Role of Extrinsic Apoptotic Signaling Pathway during Definitive Erythropoiesis in Normal Patients and in Patients with β-Thalassemia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
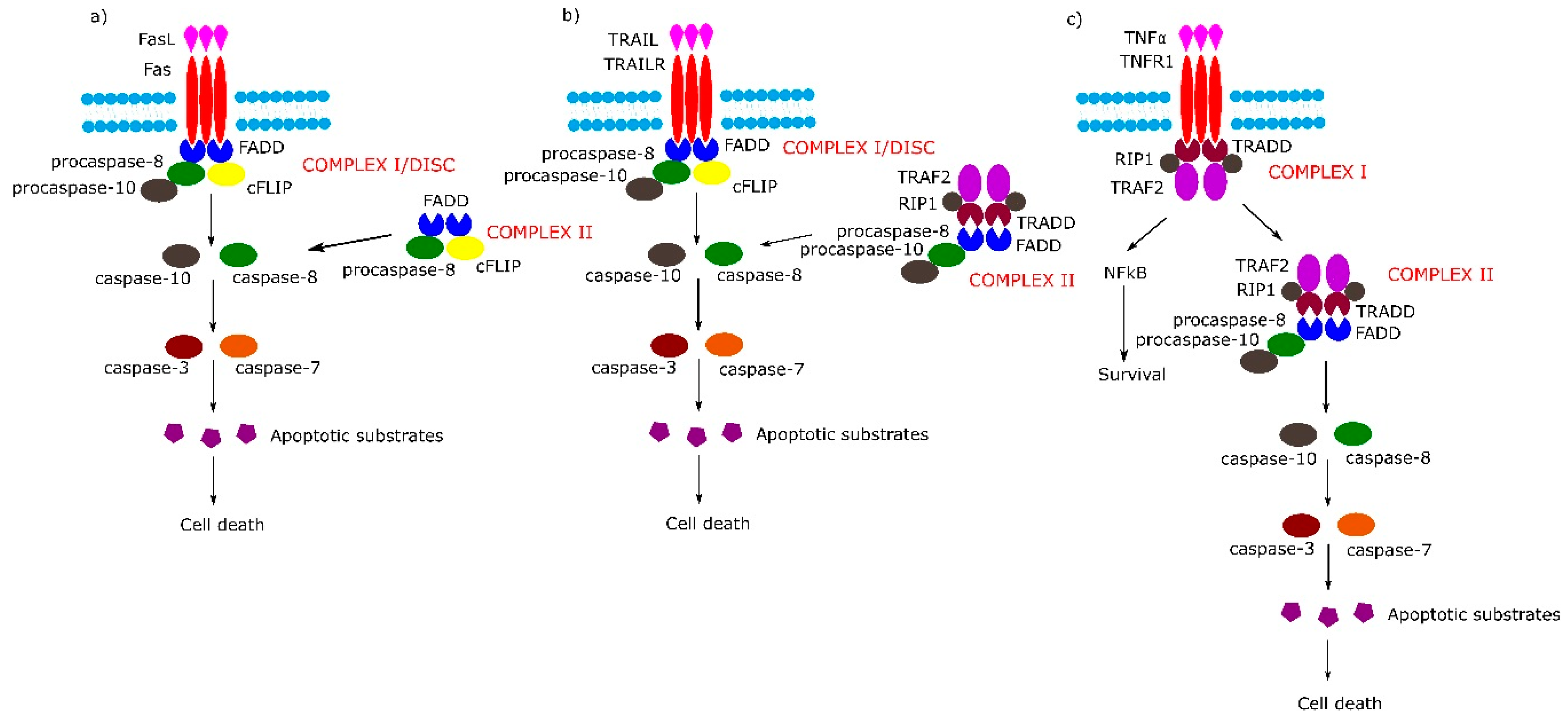
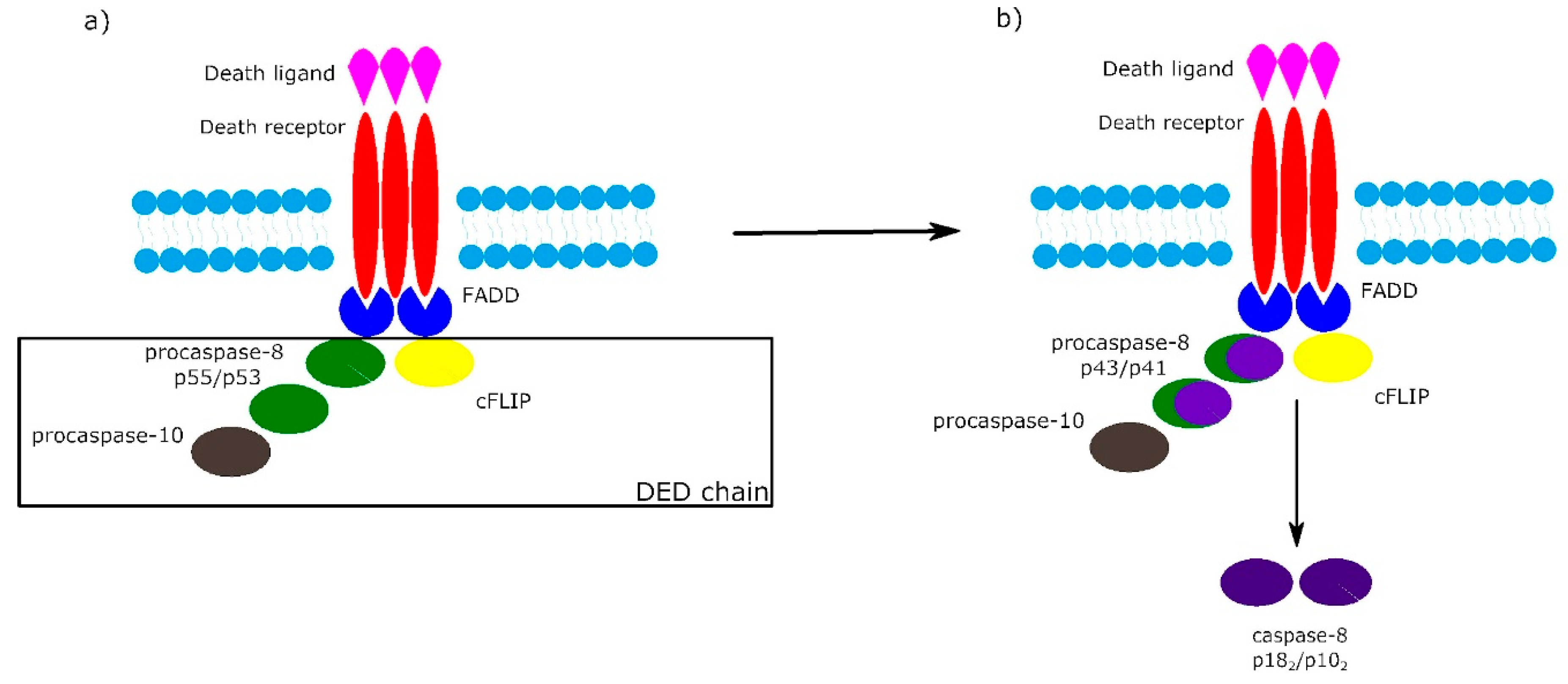
2. Receptor-Dependent Apoptotic Pathway
3. Erythropoiesis and Extrinsic Apoptotic Pathway
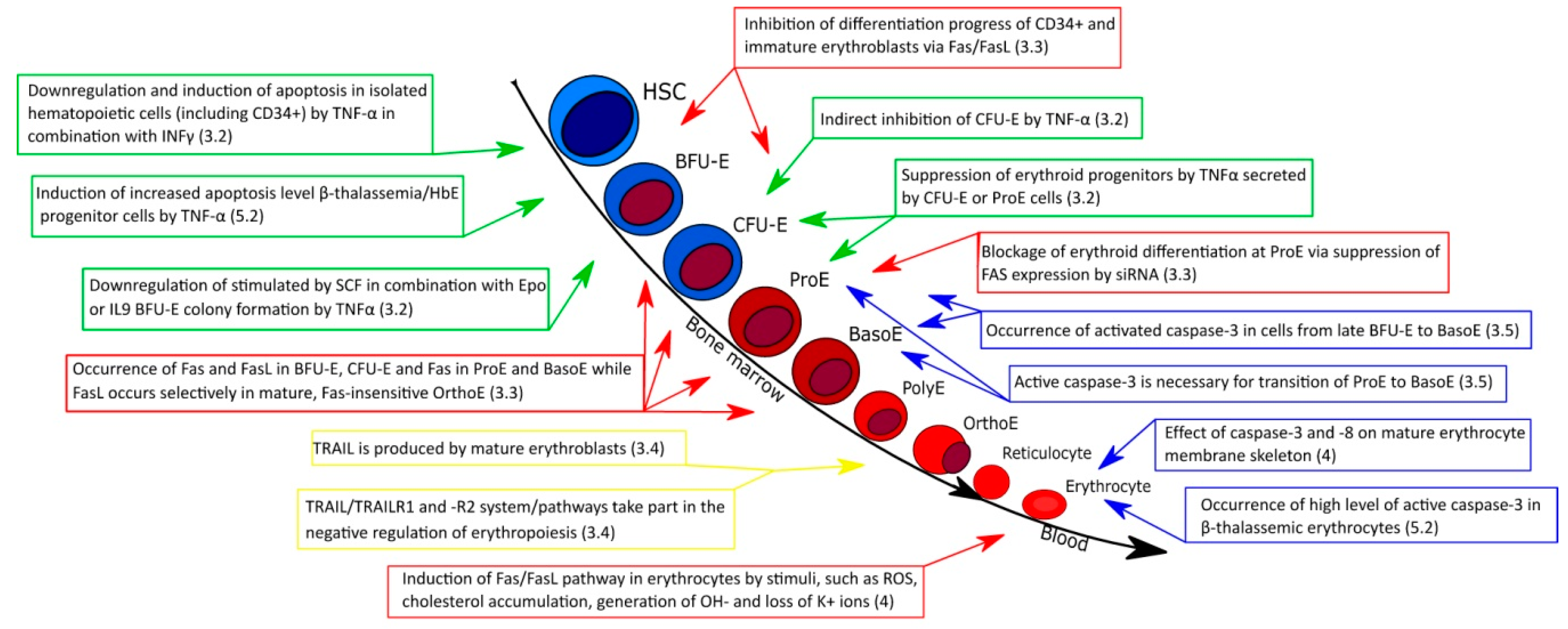
3.1. Erythropoiesis
3.2. Role of TNF-α in Normal Erythropoiesis
3.3. Fas/FasL in Normal Erythropoiesis
3.4. Role of TRAIL in Normal Erythropoiesis
3.5. Effect of Caspases on Normal Erythroid Maturation
4. Mature Erythrocytes and Extrinsic Apoptotic Pathway
5. β-Thalassemia and Extrinsic Apoptotic Pathway
5.1. β-Thalassemia
5.2. Role of Extrinsic Apoptotic Pathway in Different Types of β-Thalassemia
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BasoE | basophilic erythroblasts |
| IAP | inhibitors of apoptosis |
| AMPK | AMP-activated kinase |
| Apaf-1 | apoptotic protease activating factor 1 |
| BAD | BCL2 associated death promoter |
| BAK | BCL2 antagonist/killer |
| BAX | BCL2-associated X protein |
| BCL2 | B-cell lymphoma 2 |
| BCLB | BCL-2 like protein 10 |
| BCLW | BCL2-like protein 2 |
| BFL1 | BCL2-related protein A1 |
| BFU-E | burst-forming unit-erythroid |
| BH | BCL2 homology |
| BID | BH3 interacting-domain death agonist |
| BIK | BCL2 interacting killer |
| BIM | BCL2 interacting mediator of cell death |
| BIR | baculovirus IAP repeat domains |
| BMF | BCL2 modifying factor |
| CARD | caspase recruitment domain |
| c-FLIPs | cellular FLICE (FADD-like IL-1β-converting enzyme) inhibitory proteins |
| CFU-E | colony-forming unit-erythroid |
| cIAP1 and cIAP2 | cellular inhibitor of apoptosis protein 1 and 2 |
| CRD | cysteine-rich domain |
| DcRs | decoy receptors |
| DD | death domain |
| DED | death effector domain |
| DISC | death-inducing signaling complex |
| DRM | detergent-resistant membrane |
| DRs | death receptors |
| Epo | erythropoietin |
| FADD | Fas-associated protein with death domain |
| Fas | TNF receptor superfamily member 6 |
| FasL | tumor necrosis factor ligand superfamily member 6 |
| HRK | activator of apoptosis harakiri |
| IAPs | inhibitors of apoptosis |
| INFγ | interferon γ |
| JAK3 | Janus kinase 3 |
| KLF1 | Kruppel-like factor 1 |
| lncRNAs | long non-coding RNAs |
| LT-HSCs | long-term repopulating HSCs |
| LUBAC | linear ubiquitin chain assembly complex |
| MAPK | Shc/Ras/mitogen-activated kinase |
| MCL1 | myeloid cell leukemia |
| MPCs | multipotent progenitor cells |
| NGS | Next Generation Sequencing |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NOXA | Phorbol-12-myristate-13-acetate-induced protein 1 |
| OrthoE | orthochromatic erythroblasts |
| PAK2 | p21-activated kinase 2 |
| PI3K | phosphatidylinositol-3-kinase |
| PKC | protein kinase C |
| PKCε | protein kinase Cε |
| PKR | protein kinase R |
| PLAD | pre-ligand assembly domain |
| PolyE | polychromatophilic erythroblasts |
| ProE | Proerythroblasts |
| ProEs | Proerythroblasts |
| PUMA | p53 upregulated modulator of apoptosis |
| RING | “really interesting new gene” domain |
| RNF31 | ring finger protein 31, also known as HOIP |
| ROS | reactive oxygen species |
| SCF | stem cell factor |
| SDF-1α | stroma-derived factor 1α |
| Stat5 | signal transducer and activator of transcription 5 |
| ST-HSCs | short-term repopulating HSCs |
| TAB1 | TGFβ-activated kinase 1 (TAK1)-binding protein 1 |
| THD | TNF homology domain |
| TNFR1 | TNF-α receptor 1 |
| TNFR2 | TNF-α receptor 2 |
| TNFRSF | tumor necrosis factor receptor superfamily |
| TNFSF | TNF superfamily |
| TNFα | tumor necrosis factor α |
| TRADD | TNFR1-associated protein with death domain |
| TRAF | TNF receptor-associated factor |
| TRAILR1/TRAILR2 | TNF-related apoptosis-inducing ligand receptor 1 and 2 |
| UBA | UB-associated domain |
| XIAP | X-linked IAP |
References
- Ghashghaeinia, M.; Cluitmans, J.C.A.; Akel, A.; Dreischer, P.; Toulany, M.; Köberle, M.; Skabytska, Y.; Saki, M.; Biedermann, T.; Duszenko, M.; et al. The impact of erythrocyte age on eryptosis. Br. J. Haematol. 2012, 157, 606–614. [Google Scholar] [CrossRef]
- Repsold, L.; Joubert, A.M. Eryptosis: An Erythrocyte’s Suicidal Type of Cell Death. BioMed Res. Int. 2018, 2018, 9405617. [Google Scholar] [CrossRef]
- Lang, P.A.; Kempe, D.S.; Myssina, S.; Tanneur, V.; Birka, C.; Laufer, S.; Lang, F.; Wieder, T.; Huber, S.M. PGE2 in the regulation of programmed erythrocyte death. Cell Death Differ. 2005, 12, 415–428. [Google Scholar] [CrossRef]
- Myssina, S.; Huber, S.M.; Birka, C.; Lang, P.A.; Lang, K.S.; Friedrich, B.; Risler, T.; Wieder, T.; Lang, F. Inhibition of Erythrocyte Cation Channels by Erythropoietin. J. Am. Soc. Nephrol. 2003, 14, 2750–2757. [Google Scholar] [CrossRef] [Green Version]
- Qadri, S.M.; Bissinger, R.; Solh, Z.; Oldenborg, P.A. Eryptosis in health and disease: A paradigm shift towards understanding the (patho)physiological implications of programmed cell death of erythrocytes. Blood Rev. 2017, 31, 349–361. [Google Scholar] [CrossRef]
- Lang, F.; Qadri, S.M. Mechanisms and significance of eryptosis, the suicidal death of erythrocytes. Blood Purif. 2012, 33, 125–130. [Google Scholar] [CrossRef]
- Lang, E.; Lang, F. Triggers, Inhibitors, Mechanisms, and Significance of Eryptosis: The Suicidal Erythrocyte Death. BioMed Res. Int. 2015, 2015, 513518. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, S.K.; Ulirsch, J.C.; Sankaran, V.G. Advances in understanding erythropoiesis: Evolving perspectives. Br. J. Haematol. 2016, 173, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Testa, U. Apoptotic mechanisms in the control of erythropoiesis. Leukemia 2004, 18, 1176–1199. [Google Scholar] [CrossRef] [Green Version]
- Dzierzak, E.; Philipsen, S. Erythropoiesis: Development and Differentiation. Cold Spring Harb. Perspect. Med. 2013, 3, a011601. [Google Scholar] [CrossRef]
- Dai, H.; Meng, W.; Kaufmann, S.H. BCL2 Family, Mitochondrial Apoptosis, and Beyond. Cancer Transl. Med. 2016, 2, 7. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Motoyama, N.; Kimura, T.; Takahashi, T.; Watanabe, T.; Nakano, T. bcl-x Prevents apoptotic cell death of both primitive and definitive erythrocytes at the end of maturation. J. Exp. Med. 1999, 189, 1691–1698. [Google Scholar] [CrossRef] [Green Version]
- Dolznig, H.; Habermann, B.; Stangl, K.; Deiner, E.M.; Moriggl, R.; Beug, H.; Müllner, E.W. Apoptosis protection by the Epo target Bcl-XL allows factor-independent differentiation of primary erythroblasts. Curr. Biol. 2002, 12, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Mason, K.D.; Lin, A.; Robb, L.; Josefsson, E.C.; Henley, K.J.; Gray, D.H.D.; Kile, B.T.; Roberts, A.W.; Strasser, A.; Huang, D.C.S.; et al. Proapoptotic Bak and Bax guard against fatal systemic and organ-specific autoimmune disease. Proc. Natl. Acad. Sci. USA 2013, 110, 2599–2604. [Google Scholar] [CrossRef] [Green Version]
- Delbridge, A.R.D.; Opferman, J.T.; Grabow, S.; Strasser, A. Antagonism between MCL-1 and PUMA governs stem/progenitor cell survival during hematopoietic recovery from stress. Blood 2015, 125, 3273–3280. [Google Scholar] [CrossRef] [Green Version]
- Opferman, J.T.; Iwasaki, H.; Ong, C.C.; Suh, H.; Mizuno, S.I.; Akashi, K.; Korsmeyer, S.J. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005, 307, 1101–1104. [Google Scholar] [CrossRef]
- Delbridge, A.R.D.; Aubrey, B.J.; Hyland, C.; Bernardini, J.P.; Di Rago, L.; Garnier, J.M.; Lessene, G.; Strasser, A.; Alexander, W.S.; Grabow, S. The BH3-only proteins BIM and PUMA are not critical for the reticulocyte apoptosis caused by loss of the pro-survival protein BCL-XL. Cell Death Dis. 2017, 8, e2914-9. [Google Scholar] [CrossRef] [Green Version]
- Abutin, R.M.; Chen, J.; Lung, T.K.; Lloyd, J.A.; Sawyer, S.T.; Harada, H. Erythropoietin-Induced Phosphorylation/Degradation of BIM Contributes to Survival of Erythroid Cells. Exp. Hematol. 2009, 37, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Wensveen, F.M.; Geest, C.R.; Libregts, S.F.W.M.; Derks, I.A.M.; Ekert, P.G.; Labi, V.; Villunger, A.; Nolte, M.A.; Eldering, E. BH3-only protein Noxa contributes to apoptotic control of stress-erythropoiesis. Apoptosis 2013, 18, 1306–1318. [Google Scholar] [CrossRef] [Green Version]
- Schug, Z.T.; Gonzalvez, F.; Houtkooper, R.H.; Vaz, F.M.; Gottlieb, E. BID is cleaved by caspase-8 within a native complex on the mitochondrial membrane. Cell Death Differ. 2011, 18, 538–548. [Google Scholar] [CrossRef]
- Milhas, D.; Cuvillier, O.; Therville, N.; Clavé, P.; Thomsen, M.; Levade, T.; Benoist, H.; Ségui, B. Caspase-10 triggers bid cleavage and caspase cascade activation in fasL-induced apoptosis. J. Biol. Chem. 2005, 280, 19836–19842. [Google Scholar] [CrossRef] [Green Version]
- Peslak, S.A.; Wenger, J.; Bemis, J.C.; Kingsley, P.D.; Frame, J.M.; Koniski, A.D.; Chen, Y.; Williams, J.P.; Mcgrath, K.E.; Dertinger, S.D.; et al. Sublethal radiation injury uncovers a functional transition during erythroid. Exp. Hematol. 2011, 39, 434–445. [Google Scholar] [CrossRef] [Green Version]
- Lamarque, M.; Guillerm, F.; Belaid, Z.; Hermine, O.; Courtois, G. BID Cleavage Pattern Is a Novel Check-Point That Determines the Fate of Human Erythroid Progenitors (Differentiation or Apoptosis). Blood 2016. [Google Scholar] [CrossRef]
- Siegmund, D.; Lang, I.; Wajant, H. Cell death-independent activities of the death receptors CD95, TRAILR1, and TRAILR2. FEBS J. 2017, 284, 1131–1159. [Google Scholar] [CrossRef]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 2015, 22, 1727–1741. [Google Scholar] [CrossRef] [Green Version]
- Tourneur, L.; Chiocchia, G. FADD: A regulator of life and death. Trends Immunol. 2010, 31, 260–269. [Google Scholar] [CrossRef]
- Schleich, K.; Warnken, U.; Fricker, N.; Öztürk, S.; Richter, P.; Kammerer, K.; Schnölzer, M.; Krammer, P.H.; Lavrik, I.N. Stoichiometry of the CD95 Death-Inducing Signaling Complex: Experimental and Modeling Evidence for a Death Effector Domain Chain Model. Mol. Cell 2012, 47, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Schleich, K.; Buchbinder, J.H.; Pietkiewicz, S.; Kähne, T.; Warnken, U.; Öztürk, S.; Schnölzer, M.; Naumann, M.; Krammer, P.H.; Lavrik, I.N. Molecular architecture of the DED chains at the DISC: Regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ. 2016, 23, 681–694. [Google Scholar] [CrossRef]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.R.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Silke, J.; Meier, P. Inhibitor o fApoptosis (IAP) Proteins-Modulators of Cell Death and Inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–19. [Google Scholar] [CrossRef]
- Lu, M.; Lin, S.C.; Huang, Y.; Kang, Y.J.; Rich, R.; Lo, Y.C.; Myszka, D.; Han, J.; Wu, H. XIAP Induces NF-κB Activation via the BIR1/TAB1 Interaction and BIR1 Dimerization. Mol. Cell 2007, 26, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Wong, W.W.L.; Khan, N.; Feltham, R.; Chau, D.; Ahmed, A.U.; Benetatos, C.A.; Chunduru, S.K.; Condon, S.M.; McKinlay, M.; et al. IAP Antagonists Target cIAP1 to Induce TNFα-Dependent Apoptosis. Cell 2007, 131, 682–693. [Google Scholar] [CrossRef] [Green Version]
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef]
- Eckelman, B.P.; Salvesen, G.S. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J. Biol. Chem. 2006, 281, 3254–3260. [Google Scholar] [CrossRef] [Green Version]
- Berthelet, J.; Dubrez, L. Regulation of Apoptosis by Inhibitors of Apoptosis (IAPs). Cells 2013, 2, 163–187. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.E.; Butterworth, M.; Malladi, S.; Duckett, C.S.; Cohen, G.M.; Bratton, S.B. The E3 ubiquitin ligase cIAP1 binds and ubiquitinates caspase-3 and -7 via unique mechanisms at distinct steps in their processing. J. Biol. Chem. 2009, 284, 12772–12782. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.K.; Joazeiro, C.A.P.; Bonfoco, E.; Kamada, S.; Leverson, J.D.; Hunter, T. The inhibitor of apoptosis, cIAP2, functions as a ubiquitin-protein ligase and promotes in vitro monoubiquitination of caspases 3 and 7. J. Biol. Chem. 2000, 275, 26661–26664. [Google Scholar]
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.L.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef]
- Ikeda, F.; Deribe, Y.L.; Skånland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; Van Wijk, S.J.L.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 2011, 471, 637–641. [Google Scholar] [CrossRef]
- Tokunaga, F.; Nakagawa, T.; Nakahara, M.; Saeki, Y.; Taniguchi, M.; Sakata, S.I.; Tanaka, K.; Nakano, H.; Iwai, K. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 2011, 471, 633–636. [Google Scholar] [CrossRef]
- Joo, D.; Tang, Y.; Blonska, M.; Jin, J.; Zhao, X. Regulation of linear ubiquitin chain assembly complex by Caspase-mediated cleveage of RNF31. Mol. Cell. Biol. 2016, 36, 3010–3018. [Google Scholar] [CrossRef] [Green Version]
- Lafont, E.; Kantari-Mimoun, C.; Draber, P.; De Miguel, D.; Hartwig, T.; Reichert, M.; Kupka, S.; Shimizu, Y.; Taraborrelli, L.; Spit, M.; et al. The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. EMBO J. 2017, 36, 1147–1166. [Google Scholar] [CrossRef]
- Lafont, E.; Hartwig, T.; Walczak, H. Paving TRAIL’s Path with Ubiquitin. Trends Biochem. Sci. 2018, 43, 44–60. [Google Scholar] [CrossRef]
- Lawson, K.A.; Meneses, J.J.; Pedersen, R.A. Clonal analysis of epiblast fate during germ layer formation in the mouse embryo. Development 1991, 113, 891–911. [Google Scholar]
- Palis, J. Primitive and definitive erythropoiesis in mammals. Front. Physiol. 2014, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Merchan, A.; Hidalgo, I.; Arranz, L.; Gonzalez, S. Insights into Stem Cell Aging. In Advances in Hematopoietic Stem Cell Research; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef] [Green Version]
- Hattangadi, S.M.; Wong, P.; Zhang, L.; Flygare, J.; Lodish, H.F. From stem cell to red cell: Regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood 2011, 118, 6258–6268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunn, H.F. Erythropoietin. Cold Spring Harb. Perspect. Med. 2013, 3, a011619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigra, A.D.; Casale, C.H.; Santander, V.S. Human erythrocytes: Cytoskeleton and its origin. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.; Pham, E.; Macdougall, I.C. Erythropoietins: A common mechanism of action. Exp. Hematol. 2008, 36, 1573–1584. [Google Scholar] [CrossRef]
- Valent, P.; Büsche, G.; Theurl, I.; Uras, I.Z.; Germing, U.; Stauder, R.; Sotlar, K.; Füreder, W.; Bettelheim, P.; Pfeilstöcker, M.; et al. Normal and pathological erythropoiesis in adults: From gene regulation to targeted treatment concepts. Haematologica 2018, 103, 1593–1603. [Google Scholar] [CrossRef]
- Listowski, M.A.; Heger, E.; Bogusławska, D.M.; Machnicka, B.; Kuliczkowski, K.; Leluk, J.; Sikorski, A.F. MicroRNAs: Fine tuning of erythropoiesis. Cell. Mol. Biol. Lett. 2012, 18, 34–46. [Google Scholar] [CrossRef]
- Means, R.T.; Dessypris, E.N.; Krantz, S.B. Inhibition of human colony-forming-unit erythroid by tumor necrosis factor requires accessory cells. J. Clin. Investig. 1990, 86, 538–541. [Google Scholar] [CrossRef]
- Means, R.T.; Krantz, S.B. Inhibition of human erythroid colony-forming units by tumor necrosis factor requires beta interferon. J. Clin. Investig. 1993, 91, 416–419. [Google Scholar] [CrossRef] [Green Version]
- Backx, B.; Broeders, L.; Bot, F.J.; Lowenberg, B. Positive and negative effects of tumor necrosis factor on colony growth from highly purified normal marrow progenitors. Leukemia 1991, 5, 66. [Google Scholar]
- Caux, C.; Saeland, S.; Favre, C.; Duvert, V.; Mannoni, P.; Banchereau, J. Tumor Necrosis Factor-alpha Strongly Potentiates Interleukin-3 and Granulocyte-Macrophage Colony-Stimulating Factor-Induced Proliferation of Human CD34+ Hematopoietic Progenitor Cells. Blood 1990, 75, 2292–2298. [Google Scholar] [CrossRef]
- Rusten, L.S.; Jacobsen, S.E.W. Tumor Necrosis Factor (TNF)-a Directly Inhibits Human Erythropoiesis In Vitro: Role. J. Biol. Chem. 1995, 85, 989–996. [Google Scholar]
- Dicato, M.; Morceau, F.; Diederich, M. Pro-inflammatory cytokine-mediated anemia: Regarding molecular mechanisms of erythropoiesis. Mediators Inflamm. 2009, 2009, 405016. [Google Scholar]
- Li, J.; Yin, Q.; Wu, H. Structural Basis of Signal Transduction in the TNF Receptor Superfamily. Adv. Immunol. 2013, 119, 135–153. [Google Scholar]
- Xiao, W.; Koizumi, K.; Nishio, M.; Endo, T.; Osawa, M.; Fujimoto, K.; Sato, I.; Sakai, T.; Koike, T.; Sawada, K.I. Tumor necrosis factor-α inhibits generation of glycophorin A+ cells by CD34+ cells. Exp. Hematol. 2002, 30, 1238–1247. [Google Scholar] [CrossRef]
- De Maria, R.; Zeuner, A.; Eramo, A.; Domenichelli, C.; Bonci, D.; Grignani, F.; Srinivasula, S.M.; Alnemri, E.S.; Testa, U.; Peschle, C. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature 1999, 401, 489–493. [Google Scholar] [CrossRef]
- Chasis, J.A.; Mohandas, N. Erythroblastic islands: Niches for erythropoiesis. Blood 2008, 112, 470–478. [Google Scholar] [CrossRef] [Green Version]
- Jacobs-Helber, S.M.; Roh, K.; Bailey, D.; Dessypris, E.N.; Ryan, J.J.; Chen, J.; Wickrema, A.; Barber, D.L.; Dent, P.; Sawyer, S.T. Tumor necrosis factor-alpha expressed constitutively in erythroid cells or induced by erythropoietin has negative and stimulatory roles in normal erythropoiesis and erythroleukemia. Blood 2003, 101, 524–531. [Google Scholar] [CrossRef]
- Grigorakaki, C.; Morceau, F.; Chateauvieux, S.; Dicato, M.; Diederich, M. Tumor necrosis factor alpha-mediated inhibition of erythropoiesis involves GATA-1/GATA-2 balance impairment and PU.1 over-expression. Biochem. Pharmacol. 2011, 82, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Selleri, C.; Sato, T.; Anderson, S.; Young, N.S.; Maciejewski, J.P. Interferon-gamma and tumor necrosis factor-alpha suppress both early and late stages of hematopoiesis and induce programmed cell death. J. Cell. Physiol. 1995, 165, 538–546. [Google Scholar] [CrossRef]
- Sharma, B.; Altman, J.K.; Goussetis, D.J.; Verma, A.K.; Platanias, L.C. Protein kinase R as mediator of the effects of interferon (IFN) γ and tumor necrosis factor (TNF) α on normal and dysplastic hematopoiesis. J. Biol. Chem. 2011, 286, 27506–27514. [Google Scholar] [CrossRef] [Green Version]
- Maciejewski, B.J.; Selleri, C.; Anderson, S.; Young, N.S. Fas antigen expression on CD34+ human marrow cells is induced by interferon gamma and tumor necrosis factor alpha and potentiates cytokine-mediated hematopoietic suppression in vitro. Blood 1995, 85, 3183–3190. [Google Scholar] [CrossRef]
- Gibellini, D.; Bassini, A.; Re, M.C.; Ponti, C.; Miscia, S.; Gonelli, A.; La Placa, M.; Zauli, G. Stroma-derived factor 1alpha induces a selective inhibition of human erythroid development via the functional upregulation of Fas/CD95 ligand. Br. J. Haematol. 2000, 111, 432–440. [Google Scholar] [CrossRef]
- De Maria, R.; Testa, U.; Luchetti, L.; Zeuner, A.; Stassi, G.; Pelosi, E.; Riccioni, R.; Felli, N.; Samoggia, P.; Peschle, C. Apoptotic role of Fas/Fas ligand system in the regulation of erythropoiesis. Blood 1999, 93, 796–803. [Google Scholar] [CrossRef]
- Zeuner, A.; Eramo, A.; Testa, U.; Felli, N.; Pelosi, E.; Mariani, G.; Srinivasula, S.M.; Alnemri, E.S.; Condorelli, G.; Peschle, C.; et al. Control of erythroid cell production via caspase-mediated cleavage of transcription factor SCL/Tal-1. Cell Death Differ. 2003, 10, 905–913. [Google Scholar] [CrossRef] [Green Version]
- Gregory, T.; Yu, C.; Ma, A.; Orkin, S.H.; Blobel, G.; Weiss, M.J. GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood 1999, 94, 87–96. [Google Scholar] [CrossRef]
- Vandekerkhove, J.; Courtois, G.; Coulon, S.; Ribeli, J.A.; Hermine, O. Regulation of erythropoesis. ESH Handb. Disord. Iron Metab. Eur. Sch. Hematol. 2009, 2, 44–87. [Google Scholar]
- Carlile, G.W.; Smith, D.H.; Wiedmann, M. A non-apoptotic role for Fas/FasL in erythropoiesis. FEBS Lett. 2009, 583, 848–854. [Google Scholar] [CrossRef] [Green Version]
- Nishio, M.; Oda, A.; Koizumi, K.; Satoh, I.; Sato, Y.; Endoh, T.; Tsutsumi, A.; Fujihara, M.; Ikebuchi, K.; Ikeda, H.; et al. Stem cell factor prevents Fas-mediated apoptosis of human erythroid precursor cells with Src-family kinase dependency. Exp. Hematol. 2001, 29, 19–29. [Google Scholar] [CrossRef]
- Oda, A.; Nishio, M.; Sawada, K.I. Stem Cell Factor Regulation of Fas-Mediated Apoptosis of human erythroid precursor cell. J. Hematother. 2001, 10, 595–600. [Google Scholar] [CrossRef]
- Chung, I.J.; Dai, C.; Krantz, S.B. Stem cell factor increases the expression of FLIP that inhibits IFNgamma -induced apoptosis in human erythroid progenitor cells. Blood 2003, 101, 1324–1328. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Whartenby, K.A.; Georgantas, R.W.; Wingard, J.; Civin, C.I. Human CD34+ Hematopoietic Stem/Progenitor Cells Express High Levels of FLIP and Are Resistant to Fas-Mediated Apoptosis. Stem Cells 2002, 20, 174–182. [Google Scholar] [CrossRef] [PubMed]
- De Yan, M.; Hong, C.C.; Lai, G.M.; Cheng, A.L.; Lin, Y.W.; Chuang, S.E. Identification and characterization of a novel gene Saf transcribed from the opposite strand of Fas. Hum. Mol. Genet. 2005, 14, 1465–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulczyńska, K.; Siatecka, M. A regulatory function of long non-coding RNAs in red blood cell development. Acta Biochim. Pol. 2016, 63, 675–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villamizar, O.; Chambers, C.B.; Mo, Y.Y.; Torry, D.S.; Hofstrand, R.; Riberdy, J.M.; Persons, D.A.; Wilber, A. Fas-antisense long noncoding RNA is differentially expressed during maturation of human erythrocytes and confers resistance to Fas-mediated cell death. Blood Cells Mol. Dis. 2016, 58, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Villamizar, O.; Chambers, C.B.; Mo, Y.Y.; Torry, D.S.; Hofstrand, R.; Riberdy, J.M.; Persons, D.A.; Wilber, A. Data in support of transcriptional regulation and function of Fas-antisense long noncoding RNA during human erythropoiesis. Data Br. 2016, 7, 1288–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secchiero, P.; Melloni, E.; Heikinheimo, M.; Mannisto, S.; Di Pietro, R.; Iacone, A.; Zauli, G. TRAIL regulates normal erythroid maturation through an ERK-dependent pathway. Blood 2004, 103, 517–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamai, L.; Secchiero, P.; Pierpaoli, S.; Bassini, A.; Papa, S.; Alnemri, E.S.; Guidotti, L.; Vitale, M.; Zauli, G. TNF-related apoptosis-inducing ligand (TRAIL) as a negative regulator of normal human erythropoiesis. Blood 2000, 95, 3716–3724. [Google Scholar]
- Felli, N.; Pedini, F.; Zeuner, A.; Petrucci, E.; Testa, U.; Conticello, C.; Biffoni, M.; Di Cataldo, A.; Winkles, J.A.; Peschle, C.; et al. Multiple Members of the TNF Superfamily Contribute to IFN-γ-Mediated Inhibition of Erythropoiesis. J. Immunol. 2005, 175, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Mirandola, P.; Gobbi, G.; Ponti, C.; Sponzilli, I.; Cocco, L.; Vitale, M. PKCε controls protection against TRAIL in erythroid progenitors. Blood 2006, 107, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Vitale, M.; Gobbi, G.; Mirandola, P.; Ponti, C.; Sponzilli, I.; Rinaldi, L.; Manzoli, F.A. TNF-related apoptosis-inducing ligand (TRAIL) and erythropoiesis: A role for PKCε. Eur. J. Histochem. 2006, 50, 15–18. [Google Scholar]
- Sancilio, S.; Giacomo, V.D.; Quaglietta, A.M.; Iacone, A.; Angelucci, D.; Tatasciore, U.; Rana, R.A.; Cataldi, A.; Zauli, G.; Pietro, R. TRAIL promotes a pro-survival signal in erythropoietin-deprived human erythroblasts throught the activation of an NF-κB/IκBα pathway. J. Biol. Regul. Homeost. Agents 2011, 25, 375–386. [Google Scholar] [PubMed]
- Zermati, Y.; Garrido, C.; Amsellem, S.; Fishelson, S.; Bouscary, D.; Valensi, F.; Varet, B.; Solary, E.; Hermine, O. Caspase activation is required for terminal erythroid differentiation. J. Exp. Med. 2001, 193, 247–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregoli, P.A.; Bondurant, M.C. Function of caspases in regulating apoptosis caused by erythropoietin deprivation in erythroid progenitors. J. Cell. Physiol. 1999, 178, 133–143. [Google Scholar] [CrossRef]
- Boehm, D.; Mazurier, C.; Giarratana, M.C.; Darghouth, D.; Faussat, A.M.; Harmand, L.; Douay, L. Caspase-3 Is Involved in the Signalling in Erythroid Differentiation by Targeting Late Progenitors. PLoS ONE 2013, 8, e62303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlile, G.W.; Smith, D.H.; Wiedmann, M. Caspase-3 has a nonapoptotic function in erythroid maturation. Blood 2004, 103, 4310–4316. [Google Scholar] [CrossRef] [Green Version]
- Krauss, S.W.; Lo, A.J.; Short, S.A.; Koury, M.J.; Mohandas, N.; Chasis, J.A. Nuclear substructure reorganization during late-stage erythropoiesis is selective and does not involve caspase cleavage of major nuclear substructural proteins. Blood 2005, 106, 2200–2206. [Google Scholar] [CrossRef]
- Ribeil, J.-A.; Zermati, Y.; Vandekerckhove, J.; Cathelin, S.; Kersual, J.; Dussiot, M.; Coulon, S.; Moura, I.C.; Zeuner, A.; Kirkegaard-Sørensen, T.; et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature 2007, 445, 102–105. [Google Scholar] [CrossRef]
- Hermine, O.; Arlet, J.B.; Ribeil, J.A.; Guillerm, F.; Vandekerkhove, J.; Courtois, G. HSP70, an erythropoiesis regulator that determines the fate of erythroblasts between death and differenciation. Transfus. Clin. Biol. 2013, 20, 144–147. [Google Scholar] [CrossRef]
- Berg, C.P.; Engels, I.H.; Rothbart, A.; Lauber, K.; Renz, A.; Schlosser, S.F.; Schulze-Osthoff, K.; Wesselborg, S. Human mature red blood cells express caspase-3 and caspase-8, but are devoid of mitochondrial regulators of apoptosis. Cell Death Differ. 2001, 8, 1197–1206. [Google Scholar] [CrossRef]
- Mandal, D.; Mazumder, A.; Das, P.; Kundu, M.; Basu, J. Fas-, caspase 8-, and caspase 3-dependent signaling regulates the activity of the aminophospholipid translocase and phosphatidylserine externalization in human erythrocytes. J. Biol. Chem. 2005, 280, 39460–39467. [Google Scholar] [CrossRef] [Green Version]
- Weil, M.; Jacobson, M.D.; Davies, T.J.; Gardner, R.L.; Raft, K.D.; Raft, M.C. Constitutive Expression of the Machinery for Programmed Cell Death. J. Cell Biol. 1996, 133, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sagan, D.; Jermnim, N.; Tangvarasittichai, O. CD95 is not functional in human erythrocytes. Int. J. Lab. Hematol. 2010, 32, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.; Banerjee, M.; Sen, G.; Das, J.K.; Banerjee, A.; Sau, T.J.; Pandit, S.; Giri, A.K.; Biswas, T. Mechanism of erythrocyte death in human population exposed to arsenic through drinking water. Toxicol. Appl. Pharmacol. 2008, 230, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.; Sen, G.; Sarkar, A.; Biswas, T. Atorvastatin acts synergistically with N-acetyl cysteine to provide therapeutic advantage against Fas-activated erythrocyte apoptosis during chronic arsenic exposure in rats. Toxicol. Appl. Pharmacol. 2011, 250, 39–53. [Google Scholar] [CrossRef]
- Mandal, S.; Mukherjee, S.; Chowdhury, K.D.; Sarkar, A.; Basu, K.; Paul, S.; Karmakar, D.; Chatterjee, M.; Biswas, T.; Sadhukhan, G.C.; et al. S-allyl cysteine in combination with clotrimazole downregulates Fas induced apoptotic events in erythrocytes of mice exposed to lead. Biochim. Biophys. Acta-Gen. Subj. 2012, 1820, 9–23. [Google Scholar] [CrossRef]
- Toporkiewicz, M.; Grzybek, M.; Meissner, J.; Michalczyk, I.; Dubielecka, P.M.; Korycka, J.; Seweryn, E.; Sikorski, A.F. Release of an ~55kDa fragment containing the actin-binding domain of β-spectrin by caspase-8 during FND-induced apoptosis depends on the presence of protein 4.1. Arch. Biochem. Biophys. 2013, 535, 205–213. [Google Scholar] [CrossRef]
- Mandal, D.; Baudin-Creuza, V.; Bhattacharyya, A.; Pathak, S.; Delaunay, J.; Kundu, M.; Basu, J. Caspase 3-mediated Proteolysis of the N-terminal Cytoplasmic Domain of the Human Erythroid Anion Exchanger 1 (Band 3). J. Biol. Chem. 2003, 278, 52551–52558. [Google Scholar] [CrossRef] [Green Version]
- Machnicka, B.; Grochowalska, R.; Bogusławska, D.M.; Sikorski, F.; Lecomte, M.C. Spectrin-based skeleton as an actor in cell signaling. Cell. Mol. Life Sci. 2012, 69, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Machnicka, B.; Czogalla, A.; Hryniewicz-Jankowska, A.; Bogusławska, D.M.; Grochowalska, R.; Heger, E.; Sikorski, A.F. Spectrins: A structural platform for stabilization and activation of membrane channels, receptors and transporters. Biochim. Biophys. Acta 2014, 1838, 620–634. [Google Scholar] [CrossRef] [Green Version]
- Machnicka, B.; Grochowalska, R.; Bogusławska, D.M.; Sikorski, A.F. The role of spectrin in cell adhesion and cell–cell contact. Exp. Biol. Med. 2019, 1303–1312. [Google Scholar] [CrossRef]
- Rinalducci, S.; Ferru, E.; Blasi, B.; Turrini, F.; Zolla, L. Oxidative stress and caspase-mediated fragmentation of cytoplasmic domain of erythrocyte band 3 during blood storage. Blood Transfus. 2012, 10, 55–62. [Google Scholar]
- Pietraforte, D.; Matarrese, P.; Straface, E.; Gambardella, L.; Metere, A.; Scorza, G.; Leto, T.L.; Malorni, W.; Minetti, M. Two different pathways are involved in peroxynitrite-induced senescence and apoptosis of human erythrocytes. Free Radic. Biol. Med. 2007, 42, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Tazawa, T.; Hirano, K.; Matsushima, H.; Kumamoto, S.; Hamasaki, N.; Yamaguchi, T.; Beppu, M. Clearance of oxidized erythrocytes by macrophages: Involvement of caspases in the generation of clearance signal at band 3 glycoprotein. Biochem. Biophys. Res. Commun. 2007, 363, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Moitra, P.K.; Saha, S.; Basu, J. Caspase 3 regulates phosphatidylserine externalization and phagocytosis of oxidatively stressed erythrocytes. FEBS Lett. 2002, 513, 184–188. [Google Scholar] [CrossRef] [Green Version]
- Ingram, V.M.; Stretton, A.O.W. Genetic basis of the thalassemia diseases. Nature 1959, 184, 632–633. [Google Scholar] [CrossRef] [PubMed]
- Weatherall, D.J.; Williams, T.N.; Allen, S.J.; O’Donnell, A. The population genetics and dynamics of the thalassemias. Hematol. Oncol. Clin. N. Am. 2010, 24, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Modell, B.; Darlison, M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 2008, 86, 480–487. [Google Scholar] [CrossRef]
- Origa, R. β-Thalassemia. Genet. Med. 2017, 19, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Tari, K.; Valizadeh Ardalan, P.; Abbaszadehdibavar, M.; Atashi, A.; Jalili, A.; Gheidishahran, M. Thalassemia an update: Molecular basis, clinical features and treatment. Int. J. Biomed. Public Heal. 2018, 1, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Musallam, K.M.; Rivella, S.; Vichinsky, E.; Rachmilewitz, E.A. Non-transfusion-dependent thalassemias. Haematologica 2013, 98, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, N.; Pakbaz, Z.; Vichinsky, E. Hb E/beta-thalassaemia: A common & clinically diverse disorder. Indian J. Med. Res. 2011, 134, 522–531. [Google Scholar] [PubMed]
- Ponnikorn, S.; Kong, S.P.; Thitivirachawat, S.; Tanjasiri, C.; Tungpradabkul, S.; Hongeng, S. Proteomic Analysis of β Thalassemia/HbE: A perspective from Hematopoietic Stem Cells (HSCs). In Proteomics Technologies and Applications; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Fucharoen, S.; Weatherall, D.J. The hemoglobin E thalassemias. Cold Spring Harb. Perspect. Med. 2012, 2, a011734. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.A.; Deubelbeiss, K.; Cook, J.D.; Eschbach, J.W.; Harker, L.A.; Funk, D.D.; Mrsaglia, G.; Hillman, R.S.; Slichter, S.; Adamson, J.W.; et al. Ferrokinetics in man. Medicine 1970, 49, 17–53. [Google Scholar] [CrossRef] [PubMed]
- Centis, F.; Tabellini, L.; Lucarelli, G.; Buffi, O.; Tonucci, P.; Persini, B.; Annibali, M.; Emiliani, R.; Iliescu, A.; Rapa, S.; et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with beta-thalassemia major. Blood 2000, 96, 3624–3629. [Google Scholar] [CrossRef]
- Yuan, J.; Angelluci, E.; Lucarelli, G.; Aljurf, M.; Sznder, L.M.; Kiefer, C.R.; Ma, L.; Schrier, S.L. Accelerated Programmed Cell Death (Apoptosis) in Erythroid Precursors of Patients With Severe β-Thalassemia (Cooley’s Anemia). Blood 1993, 82, 374–377. [Google Scholar] [CrossRef] [Green Version]
- Pootrakul, P.; Sirankapracha, P.; Hemsorach, S.; Moungsub, W.; Kumbunlue, R.; Piangitjagum, A.; Wasi, P.; Ma, L.; Schrier, S.L. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in thai patients with thalassemia. Blood 2000, 96, 2606–2612. [Google Scholar] [CrossRef]
- Mathias, L.A.; Fisher, T.C.; Zeng, L.; Meiselman, H.J.; Weinberg, K.I.; Hiti, A.L.; Malik, P. Ineffective erythropoiesis in β-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp. Hematol. 2000, 28, 1343–1353. [Google Scholar] [CrossRef]
- Angelucci, E.; Bai, H.; Centis, F.; Bafti, M.S.; Lucarelli, G.; Ma, L.; Schrier, S. Enhanced macrophagic attack on β-thalassemia major erythroid precursors. Haematologica 2002, 87, 578–583. [Google Scholar]
- Schrier, S.L. Pathophysiology of thalassemia. Curr. Opin. Hematol. 2002, 9, 123–126. [Google Scholar] [CrossRef]
- Schrier, S.L.; Centis, F.; Verneris, M.; Ma, L.; Angelucci, E. The role of oxidant injury in the pathophysiology of human thalassemias. Redox Rep. 2003, 8, 241–245. [Google Scholar] [CrossRef]
- Ponnikorn, S.; Panichakul, T.; Sresanga, K.; Wongborisuth, C.; Roztrakul, S.; Hongeng, S.; Tungpradabkul, S. Phosphoproteomic analysis of apoptotic hematopoietic stem cells from hemoglobin E/β-thalassemia. J. Transl. Med. 2011, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickramasinghe, S.N.; Wiener, E.; Siripanyaphinyo, U.; Chinprasertsuk, S.; Mawas, F.; Fucharoen, S.; Wickramasinghe, S.N.; Pootrakul, P.; Visudhiphan, S. Serum Levels of Tumor Necrosis Factor alpha, Interleukin-1, and Interferon gamma in Betta-Thalassemia/HbE and Their Clinical Significance. Viral Immunol. 1999, 111, 75–79. [Google Scholar]
- Abo Shanab, A.M.; El-Desouky, M.A.; Kholoussi, N.; El-Kamah, G.; Fahmi, A.A. Evaluation of neopterin as a prognostic factor in patients with beta-thalassemia, in comparison with cytokines and immunoglobulins. Arch. Hell. Med. 2015, 32, 60–65. [Google Scholar]
- Tanyong, D.I.; Panichob, P.; Kheansaard, W.; Fucharoen, S. Effect of Tumor Necrosis Factor-Alpha on Erythropoietin and Erythropoietin Receptor-Induced Erythroid Progenitor Cell Proliferation in β-Thalassemia/Hemoglobin E Patients. Turkish J. Haematol. Off. J. Turkish Soc. Haematol. 2015, 32, 304–310. [Google Scholar] [CrossRef]
- Fadhil, R.; Mohammed, H.; Faraj, S. Evaluation of cellular immunity for β-thalassemia major patients in waist thalassemia center. Int. J. Microbiol. Genet. Mol. Biol. Res. 2017, 3, 1–8. [Google Scholar]
- Kheansaard, W.; Panichob, P.; Fucharoen, S.; Tanyong, D.I. Cytokine-induced apoptosis of beta-thalassemia/hemoglobin e erythroid progenitor cells via nitric oxide-mediated process in vitro. Acta Haematol. 2011, 126, 224–230. [Google Scholar] [CrossRef]
- Gupta, U.; Fucharoen, P.; Tanyong, D.; Al, E. Interleukin-1 involved in apoptosis of Beta-thalassemia/Hemoglobin E erythroid progenitor cells. Asian J. Med. Sci. 2012, 3, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Ficarra, S.; Tellone, E.; Giardina, B.; Scatena, R.; Russo, A.; Misiti, F.; Clementi, M.E.; Colucci, D.; Bellocco, E.; Laganà, G.; et al. Derangement of erythrocytic AE1 in beta-thalassemia by caspase 3: Pathogenic mechanisms and implications in red blood cell senescence. J. Membr. Biol. 2009, 228, 43–49. [Google Scholar] [CrossRef]
- Arlet, J.B.; Ribeil, J.A.; Guillem, F.; Negre, O.; Hazoume, A.; Marcion, G.; Beuzard, Y.; Dussiot, M.; Moura, I.C.; Demarest, S.; et al. HSP70 sequestration by free α-globin promotes ineffective erythropoiesis in β-thalassaemia. Nature 2014, 514, 242–246. [Google Scholar] [CrossRef]
- Guillem, F.; Dussiot, M.; Colin, E.; Suriyun, T.; Arlet, J.B.; Goudin, N.; Marcion, G.; Seigneuric, R.; Causse, S.; Gonin, P.; et al. XPO1 regulates erythroid differentiation and is a new target for the treatment of β-thalassemia. Haematologica 2018, 210054. [Google Scholar] [CrossRef] [Green Version]
- Ribeil, J.; Arlet, J.; Dussiot, M.; Al, E. Ineffective Erythropoiesis in β-Thalassemia. Hindawi Publ. Corp. 2013, 2013, 1–11. [Google Scholar]
- Gautier, E.F.; Ducamp, S.; Leduc, M.; Salnot, V.; Guillonneau, F.; Dussiot, M.; Hale, J.; Giarratana, M.C.; Raimbault, A.; Douay, L.; et al. Comprehensive Proteomic Analysis of Human Erythropoiesis. Cell Rep. 2016, 16, 1470–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, X.; Schulz, V.P.; Li, J.; Wu, K.; Liu, J.; Xue, F.; Hu, J.; Mohandas, N.; Gallagher, P.G. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 2014, 123, 3466–3477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennrich, M.L.; Romanov, N.; Horn, P.; Jaeger, S.; Eckstein, V.; Steeples, V.; Ye, F.; Ding, X.; Poisa-Beiro, L.; Lai, M.C.; et al. Cell-specific proteome analyses of human bone marrow reveal molecular features of age-dependent functional decline. Nat. Commun. 2018, 9, 4004. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raducka-Jaszul, O.; Bogusławska, D.M.; Jędruchniewicz, N.; Sikorski, A.F. Role of Extrinsic Apoptotic Signaling Pathway during Definitive Erythropoiesis in Normal Patients and in Patients with β-Thalassemia. Int. J. Mol. Sci. 2020, 21, 3325. https://doi.org/10.3390/ijms21093325
Raducka-Jaszul O, Bogusławska DM, Jędruchniewicz N, Sikorski AF. Role of Extrinsic Apoptotic Signaling Pathway during Definitive Erythropoiesis in Normal Patients and in Patients with β-Thalassemia. International Journal of Molecular Sciences. 2020; 21(9):3325. https://doi.org/10.3390/ijms21093325
Chicago/Turabian StyleRaducka-Jaszul, Olga, Dżamila M. Bogusławska, Natalia Jędruchniewicz, and Aleksander F. Sikorski. 2020. "Role of Extrinsic Apoptotic Signaling Pathway during Definitive Erythropoiesis in Normal Patients and in Patients with β-Thalassemia" International Journal of Molecular Sciences 21, no. 9: 3325. https://doi.org/10.3390/ijms21093325