspase-3 and Caspase-8 Activation Analysis
2.8. Cleavage Poly (ADP-ribose) Polymerase 1 (PARP-1) Activation Analysis
2.9. Mitochondria Membrane Potential Analysis
2.10. FAS/FASL Double Staining for Flow Cytometry Assay
2.11. Invasion Assay
2.12. In Vitro and Ex Vivo Western Blot
2.13. Animal Experiment and U-2 OS Xenograft Animal Model
2.14. Animal Bioluminescent Imaging
2.15. Histological Analysis
2.16. Statistical Analysis
3. Results
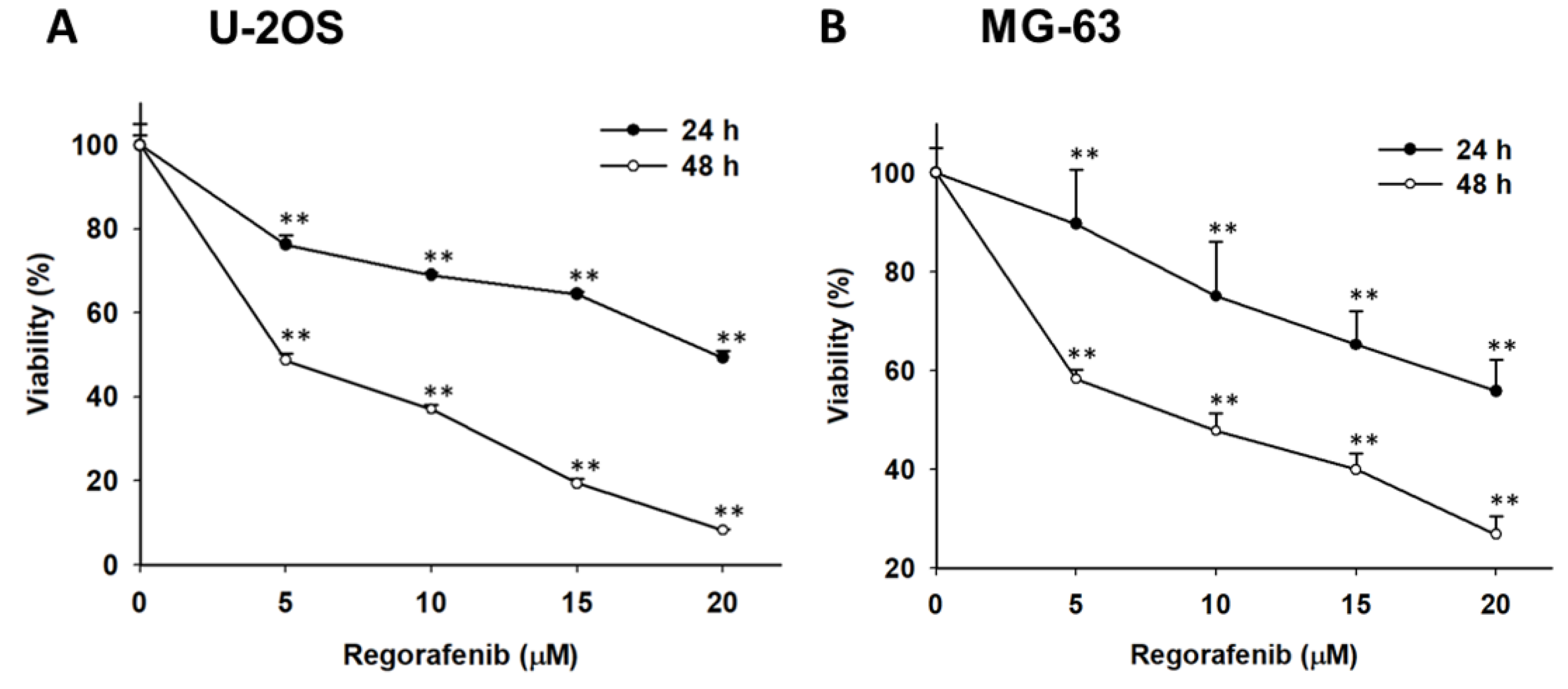
3.1. Regorafenib-Induced Cytotoxicity of U-2 OS Human Osteosarcoma Cells
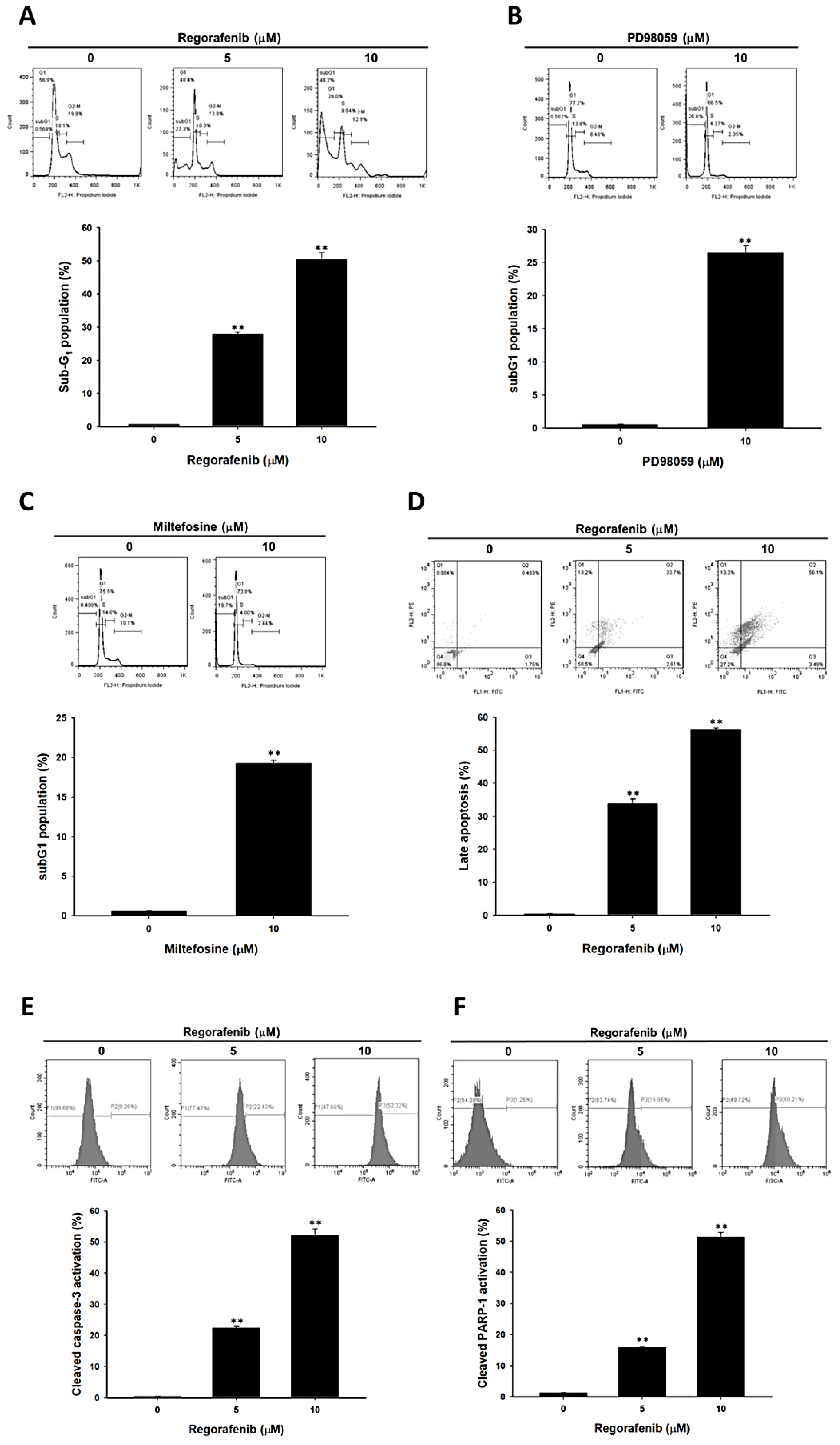
3.2. Regorafenib-Induced Apoptosis and DNA Damage of U-2 OS Human Osteosarcoma Cells
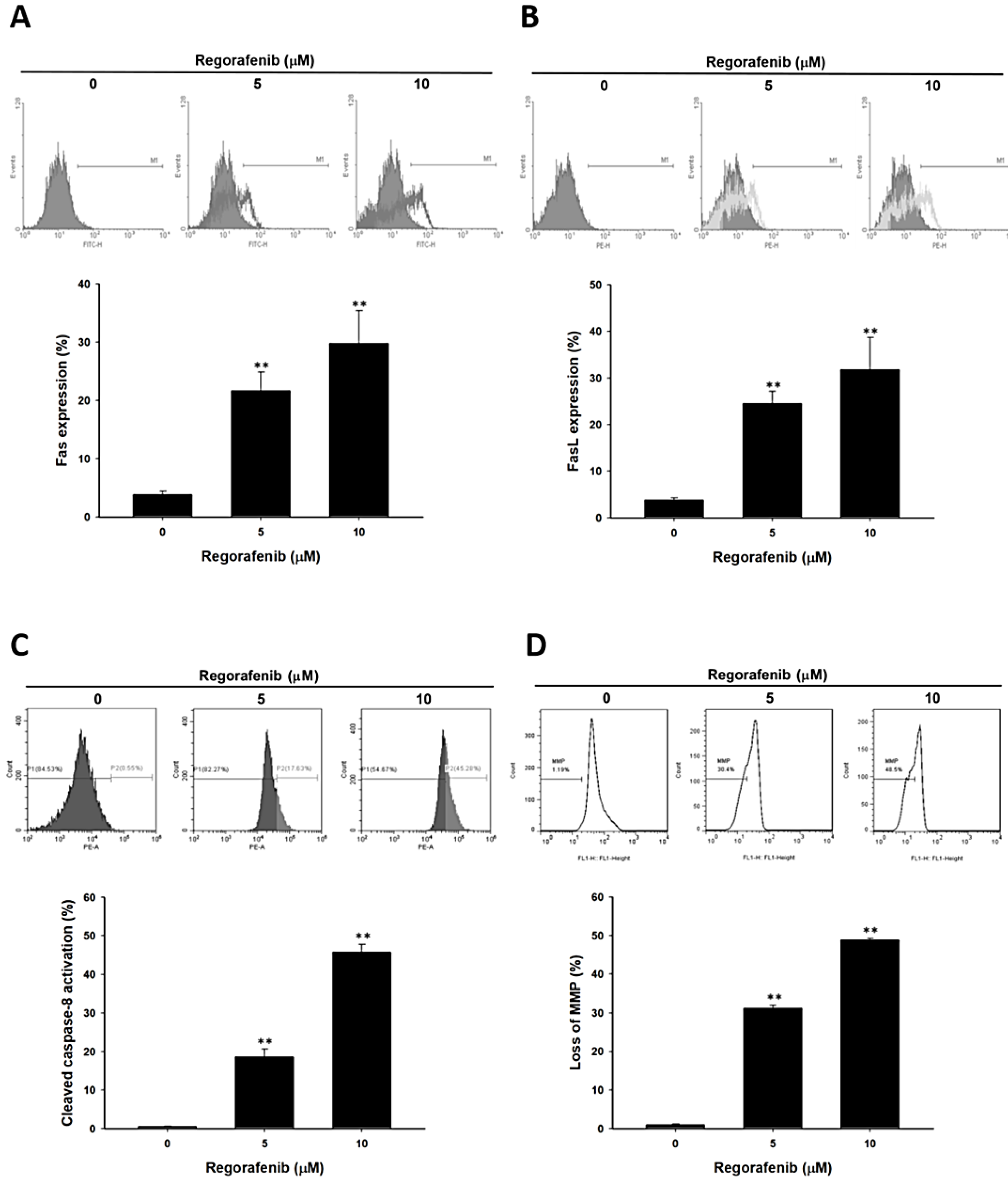
3.3. Regorafenib-Induced Apoptosis was Dependent on Both Extrinsic and Intrinsic Pathways
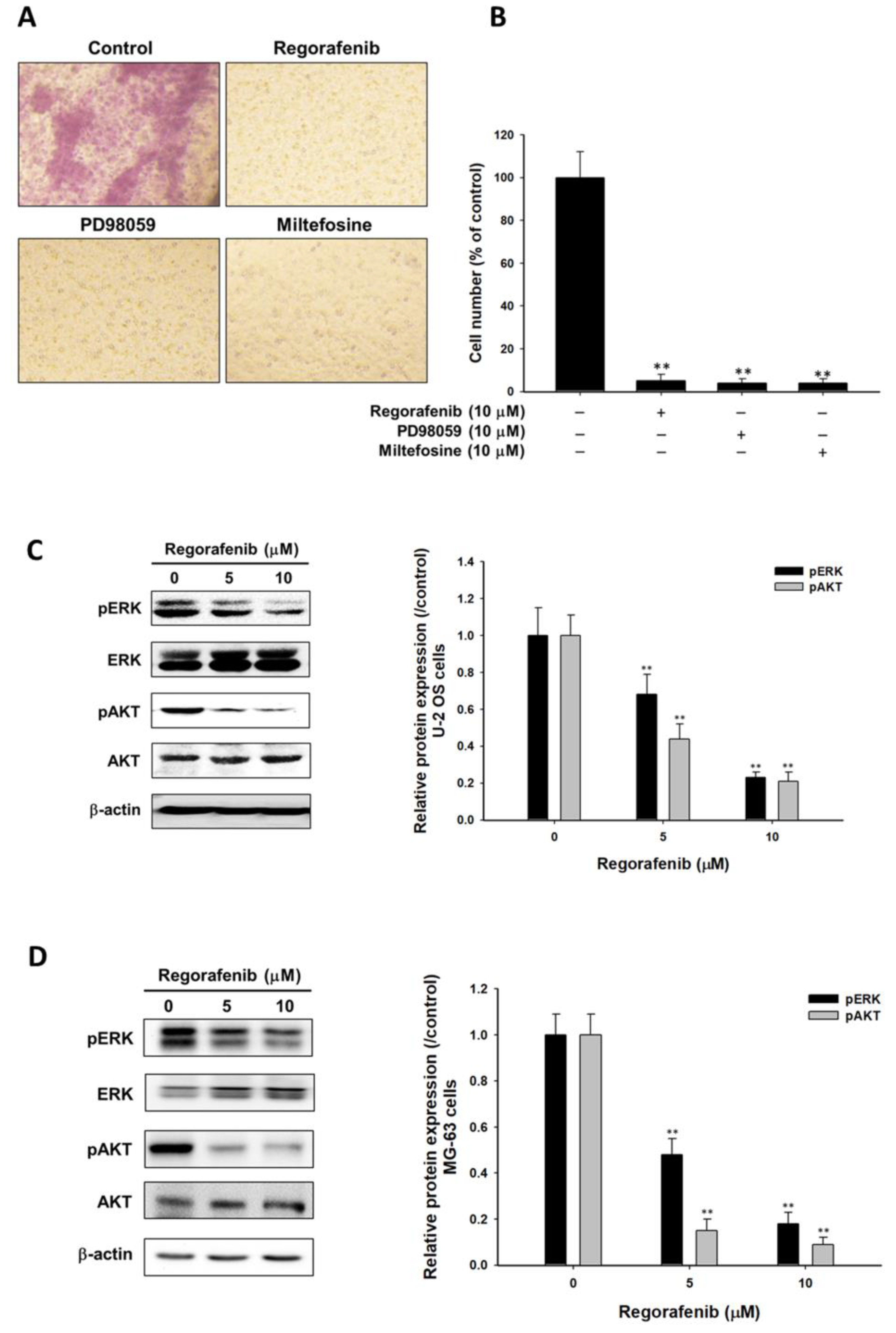
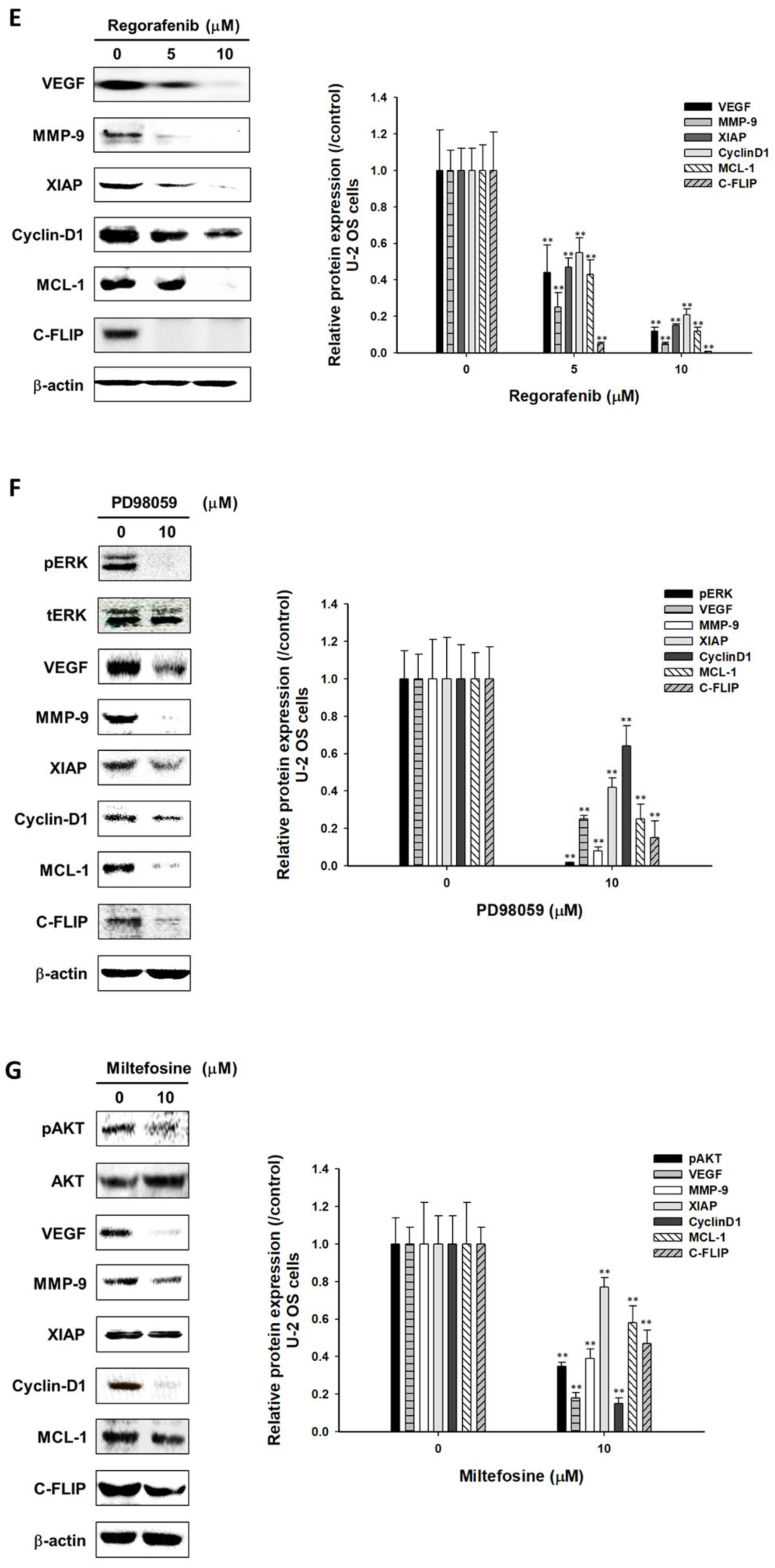
3.4. Regorafenib Suppressed Tumor Progression via Blocking ERK and AKT Signaling Transduction
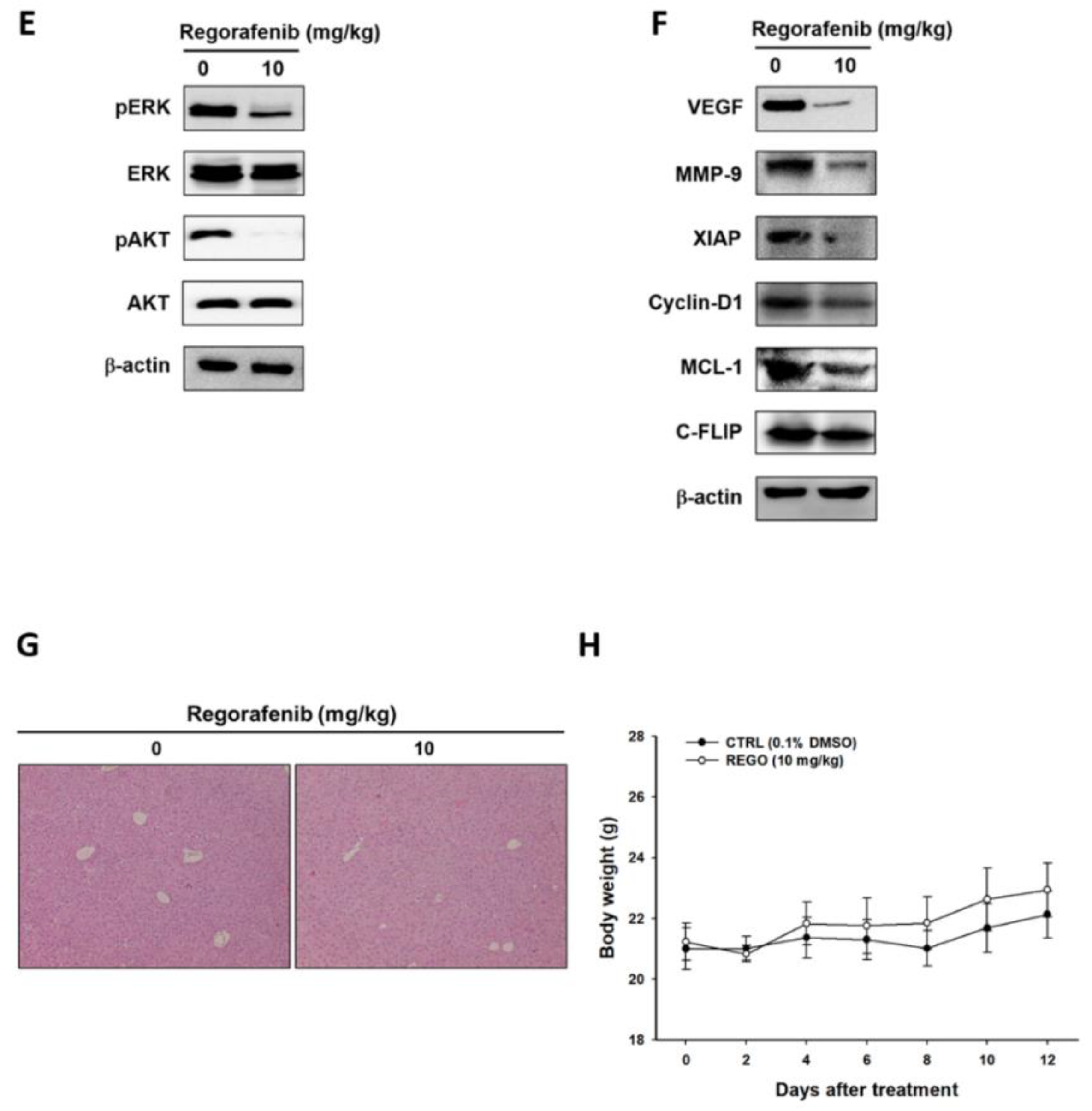
3.5. Regorafenib Markedly Repressed the Growth of U-2 OS Human Osteosarcoma
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Heymann, M.F.; Stresing, V.; Mori, K.; Redini, F.; Heymann, D. Current therapeutic strategies and novel approaches in osteosarcoma. Cancers 2013, 5, 591–616. [Google Scholar] [CrossRef] [PubMed]
- Endo-Munoz, L.; Cai, N.; Cumming, A.; Macklin, R.; Merida de Long, L.; Topkas, E.; Mukhopadhyay, P.; Hill, M.; Saunders, N.A. Progression of Osteosarcoma from a Non-Metastatic to a Metastatic Phenotype Is Causally Associated with Activation of an Autocrine and Paracrine uPA Axis. PLoS ONE 2015, 10, e0133592. [Google Scholar] [CrossRef]
- Fleuren, E.D.; Versleijen-Jonkers, Y.M.; Boerman, O.C.; van der Graaf, W.T. Targeting receptor tyrosine kinases in osteosarcoma and Ewing sarcoma: Current hurdles and future perspectives. Biochim. Biophys. Acta 2014, 1845, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Bishop, M.W.; Janeway, K.A.; Gorlick, R. Future directions in the treatment of osteosarcoma. Curr. Opin. Pediatr. 2016, 28, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, I.T.; Liu, Y.C.; Wang, W.H.; Hsu, F.T.; Chen, H.W.; Lin, W.J.; Chang, W.Y.; Hwang, J.J. Sorafenib inhibits TPA-induced MMP-9 and VEGF expression via suppression of ERK/NF-kappaB pathway in hepatocellular carcinoma cells. In Vivo 2012, 26, 671–681. [Google Scholar]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Dileo, P.; Asaftei, S.D.; D’Ambrosio, L.; Pignochino, Y.; Mercuri, M.; Picci, P.; Fagioli, F.; Casali, P.G.; et al. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: An Italian Sarcoma Group study. Ann. Oncol. 2012, 23, 508–516. [Google Scholar] [CrossRef]
- Tsai, J.J.; Pan, P.J.; Hsu, F.T. Regorafenib induces extrinsic and intrinsic apoptosis through inhibition of ERK/NF-kappaB activation in hepatocellular carcinoma cells. Oncol. Rep. 2017, 37, 1036–1044. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Duffaud, F.; Mir, O.; Boudou-Rouquette, P.; Piperno-Neumann, S.; Penel, N.; Bompas, E.; Delcambre, C.; Kalbacher, E.; Italiano, A.; Collard, O.; et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: A non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2019, 20, 120–133. [Google Scholar] [CrossRef]
- Pignochino, Y.; Grignani, G.; Cavalloni, G.; Motta, M.; Tapparo, M.; Bruno, S.; Bottos, A.; Gammaitoni, L.; Migliardi, G.; Camussi, G.; et al. Sorafenib blocks tumour growth, angiogenesis and metastatic potential in preclinical models of osteosarcoma through a mechanism potentially involving the inhibition of ERK1/2, MCL-1 and ezrin pathways. Mol. Cancer 2009, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.H.; Liu, K.L.; Shih, Y.L.; Chuang, Y.Y.; Chou, J.; Lu, H.F.; Jair, H.W.; Lee, M.Z.; Au, M.K.; Chung, J.G. Ouabain Induces Apoptotic Cell Death Through Caspase- and Mitochondria-dependent Pathways in Human Osteosarcoma U-2 OS Cells. Anticancer Res. 2018, 38, 169–178. [Google Scholar]
- Hsu, F.T.; Sun, C.C.; Wu, C.H.; Lee, Y.J.; Chiang, C.H.; Wang, W.S. Regorafenib Induces Apoptosis and Inhibits Metastatic Potential of Human Bladder Carcinoma Cells. Anticancer Res. 2017, 37, 4919–4926. [Google Scholar] [PubMed]
- Chen, S.; Cheng, A.-C.; Wang, M.-S.; Peng, X. Detection of apoptosis induced by new type gosling viral enteritis virus in vitro through fluorescein annexin V-FITC/PI double labeling. World J. Gastroenterol. 2008, 14, 2174–2178. [Google Scholar] [CrossRef]
- Kuan, L.Y.; Chen, W.L.; Chen, J.H.; Hsu, F.T.; Liu, T.T.; Chen, W.T.; Wang, K.L.; Chen, W.C.; Liu, Y.C.; Wang, W.S. Magnolol Induces Apoptosis and Inhibits ERK-modulated Metastatic Potential in Hepatocellular Carcinoma Cells. In Vivo 2018, 32, 1361–1368. [Google Scholar] [CrossRef] [Green Version]
- Kunzmann, A.; Liu, D.; Annett, K.; Malaisé, M.; Thaa, B.; Hyland, P.; Barnett, Y.; Bürkle, A. Flow-cytometric assessment of cellular poly(ADP-ribosyl)ation capacity in peripheral blood lymphocytes. Immun. Ageing 2006, 3, 8. [Google Scholar] [CrossRef]
- Lee, C.F.; Chiang, N.N.; Lu, Y.H.; Huang, Y.S.; Yang, J.S.; Tsai, S.H.; Lu, C.C.; Chen, F.A. Benzyl isothiocyanate (BITC) triggers mitochondria-mediated apoptotic machinery in human cisplatin-resistant oral cancer CAR cells. Biomedicine 2018, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Krzyzowska, M.; Shestakov, A.; Eriksson, K.; Chiodi, F. Role of Fas/FasL in regulation of inflammation in vaginal tissue during HSV-2 infection. Cell Death Dis. 2011, 2, e132. [Google Scholar] [CrossRef]
- Lee, K.C.; Tsai, J.J.; Tseng, C.W.; Kuo, Y.C.; Chuang, Y.C.; Lin, S.S.; Hsu, F.T. Amentoflavone Inhibits ERK-modulated Tumor Progression in Hepatocellular Carcinoma In Vitro. In Vivo 2018, 32, 549–554. [Google Scholar] [CrossRef]
- Cain, J.E.; McCaw, A.; Jayasekara, W.S.N.; Rossello, F.J.; Marini, K.D.; Irving, A.T.; Kansara, M.; Thomas, D.M.; Ashley, D.M.; Watkins, D.N. Sustained Low-Dose Treatment with the Histone Deacetylase Inhibitor LBH589 Induces Terminal Differentiation of Osteosarcoma Cells. Sarcoma 2013, 2013, 608964. [Google Scholar] [CrossRef]
- Weng, M.C.; Wang, M.H.; Tsai, J.J.; Kuo, Y.C.; Liu, Y.C.; Hsu, F.T.; Wang, H.E. Regorafenib inhibits tumor progression through suppression of ERK/NF-kappaB activation in hepatocellular carcinoma bearing mice. Biosci Rep. 2018, 38, BSR20171264. [Google Scholar] [CrossRef]
- Salas, S.; Jiguet-Jiglaire, C.; Campion, L.; Bartoli, C.; Frassineti, F.; Deville, J.-L.; Maues De Paula, A.; Forest, F.; Jézéquel, P.; Gentet, J.-C.; et al. Correlation between ERK1 and STAT3 expression and chemoresistance in patients with conventional osteosarcoma. BMC Cancer 2014, 14, 606. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, X.H.; Yan, Y.G.; Wang, C.; Wang, W.J. PI3K/Akt signaling in osteosarcoma. Clin. Chim. Acta 2015, 444, 182–192. [Google Scholar] [CrossRef]
- Berlanga, P.; Munoz, L.; Piqueras, M.; Sirerol, J.A.; Sanchez-Izquierdo, M.D.; Hervas, D.; Hernandez, M.; Llavador, M.; Machado, I.; Llombart-Bosch, A.; et al. miR-200c and phospho-AKT as prognostic factors and mediators of osteosarcoma progression and lung metastasis. Mol. Oncol. 2016, 10, 1043–1053. [Google Scholar] [CrossRef]
- Chandhanayingyong, C.; Kim, Y.; Staples, J.R.; Hahn, C.; Lee, F.Y. MAPK/ERK Signaling in Osteosarcomas, Ewing Sarcomas and Chondrosarcomas: Therapeutic Implications and Future Directions. Sarcoma 2012, 2012, 404810. [Google Scholar] [CrossRef]
- Du, L.; Li, X.; Zhen, L.; Chen, W.; Mu, L.; Zhang, Y.; Song, A. Everolimus inhibits breast cancer cell growth through PI3K/AKT/mTOR signaling pathway. Mol. Med. Rep. 2018, 17, 7163–7169. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Ferraresi, V.; D’Ambrosio, L.; Bertulli, R.; Asaftei, S.D.; Tamburini, A.; Pignochino, Y.; Sangiolo, D.; Marchesi, E.; et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: A non-randomised phase 2 clinical trial. Lancet Oncol. 2015, 16, 98–107. [Google Scholar] [CrossRef]
- Kaya, M.; Wada, T.; Akatsuka, T.; Kawaguchi, S.; Nagoya, S.; Shindoh, M.; Higashino, F.; Mezawa, F.; Okada, F.; Ishii, S. Vascular endothelial growth factor expression in untreated osteosarcoma is predictive of pulmonary metastasis and poor prognosis. Clin. Cancer Res. 2000, 6, 572–577. [Google Scholar]
- Zhou, J.; Liu, T.; Wang, W. Prognostic significance of matrix metalloproteinase 9 expression in osteosarcoma: A meta-analysis of 16 studies. Medicine 2018, 97, e13051. [Google Scholar] [CrossRef]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, Y.L.; Zhao, J.L.; Zhen, O.; Yu, C.; Yang, B.H.; Yu, X.R. Hypoxia-inducible factor-1 promotes cancer progression through activating AKT/Cyclin D1 signaling pathway in osteosarcoma. Biomed. Pharm. 2018, 105, 1–9. [Google Scholar] [CrossRef]
- Chiang, I.T.; Chen, W.T.; Tseng, C.W.; Chen, Y.C.; Kuo, Y.C.; Chen, B.J.; Weng, M.C.; Lin, H.J.; Wang, W.S. Hyperforin Inhibits Cell Growth by Inducing Intrinsic and Extrinsic Apoptotic Pathways in Hepatocellular Carcinoma Cells. Anticancer Res. 2017, 37, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Rao-Bindal, K.; Rao, C.K.; Yu, L.; Kleinerman, E.S. Expression of c-FLIP in pulmonary metastases in osteosarcoma patients and human xenografts. Pediatr. Blood Cancer 2013, 60, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Guerrero, A.D.; Huang, L.; Shabier, Z.; Pan, M.; Tan, T.H.; Wang, J. Caspase-9-induced mitochondrial disruption through cleavage of anti-apoptotic BCL-2 family members. J. Biol. Chem. 2007, 282, 33888–33895. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Xia, P.; Zhang, S.; Pan, S.; Zhao, J. Silencing XIAP suppresses osteosarcoma cell growth, and enhances the sensitivity of osteosarcoma cells to doxorubicin and cisplatin. Oncol. Rep. 2015, 33, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Pang, L.; Yang, T.; Liu, P. lncRNA LINC01296 regulates the proliferation, metastasis and cell cycle of osteosarcoma through cyclin D1. Oncol. Rep. 2018, 40, 2507–2514. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, X. A Single Nucleotide Polymorphism (rs1056629) in 3′-UTR of MMP-9 is Responsible for a Decreased Risk of Metastatic Osteosarcoma by Compromising its Interaction with microRNA-491–5p. Cell. Physiol. Biochem. 2016, 38, 1415–1424. [Google Scholar] [CrossRef]
- Peng, N.; Gao, S.; Guo, X.; Wang, G.; Cheng, C.; Li, M.; Liu, K. Silencing of VEGF inhibits human osteosarcoma angiogenesis and promotes cell apoptosis via VEGF/PI3K/AKT signaling pathway. Am. J. Transl. Res. 2016, 8, 1005–1015. [Google Scholar]
- Zhang, Y.; Shi, C.; Yin, L.; Zhou, W.; Wang, H.; Seng, J.; Li, W. Inhibition of MCL-1 enhances Pevonedistat-triggered apoptosis in osteosarcoma cells. Exp. Cell Res. 2017, 358, 234–241. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, P.-J.; Liu, Y.-C.; Hsu, F.-T. Protein Kinase B and Extracellular Signal-Regulated Kinase Inactivation is Associated with Regorafenib-Induced Inhibition of Osteosarcoma Progression In Vitro and In Vivo. J. Clin. Med. 2019, 8, 900. https://doi.org/10.3390/jcm8060900
Pan P-J, Liu Y-C, Hsu F-T. Protein Kinase B and Extracellular Signal-Regulated Kinase Inactivation is Associated with Regorafenib-Induced Inhibition of Osteosarcoma Progression In Vitro and In Vivo. Journal of Clinical Medicine. 2019; 8(6):900. https://doi.org/10.3390/jcm8060900
Chicago/Turabian StylePan, Po-Jung, Yu-Chang Liu, and Fei-Ting Hsu. 2019. "Protein Kinase B and Extracellular Signal-Regulated Kinase Inactivation is Associated with Regorafenib-Induced Inhibition of Osteosarcoma Progression In Vitro and In Vivo" Journal of Clinical Medicine 8, no. 6: 900. https://doi.org/10.3390/jcm8060900