Effect of Chemokine (C-C Motif) Ligand 7 (CCL7) and Its Receptor (CCR2) Expression on Colorectal Cancer Behaviors

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
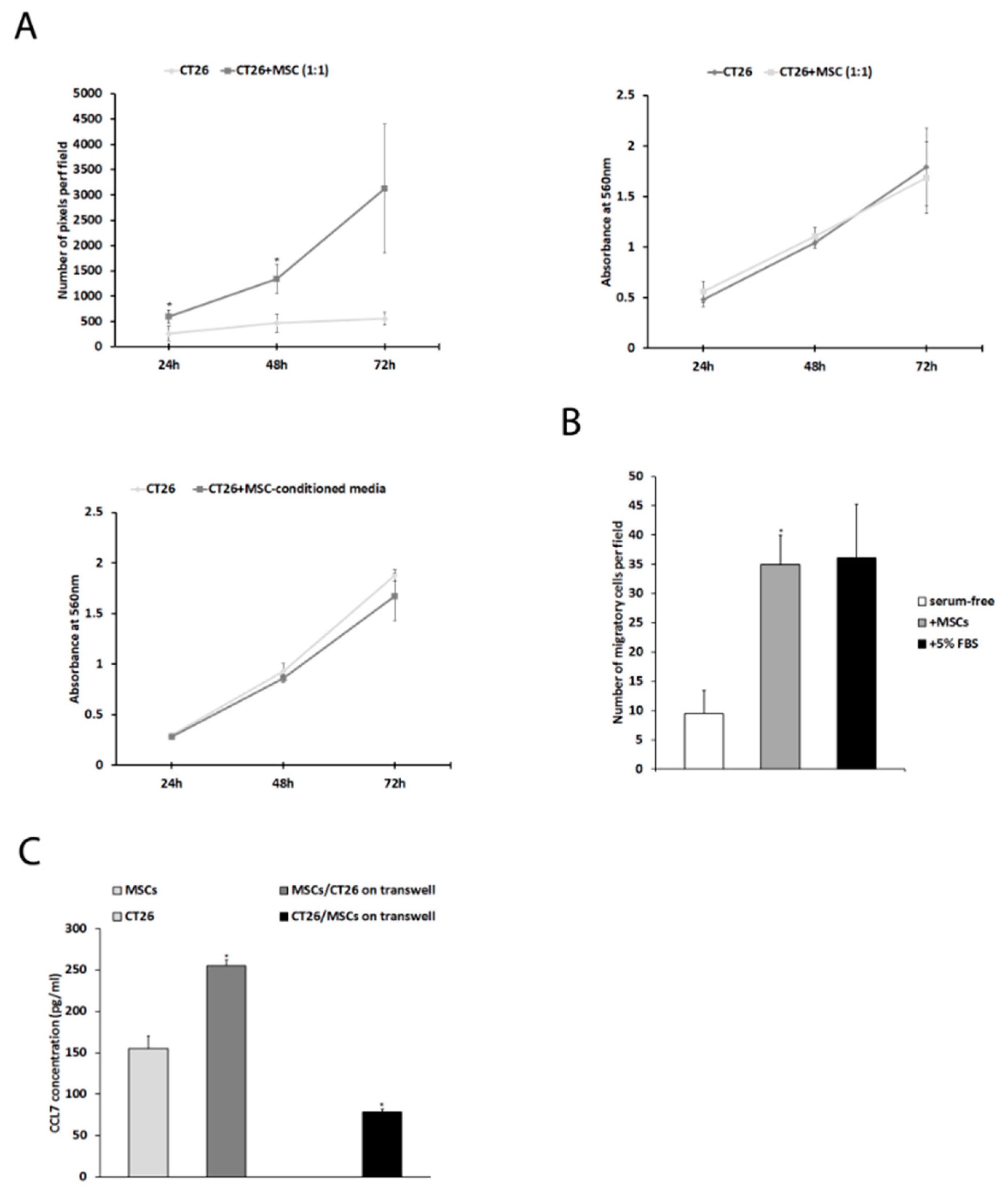
2.1. Mesenchymal Stem Cells Affect CT26 Tumor Cell Proliferation, Migration, and Expression of CCL7 In Vitro
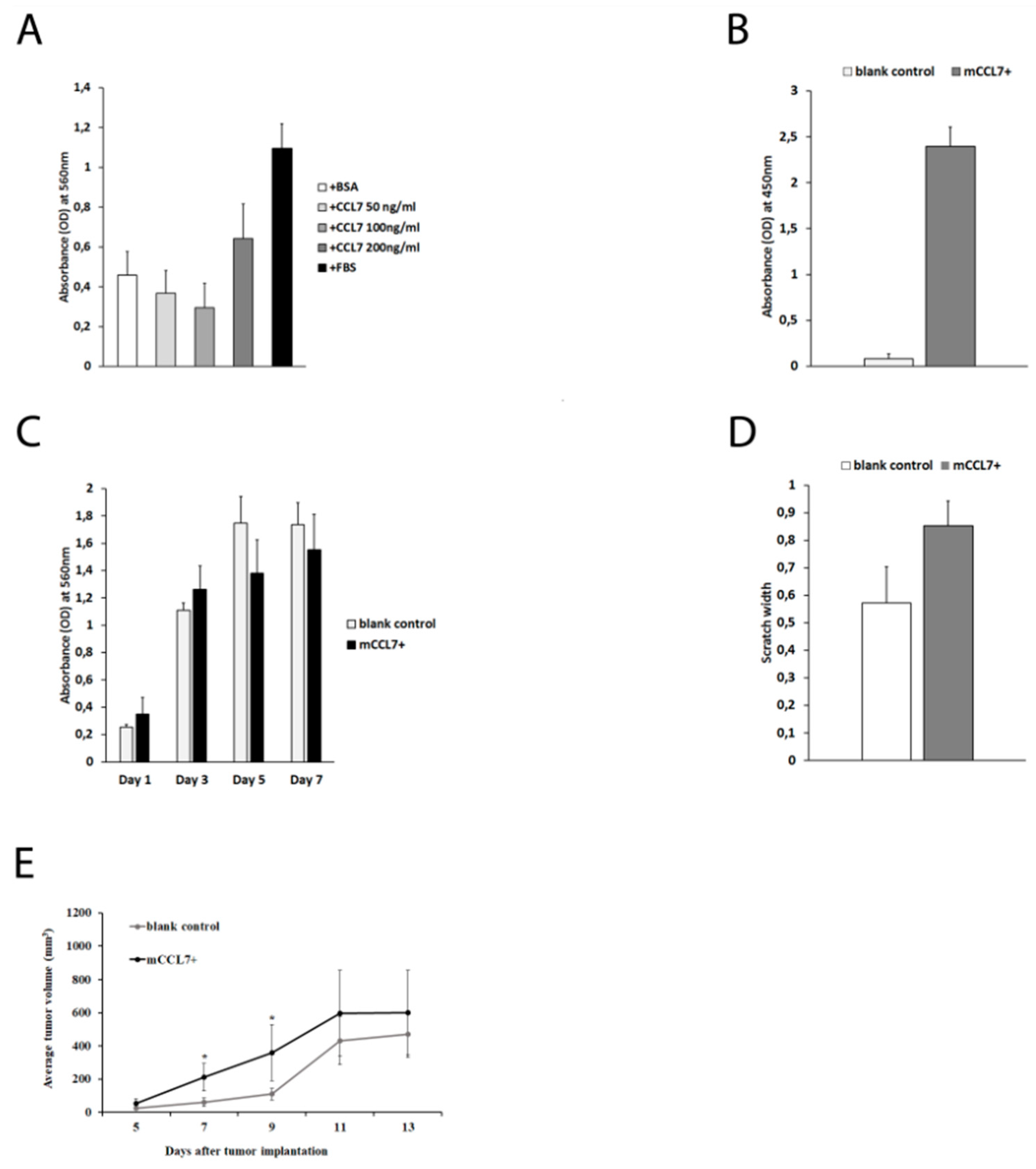
2.2. CCL7 Overexpression Accelerates Early Phase of CT26 Tumor Growth In Vivo
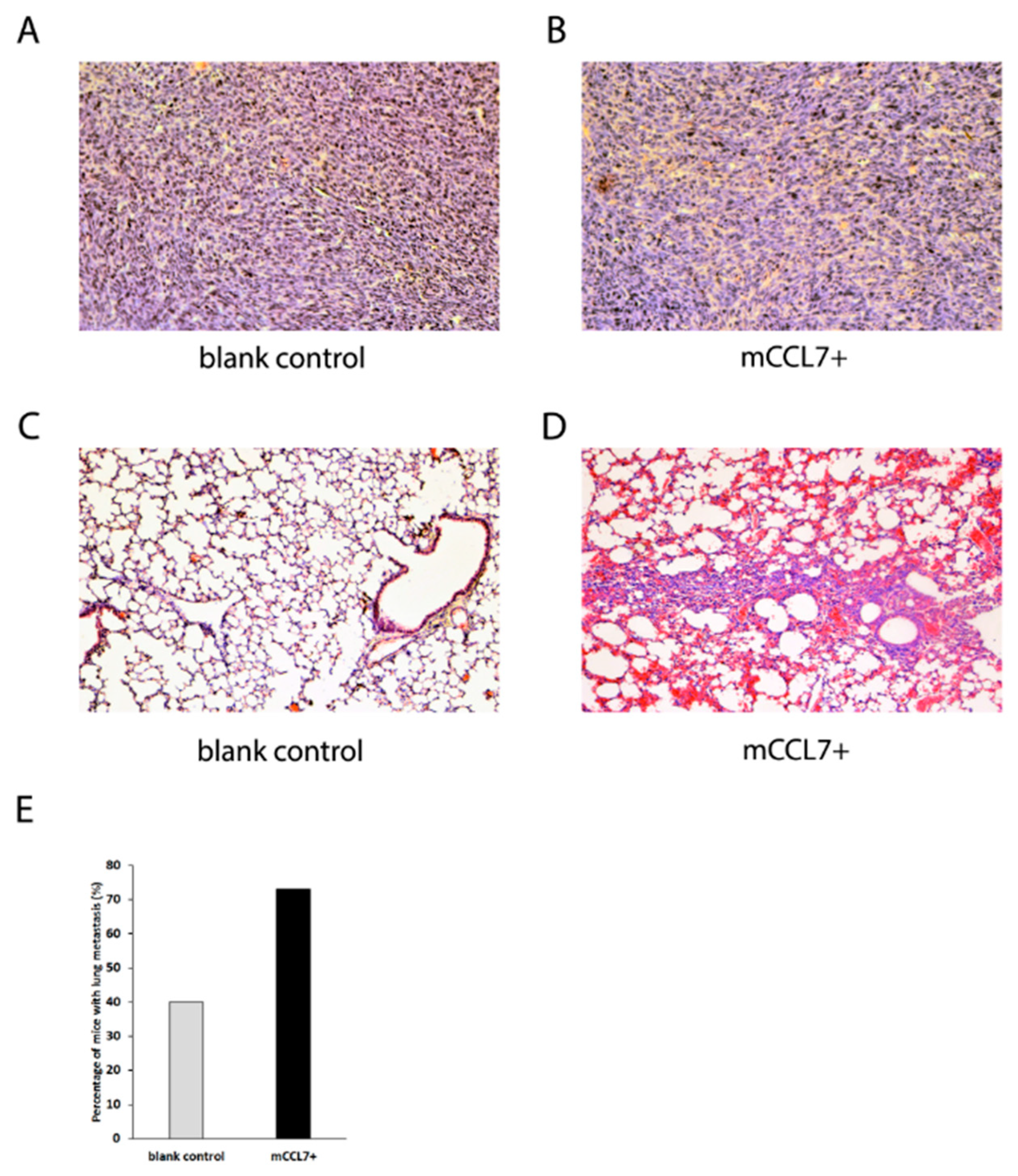
2.3. Overexpressed CCL7 Enhances Lung Metastasis Formation In Murine CT26 Tumor Model
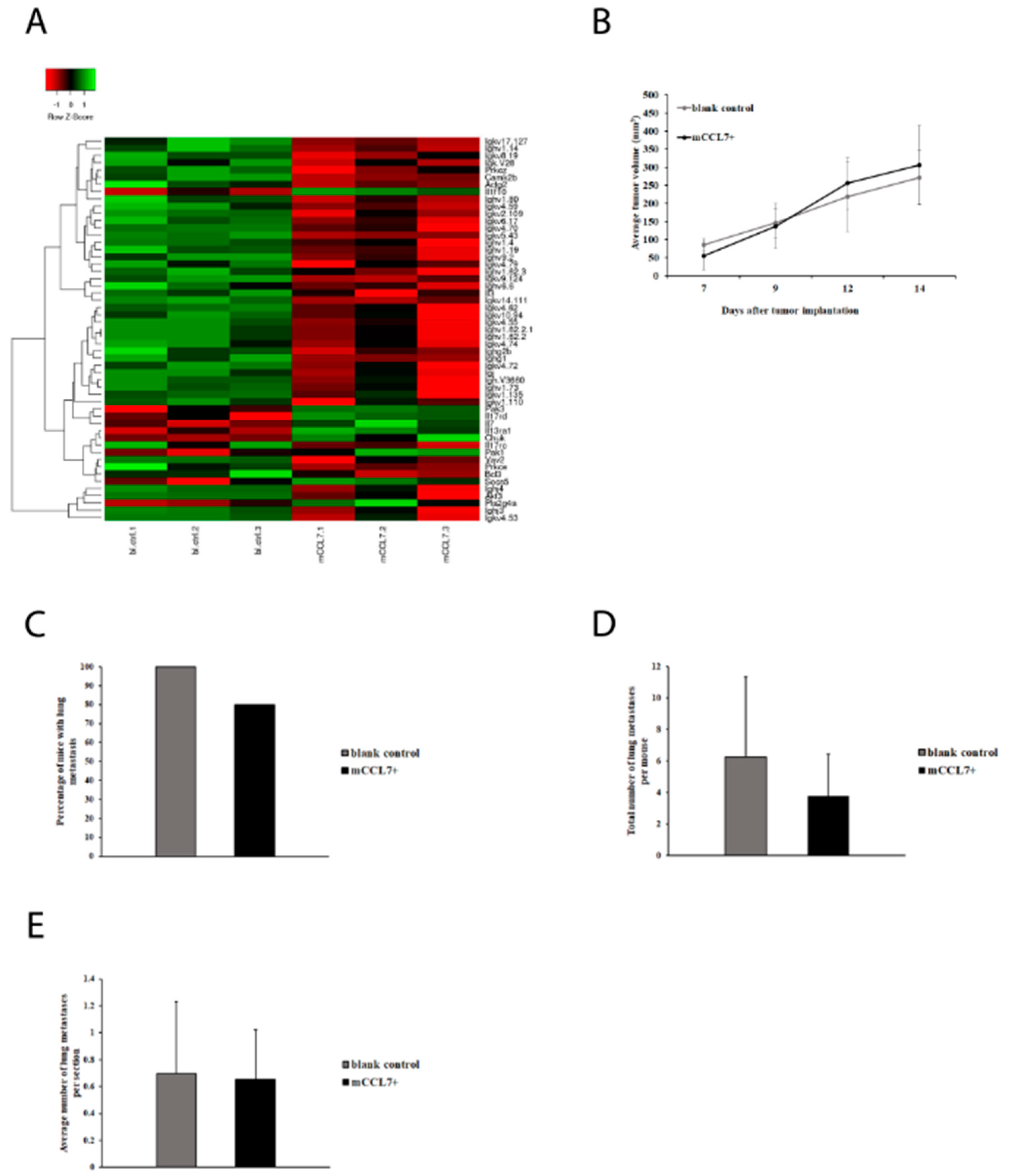
2.4. CCL7 Overexpression in CT26 Tumors Suppresses Expression of Immunoglobulins
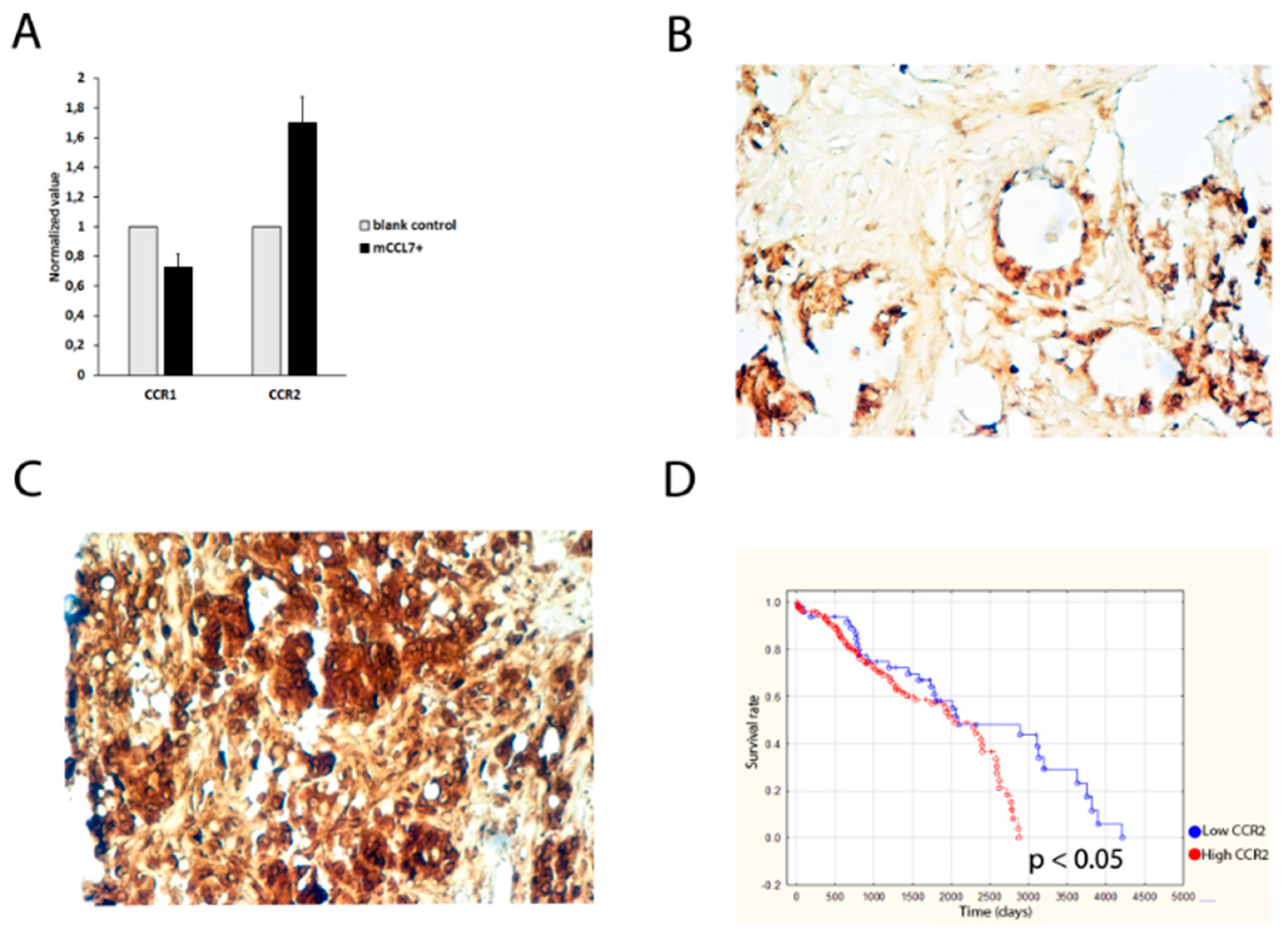
2.5. CCR2 Expression Predicts Overall Survival of Colorectal Cancer Patients
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. MTT Proliferation Assay
4.3. Migration Assays
4.4. Microarray Analysis
4.5. ELISA
4.6. Mouse Tumor Models
4.7. Immunohistochemical Stainings
4.8. Quantitative Real-Time RT-PCR
4.9. Clinical Samples of Human Colon Cancer
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CCL7 | C-C motif chemokine ligand 7 |
| CCR1 | C-C chemokine receptor type 1 |
| CCR2 | C-C chemokine receptor type 2 |
| CCR3 | C-C chemokine receptor type 3 |
| TMA | tissue microarray |
| MSC | mesenchymal stem cell |
| SD | standard deviation |
| RFS | relapse-free survival |
References
- Stewart, B.W.; Wild, C.; International Agency for Research on Cancer; World Health Organization. World Cancer Report 2014; International Agency for Research on Cancer WHO Press: Lyon, France; Geneva, Switzerland, 2014; p. xiv. 630p. [Google Scholar]
- Verdecchia, A.; Francisci, S.; Brenner, H.; Gatta, G.; Micheli, A.; Mangone, L.; Kunkler, I.; EUROCARE-4 Working Group. Recent cancer survival in Europe: A 2000-02 period analysis of EUROCARE-4 data. Lancet Oncol. 2007, 8, 784–796. [Google Scholar] [CrossRef]
- O’Malley, G.; Heijltjes, M.; Houston, A.M.; Rani, S.; Ritter, T.; Egan, L.J.; Ryan, A.E. Mesenchymal stromal cells (MSCs) and colorectal cancer: A troublesome twosome for the anti-tumour immune response? Oncotarget 2016, 7, 60752–60774. [Google Scholar] [CrossRef] [PubMed]
- Itatani, Y.; Kawada, K.; Inamoto, S.; Yamamoto, T.; Ogawa, R.; Taketo, M.M.; Sakai, Y. The Role of Chemokines in Promoting Colorectal Cancer Invasion/Metastasis. Int. J. Mol. Sci. 2016, 17, 643. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, J.; Proost, P.; Lenaerts, J.P.; Opdenakker, G. Structural and functional identification of two human, tumor-derived monocyte chemotactic proteins (MCP-2 and MCP-3) belonging to the chemokine family. J. Exp. Med. 1992, 176, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Uguccioni, M.; D’Apuzzo, M.; Loetscher, M.; Dewald, B.; Baggiolini, M. Actions of the chemotactic cytokines MCP-1, MCP-2, MCP-3, RANTES, MIP-1 alpha and MIP-1 beta on human monocytes. Eur. J. Immunol. 1995, 25, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Menten, P.; Proost, P.; Struyf, S.; Van Coillie, E.; Put, W.; Lenaerts, J.P.; Conings, R.; Jaspar, J.M.; De Groote, D.; Billiau, A.; et al. Differential induction of monocyte chemotactic protein-3 in mononuclear leukocytes and fibroblasts by interferon-alpha/beta and interferon-gamma reveals MCP-3 heterogeneity. Eur. J. Immunol. 1999, 29, 678–685. [Google Scholar] [CrossRef]
- Power, C.A.; Clemetson, J.M.; Clemetson, K.J.; Wells, T.N. Chemokine and chemokine receptor mRNA expression in human platelets. Cytokine 1995, 7, 479–482. [Google Scholar] [CrossRef]
- Xu, L.L.; McVicar, D.W.; Ben-Baruch, A.; Kuhns, D.B.; Johnston, J.; Oppenheim, J.J.; Wang, J.M. Monocyte Chemotactic Protein-3 (Mcp3) Interacts with Multiple Leukocyte Receptors—Binding and Signaling of Mcp3 through Shared as Well as Unique Receptors on Monocytes and Neutrophils. Eur. J. Immunol. 1995, 25, 2612–2617. [Google Scholar] [CrossRef]
- Fioretti, F.; Fradelizi, D.; Stoppacciaro, A.; Ramponi, S.; Ruco, L.; Minty, A.; Sozzani, S.; Garlanda, C.; Vecchi, A.; Mantovani, A. Reduced tumorigenicity and augmented leukocyte infiltration after monocyte chemotactic protein-3 (MCP-3) gene transfer: Perivascular accumulation of dendritic cells in peritumoral tissue and neutrophil recruitment within the tumor. J. Immunol. 1998, 161, 342–346. [Google Scholar]
- Hu, J.Y.; Li, G.C.; Wang, W.M.; Zhu, J.G.; Li, Y.F.; Zhou, G.H.; Sun, Q.B. Transfection of colorectal cancer cells with chemokine MCP-3 (monocyte chemotactic protein-3) gene retards tumor growth and inhibits tumor metastasis. World J. Gastroenterol. 2002, 8, 1067–1072. [Google Scholar] [CrossRef]
- Cho, Y.B.; Lee, W.Y.; Choi, S.J.; Kim, J.; Hong, H.K.; Kim, S.H.; Choi, Y.L.; Kim, H.C.; Yun, S.H.; Chun, H.K.; et al. CC chemokine ligand 7 expression in liver metastasis of colorectal cancer. Oncol. Rep. 2012, 28, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.T.; Wang, J.Y.; Chen, M.K.; Chen, H.C.; Chang, T.H.; Su, B.W.; Chang, P.J. Colon cancer mesenchymal stem cells modulate the tumorigenicity of colon cancer through interleukin 6. Exp. Cell Res. 2013, 319, 2216–2229. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.S.; Yang, S.H.; Lei, Y.P.; Tsai, C.C.; Chen, H.W.; Hsu, C.Y.; Chen, L.L.; Wang, H.W.; Miller, S.A.; Chiou, S.H.; et al. Mesenchymal stem cells promote formation of colorectal tumors in mice. Gastroenterology 2011, 141, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, K.; Kitadai, Y.; Tanaka, M.; Sumida, T.; Kodama, M.; Higashi, Y.; Tanaka, S.; Yasui, W.; Chayama, K. Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int. J. Cancer 2010, 127, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, S.Y.; Song, S.J.; Hong, H.K.; Lee, Y.; Oh, B.Y.; Lee, W.Y.; Cho, Y.B. Crosstalk between CCL7 and CCR3 promotes metastasis of colon cancer cells via ERK-JNK signaling pathways. Oncotarget 2016, 7, 36842–36853. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.W.; Che, Z.M.; Kim, J.; Kim, K.; Kim, K.Y.; Williams, D.; Kim, J. Tumor-stromal crosstalk in invasion of oral squamous cell carcinoma: A pivotal role of CCL7. Int. J. Cancer 2010, 127, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.L.; Lee, L.Y.; Wang, C.C.; Liang, Y.; Huang, S.F.; Wu, C.M. CCL7 and CCL21 overexpression in gastric cancer is associated with lymph node metastasis and poor prognosis. World J. Gastroenterol. 2012, 18, 1249–1256. [Google Scholar] [CrossRef]
- Wyler, L.; Napoli, C.U.; Ingold, B.; Sulser, T.; Heikenwälder, M.; Schraml, P.; Moch, H. Brain metastasis in renal cancer patients: Metastatic pattern, tumour-associated macrophages and chemokine/chemoreceptor expression. Br. J. Cancer 2014, 110, 686–694. [Google Scholar] [CrossRef]
- Wu, K.; Fukuda, K.; Xing, F.; Zhang, Y.; Sharma, S.; Liu, Y.; Chan, M.; Zhou, X.; Qasem, S.; Pochampally, R.; et al. Roles of the cyclooxygenase 2 matrix metalloproteinase 1 pathway in brain metastasis of breast cancer. J. Biol. Chem. 2015, 290, 9842–9854. [Google Scholar] [CrossRef]
- Rafei, M.; Hsieh, J.; Fortier, S.; Li, M.; Yuan, S.; Birman, E.; Forner, K.; Boivin, M.N.; Doody, K.; Tremblay, M.; et al. Mesenchymal stromal cell-derived CCL2 suppresses plasma cell immunoglobulin production via STAT3 inactivation and PAX5 induction. Blood 2008, 112, 4991–4998. [Google Scholar] [CrossRef]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Fujishita, T.; Loetscher, P.; Revesz, L.; Hashida, H.; Kizaka-Kondoh, S.; Aoki, M.; Taketo, M.M. Inactivation of chemokine (C-C motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 13063–13068. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Fujishita, T.; Kurimoto, K.; Miyachi, H.; Kitano, S.; Inamoto, S.; Itatani, Y.; Saitou, M.; Maekawa, T.; Taketo, M.M. CCR1-mediated accumulation of myeloid cells in the liver microenvironment promoting mouse colon cancer metastasis. Clin. Exp. Metast. 2014, 31, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Rodero, M.P.; Auvynet, C.; Poupel, L.; Combadiere, B.; Combadiere, C. Control of Both Myeloid Cell Infiltration and Angiogenesis by CCR1 Promotes Liver Cancer Metastasis Development in Mice. Neoplasia 2013, 15, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www1.heatmapper.ca (accessed on 3 December 2018).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Fold Change (CT26 vs. MSC) | Regulation (CT26 vs. MSC) |
|---|---|---|
| Ccl2 | −18,087187 | down |
| Ccl5 | −20,740484 | down |
| Ccl7 | −13,637749 | down |
| Ccl8 | −6,8785295 | down |
| Ccl27a | −19,154331 | down |
| Ccr1 | 5,850982 | up |
| Ccrl2 | −6,921777 | down |
| Cxcl1 | −13,09386 | down |
| Cxcl12 | −877,7667 | down |
| Cxcl13 | 13,571245 | up |
| Cxcl16 | −47,79881 | down |
| Cxcl16 | −24,529097 | down |
| Cxcl2 | −35,86758 | down |
| Cxcl3 | −6,8512273 | down |
| Cxcl5 | −83,25011 | down |
| Cxcr7 | −6,042842 | down |
| Ccl2 | −18,087187 | down |
| Gene Symbol | Fold Change (CT26 + MSC vs. CT26) |
|---|---|
| C5ar1 | 1,1 |
| Ccbp2 | 1,1 |
| Ccl1 | 1,1 |
| Ccl11 | −1,03 |
| Ccl12 | 1,1 |
| Ccl17 | 1,58 |
| Ccl19 | 1,1 |
| Ccl2 | 2,07 |
| Ccl20 | 2,97 |
| Ccl22 | 1,1 |
| Ccl24 | 1,1 |
| Ccl25 | 1,21 |
| Ccl26 | 1,1 |
| Ccl28 | 1,1 |
| Ccl3 | 1,1 |
| Ccl4 | 1,93 |
| Ccl5 | 1,82 |
| Ccl6 | 3,12 |
| Ccl7 | 1,46 |
| Ccl8 | 1,25 |
| Ccl9 | 2,04 |
| Ccr1 | −1,91 |
| Ccr10 | −1,09 |
| Ccr1l1 | 1,1 |
| Ccr2 | 1,1 |
| Ccr3 | 1,1 |
| Ccr4 | 1,1 |
| Ccr5 | 1,1 |
| Ccr6 | 1,1 |
| Ccr7 | 1,1 |
| Ccr8 | 1,1 |
| Ccr9 | 1,1 |
| Ccrl1 | 1,1 |
| Ccrl2 | 1,1 |
| Cmklr1 | 1,1 |
| Cmtm2a | 1,1 |
| Cmtm3 | −1,07 |
| Cmtm4 | 1,05 |
| Cmtm5 | 1,1 |
| Cmtm6 | 1,25 |
| Cx3cl1 | 1,1 |
| Cx3cr1 | 1,32 |
| Cxcl1 | −1,43 |
| Cxcl10 | 1,87 |
| Cxcl11 | 1,1 |
| Cxcl12 | 1,1 |
| Cxcl13 | −1,48 |
| Cxcl14 | 1,1 |
| Cxcl15 | 1,15 |
| Cxcl16 | 1,11 |
| Cxcl2 | 1,1 |
| Cxcl3 | 1,1 |
| Cxcl5 | −2,21 |
| Cxcl9 | 1,1 |
| Cxcr1 | 1,1 |
| Cxcr2 | 1,1 |
| Cxcr3 | 1,1 |
| Cxcr4 | −1,03 |
| Cxcr5 | 1,1 |
| Cxcr6 | 1,19 |
| Cxcr7 | 1,32 |
| Darc | 1,74 |
| Fpr1 | 1,1 |
| Gpr17 | 1,1 |
| Hif1a | 1,19 |
| Ifng | 1,1 |
| Il16 | 1,1 |
| Il1b | 1,1 |
| Il4 | 1,1 |
| Il6 | 1,51 |
| Itgam | 1,1 |
| Itgb2 | 1,04 |
| Mapk1 | 1,17 |
| Mapk14 | 1,08 |
| Pf4 | 1,21 |
| Ppbp | −1,78 |
| Slit2 | −1,13 |
| Tgfb1 | 1,07 |
| Tlr2 | 1,1 |
| Tlr4 | −1,31 |
| Tnf | 1,1 |
| Tymp | 1,03 |
| Xcl1 | 1,1 |
| Xcr1 | 1,1 |
| Gene Symbol | Fold Change (mCCL7+ vs. Blank Control) | Regulation (mCCL7+ vs. Blank Control) |
|---|---|---|
| Obox2 | 1.890295359 | up |
| H2-DMb2 | 1.765182186 | up |
| C4a | 1.674796748 | up |
| Olfr102 | 1.603092784 | up |
| Olfr907 | 1.547263682 | up |
| Mcpt4 | 1.546511628 | up |
| Rhox2h | 1.529680365 | up |
| Igh-V7183 | 1.520408163 | up |
| Akr1c19 | 1.495283019 | up |
| Ccl7 | 1.495238095 | up |
| Scgb2b6 | 1.478723404 | up |
| Olfr347 | 1.464539007 | up |
| Vmn2r28 | 1.458715596 | up |
| Ighv8-5 | 1.458563536 | up |
| Fpr2 | 1.457413249 | up |
| CCR2 Expression | Low | High |
|---|---|---|
| number of patients | 56 | 149 |
| female | 29 | 80 |
| male | 27 | 69 |
| age (years) ± SD | 68 ± 15 | 69 ± 12 |
| LN metastasis + (% of positive patients) | 25 (27%) | 69 (73%) |
| RFS (days) ± SD | 1403 ± 1279 | 981 ± 865 |
| OS (days) ± SD | 1622 ± 1197 | 1219 ± 988 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurzejamska, E.; Sacharczuk, M.; Landázuri, N.; Kovtonyuk, O.; Lazarczyk, M.; Ananthaseshan, S.; Gaciong, Z.; Religa, P. Effect of Chemokine (C-C Motif) Ligand 7 (CCL7) and Its Receptor (CCR2) Expression on Colorectal Cancer Behaviors. Int. J. Mol. Sci. 2019, 20, 686. https://doi.org/10.3390/ijms20030686
Kurzejamska E, Sacharczuk M, Landázuri N, Kovtonyuk O, Lazarczyk M, Ananthaseshan S, Gaciong Z, Religa P. Effect of Chemokine (C-C Motif) Ligand 7 (CCL7) and Its Receptor (CCR2) Expression on Colorectal Cancer Behaviors. International Journal of Molecular Sciences. 2019; 20(3):686. https://doi.org/10.3390/ijms20030686
Chicago/Turabian StyleKurzejamska, Ewa, Mariusz Sacharczuk, Natalia Landázuri, Oksana Kovtonyuk, Marzena Lazarczyk, Sharan Ananthaseshan, Zbigniew Gaciong, and Piotr Religa. 2019. "Effect of Chemokine (C-C Motif) Ligand 7 (CCL7) and Its Receptor (CCR2) Expression on Colorectal Cancer Behaviors" International Journal of Molecular Sciences 20, no. 3: 686. https://doi.org/10.3390/ijms20030686