
EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment


1
Molecular Signal Transduction Laboratories, Institute for Microscopic Anatomy and Neurobiology, Focus Program Translational Neuroscience (FTN), Rhine Mainz Neuroscience Network (rmn2), Johannes Gutenberg University, School of Medicine, 55131 Mainz, Germany
2
German Cancer Consortium (DKTK), partner site Frankfurt/Mainz, 55131 Mainz, Germany
3
German Cancer Research Center (DKFZ), 69120 Heidelberg, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(6), 1295; https://doi.org/10.3390/ijms18061295
Submission received: 30 March 2017
/
Revised: 9 June 2017
/
Accepted: 14 June 2017
/
Published: 18 June 2017
(This article belongs to the Special Issue Glioma Cell Invasion)
Abstract
:Epidermal growth factor receptor (EGFR) and the mutant EGFRvIII are major focal points in current concepts of targeted cancer therapy for glioblastoma multiforme (GBM), the most malignant primary brain tumor. The receptors participate in the key processes of tumor cell invasion and tumor-related angiogenesis and their upregulation correlates with the poor prognosis of glioma patients. Glioma cell invasion and increased angiogenesis share mechanisms of the degradation of the extracellular matrix (ECM) through upregulation of ECM-degrading proteases as well as the activation of aberrant signaling pathways. This review describes the role of EGFR and EGFRvIII in those mechanisms which might offer new combined therapeutic approaches targeting EGFR or EGFRvIII together with drug treatments against proteases of the ECM or downstream signaling to increase the inhibitory effects of mono-therapies.
1. Introduction
Enhanced angiogenesis and diffuse cell invasion are prominent hallmarks of glioblastoma multiforme (GBM) which add to its lethal character. It is classified as grade IV by the World Health Organization (WHO) classification of tumors of the central nervous system and is diagnosed based on histological parameters like tumor necrosis and, since the current update (2016 WHO Classification of Tumors of the Central Nervous System), on additional molecular features like the status of isocitrate dehydrogenase (IDH). GBMs are divided into two subgroups according to the presence of wildtype or mutated IDH. While IDH mutation occurs in about 10% of cases and corresponds mostly with secondary GBM (resulting from progression of lower grade diffuse glioma), IDH-wildtype is with about 90% of cases the most common type and corresponds frequently with primary or de novo GBM [1]. The patient prognosis is poor; the median survival time is only approximately 14 months after initial diagnosis. Optimized therapy, a combination of radiation and temozolomide, an alkylating agent, increases the percentage of patients alive 2 years after diagnosis but, unfortunately, the tumors usually recur [2].
1.1. Glioma Invasion and Angiogenesis
The major reasons that therapies fail are the proliferative and highly invasive nature of the tumor cells and the occurrence of increased angiogenesis. Glioma migration is assisted through the interaction of glioma cells with the neural microenvironment, which can be blood vessels, white matter tracts, brain parenchyma, or the subarachnoid space [3]. The invasion of the cells is a complex process of cell-cell-interaction, extracellular matrix (ECM)-cell interaction, matrix modulation, and cell motility. Tumor growth and invasion needs space, therefore processes to degrade the extracellular environment are incorporated. Through upregulation of proteases such as cysteine proteases, serine proteases, and especially matrix metalloproteases, the tumor cells actively enlarge the space for migration while a parallel activation of aberrant signaling pathways promotes cell motility and proliferation [4]. Angiogenesis, which is the formation of new blood vessels from existing ones, and tumor cell invasion share these mechanisms. The upregulation of angiogenic factors like vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), and fibroblast growth factor (FGF) leads to the secretion of proteases, which degrade the basement membrane and the ECM to enable endothelial cells to proliferate into the surrounding matrix. The inhibition of cell invasion and a reduction of angiogenesis is a favorable aim in glioma therapy to reduce tumor growth and size and to make the tumor mass more accessible to standard therapies and surgery.
1.2. The Epidermal Growth Factor and the Mutant EGFRvIII
Epidermal growth factor receptor (EGFR) gene amplification is the most frequent genetic alteration in primary GBM (about 40 percent) and high EGFR expression has been associated with primary human tumors [5]. In addition to gene amplification, a mutant form of EGFR, generally known as EGFRvIII or ΔEGFR, occurs in about 50–60 percent of EGFR-overexpressing GMBs and is exclusively expressed in tumor cells. An in-frame deletion of 267 amino acids from exon 2 to 7 in the extracellular domain of EGFR leads to the inability to bind canonical EGFR ligands. However, EGFRvIII displays constitutive tyrosine kinase activity and mediates persistent intracellular signaling, inefficient ubiquitylation, impaired internalization, and degradation, and therefore escapes downregulation [6,7,8]. EGFRvIII-positive tumors have been associated with poor prognosis and shorter life expectancy and both EGFR and EGFRvIII have been linked to the invasive behavior of glioblastomas together with increased angiogenesis [9,10,11,12,13]. It is also known that EGF promotes tumor angiogenesis, cell motility, and invasion, and that EGFR stimulates cell migration through receptor phosphorylation and subsequent activation of downstream signaling pathways [14,15,16]. However, the cellular and molecular mechanisms behind this and the involvement of the mutant EGFRvIII are still not fully resolved.
There are several treatments available (Table 1), including monoclonal antibodies like cetuximab or small molecule inhibitors like gefitinib, and a vaccination called rindopepimut was even developed to be administered to EGFRvIII-positive tumors [17,18,19]. To complicate things, glioma cells treated with those agents often show resistance mechanisms [20]. They simply overcome the inhibition through increased expression of other growth factors or they switch to different signaling pathways. The latest example of the difficulties in developing effective agents is the failure of a phase III clinical trial of the vaccine rindopepimut, which showed no significant benefit of the vaccine on the overall survival of the patients, despite promising results in a phase II trial [21,22]. A logical consequence to circumvent cell resistance is to apply combination gene therapy and target different molecules or pathways to cover non-overlapping mechanisms and to increase the inhibitory effect. Diverse combination gene therapies are already in clinical trials but there is still need for research to develop novel effective therapeutics. The purpose of this review is to provide current knowledge on how EGFR and EGFRvIII contribute to cell invasion and angiogenesis with a focus on the interaction with ECM-degrading proteases which are involved in matrix modulations and on the activation of transduction signaling pathways. In this context, we discuss the perspective on potential therapeutics and combination gene therapy.
2. ECM Remodeling: Induction of Matrix-Degrading Proteases
The controlled attachment to or the release from matrix components is inevitable for tumor cell invasion and migration of endothelial cells during angiogenesis (Figure 1). It involves increased proteolytic activity of matrix-degrading proteases which alters cell-cell and cell-ECM interaction to loosen intercellular adhesion and enable migration through the ECM. Matrix metalloproteases (MMPs), serine proteases, and cysteine proteases mediate the cleavage of various, partially shared, substrates such as gelatin, elastin, collagen, plasminogen, and laminin. As their upregulation in glioma correlates with the upregulation of EGFR and EGFRvIII, the proteases might be an interesting target to examine closer.
2.1. Matrix Metalloprotease (MMPs)
MMPs are a large group of proteolytic enzymes that degrade ECM proteins in processes associated with morphogenesis, metabolism regulation, and normal tissue turnover. A loss of activity control results in pathological conditions such as arthritis, inflammation, and vascular diseases; a variety of metalloproteases have already been linked to tumor progression in cancer. Among those, gelatinase-A (MMP-2), gelatinase-B (MMP-9), and matrix metalloproteinase-14 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas [23]. Upregulated expressions are correlated with increased glioma malignancy and the aggressiveness of the tumor. Their expression are both increased in high-grade astrocytomas and their downregulation decreases tumor invasion and tumor growth [23,24].
2.1.1. EGFR and MMPs
In glioblastoma cell line T98G, EGFR upregulates the collagenase MMP-1 via mitogen-activated protein kinase (MAPK) signaling which leads to a decreased cell invasion. An inhibition of EGFR via tyrosine kinase inhibitor AG1478 in turn suppresses EGF-induced MMP-1 expression [25]. The protease MMP-2 is not typically associated with tumor invasion but it is indicated that integrin-mediated EGFR signaling is activated through the direct binding of MMP-2 and PAK4 (a serine/threonine kinase that is involved in the regulation of cell motility, invasion, and growth). As a result, glioma cells develop resistance to anoikis, a form of ECM detachment-induced cell death, increasing their ability to survive in an anchorage-independent manner and therefore facilitating tumor cell invasion and migration [26]. Also, it is already known that EGFR activation promotes MMP-9 expression in different cell types. For example, EGF stimulates migration and MMP-9-dependent invasion in ovarian cancer cells. The process could be impaired through the inhibition of phosphoinositide 3-kinase (PI3K) by LY294002 or wortmannin and MAPK by SB202190 or PD98059, thereby blocking EGF-dependent junction dissolution [27]. The activation of MMP-9 was also correlated with EGFR expression in primary GBM tumors (tumors positive for EGFR, but negative for tumor suppressor protein p53), indicating a link between MMP-9 activation and molecular features of primary GBMs [28]. However, the associated signaling pathways that control MMP-9 remain unclear. Wang et al. discovered the impact of microRNA (miRNA)-181c on MMP-9 in the human glioblastoma cell line A-172. The upregulation of MMP-9 through EGF stimulation could be inhibited by EGFR inhibitor AG1478 or by inhibiting Akt-phosphorylation using LY294002 which blocks the PI3K/Akt pathway. Additionally, overexpression of miRNA-181c downregulates MMP-9 expression at the level of Akt phosphorylation [29].
2.1.2. EGFRvIII and MMPs
The receptor mutant EGFRvIII seems to be mainly involved in upregulation of genes promoting invasive phenotypes like MMP-1 and collagenase-3 (MMP-13), an association shown in athymic mice xenografts [10]. However, this finding stands in contrast to a quantitative proteomics analysis showing only low to no detectible secretion of MMP-1 by EGFRvIII-overexpressing U87 glioma cells [30]. EGFRvIII expression was also strongly correlated with MMP-9 expression, similar to the wild type receptor [28]. The mechanism is still not fully clear, but a regulation through the MAPK/ERK pathway is presumable as it is known that EGFRvIII activates extracellular-signal regulated kinase ERK (1/2) in glioma cells which has been shown to be a direct regulator of MMP-9 [31,32]. In addition to this, a paracrine mechanism driven by EGFRvIII promotes tumor proliferation of EGFR-expressing human glioma cells and stem-like cells (GSCs) and thereby increases tumor heterogeneity and invasiveness [33]. Treatment of EGFRvIII-expressing GSCs with EGFR inhibitors AG1478 or gefitinib promotes invasiveness by suppressing EGFR-mediated transcription factor ID3 expression and subsequently increasing MMP-3 expression [34].
2.2. Serine Proteases
The family of serine proteases contains multiple enzymes with a nucleophilic serine in the enzyme active site that cleave peptide bonds in proteins and are ubiquitously expressed in both eukaryotes and prokaryotes. Degradation of the ECM involves the activation of the serine proteases urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA) which share plasminogen as the major substrate and specific inhibitors, the plasminogen activator inhibitors (PAI) [4]. uPA and its receptor (uPAR) associate with several members of the integrin family and receptor tyrosine kinases to activate known signaling pathways like MEK-ERK1/2 and JAK-STAT signaling and are frequently expressed at high levels in GBM [35]. Studies showed that they promote glioma invasion and cell growth and protein overexpression was associated with poor prognosis [36].
2.2.1. EGFR and Serine Proteases
PAI-1 is a key regulator of the uPA-uPAR system as its binding with lipoprotein receptor-related protein (LRP) causes the internalization of the ligand-receptor complex [37]. PAI-1 as well as EGFR are highly expressed in grade IV gliomas, and both are poor prognosis markers for overall survival in glioma patients [38]. The in vitro expression of PAI-1, regulated by cytokines and growth factors like EGF, upregulates tPA and uPA in U373 astrocytoma cells followed by a slower increase in PAI-1 [39]. EGFR ligand transforming growth factor (TGFα) upregulates uPA and uPAR, while inhibition via uPAR-specific antibodies decreased the TGFα-induced cell invasiveness [40]. EGFR signaling also increases uPAR-mediated cell invasion downstream of phospholipase Cγ (PLCγ) signaling in human prostate carcinoma and binding of EGF enhances PAI-1 expression via sequential activation of c-Src, protein kinase C-δ, and sphingosine kinase 1 [41,42].
2.2.2. EGFRvIII and Serine Proteases
As of yet, the interaction with EGFRvIII is not well known. However, it was demonstrated that uPAR is required for Stat5b activation downstream of EGFRvIII in EGFRvIII-expressing U87 cells, but can also activate ERK (1/2) even if the cells were treated with tyrosine inhibitors erlotinib and gefitinib [43]. In contrast to wild type EGFR-overexpressing cell lines, PAI-1 was not detected when EGFRvIII was overexpressed [30].
2.3. Cysteine Proteases
Cathepsins belong to the papain family, one of six major families of cysteine proteases; cathepsin B is the most studied. It mediates the degradation of the ECM by itself or activates other proteases. Precursor pro-cathepsin D can be activated by several molecules such as cathepsin D and G or uPA and tPA, and cathepsin D in turn can activate uPA, MMPs, and plasmin [4]. Like the MMPs and serine proteases, cathepsin B expression is highly elevated in glioma cells [44].
2.3.1. EGFR and Cysteine Proteases
The mRNA level of cathepsin B correlates with the EGFR expression level [45]. The downregulation of cathepsin B and uPAR decreased pERK levels downstream of EGFR and integrins and increased the expression of p27, indicating an involvement in cell cycle regulation and can therefore induce cell proliferation [46].
2.3.2. EGFRvIII and Cysteine Proteases
The quantitative proteomic analysis, which also revealed the association of EGFRvIII with MMPs, showed that EGFRvIII-overexpressing U87 cells have a stronger invasive profile compared to other alterations in the U87MG cell line and secrete higher levels of cathepsin B [30].
3. ECM-Cell Interaction: Activation of Transduction Signaling Pathways
The activation of intracellular pathways is the key to pass information from the extracellular environment to the genome of the cell. Through the binding of soluble factors to transmembrane receptors or the interaction between receptors, signals can be transferred and integrated into the cell, increasing or decreasing protein activity and changing and modifying DNA transcription. As already indicated, the preferentially activated pathways by ECM-degrading proteases include the PI3K/Akt, the MAPK, and in some cases the JAK/STAT pathways, which can also be controlled by EGFR and EGFRvIII. Detailed descriptions of the EGFR signaling networks and the deregulated signaling of EGFRvIII in glioma can be found in excellent reviews [47,48]. In this review, we focus specifically on the crosstalk of EGFR- and EGFRvIII with pathways leading to increased tumor invasion and angiogenesis (Figure 2).
3.1. Integrin-FAK-ERK Signaling
The integrin family of transmembrane receptors is widely known for their contribution to tumor progression, angiogenesis, and invasion in various tumor types including glioblastomas. Their main function is to link the ECM to the cell’s cytoskeleton but they also take part in specialized cell-cell interactions. It directly binds to ECM glycoproteins and substrates of ECM-degrading proteases such as collagen, fibronectin, and laminin, but also interacts with specific ligands like EGF-like domain-containing protein 7 (EGFL7) to modulate cell adhesion and migration [49]. In addition to the structural role, the clustering of integrin receptors can generate bidirectional signaling to promote cell proliferation, survival, and migration [50,51]. The signaling is transmitted through integrin-associated proteins like talin and paxilin as integrins themselves do not have intrinsic catalytic activity. One of their interaction partners is the focal adhesion kinase FAK, a non-receptor protein-tyrosine kinase which is involved in multiple signaling pathways promoting cell migration and is upregulated in astrocytomas [52,53,54]. FAK binds and activates c-Src kinase (a proto-oncogene which is found to be over-expressed and highly activated in tumors), PI3K, PLCγ, and growth factor receptor-bound protein 2 (Grb2) which links into the MAPK pathway, resulting in the activation of several transcription factors [55,56,57,58].
3.1.1. EGFR in Integrin-FAK-ERK Signaling
The ERK class of MAP kinases can be separately activated by either clustering of integrin receptors and various growth factors but they often collaborate or synergize for a transient activation of the pathway. Phosphorylation of EGFR can not only be induced by the binding of its ligands but also through the interaction of clustered integrin receptors in the absence of EGF, leading to an activation of typical EGFR signaling pathways including the ERK (1/2) pathway [59,60]. This signaling is FAK-dependent, as FAK acts as a bridging protein that links both signals, binding with its N-terminal domain to EGFR [61]. Binding of EGF to EGFR leads to the downregulation of FAK kinase activity in human carcinoma cells overexpressing EGFR. Cells detached from the ECM and showed increased motility and invasion. In the case of reattaching mechanisms, FAK is phosphorylated by an involvement of activated integrin signaling. Therefore, integrin stimulation of FAK promotes adhesion and growth once the tumor cells reattach [62].
3.1.2. EGFRvIII in Integrin-FAK-ERK Signaling
It has been demonstrated that EGFRvIII constitutively activates the MAPK pathway in human glioma cells [32,63]. Substrates of phosphatase and tensin homolog PTEN, a tumor suppressor in many cancer types, includes FAK and inhibits integrin- and growth factor-mediated MAPK signaling [64]. PTEN phosphatase activity suppresses the invasion of EGFRvIII-expressing glioma cells. EGFRvIII could enhance the phosphorylation levels of FAK at Tyr-397 in glioma cells while forming a complex, which correlates with increased catalytic activity of FAK comparable to stimulation by growth factors or integrins. The knockdown of FAK expression reduced the phosphorylation level of ERK (1/2) and inhibited EGFRvIII-induced cell migration in U87ΔEGFR glioma cells [65,66].
3.2. SFK Signaling
The nine members of Src family kinase (SFK) are non-receptor tyrosine kinases which are engaged in the modulation of multiple cancer cell processes like cell adhesion, migration, and invasiveness. They are activated by cytokine receptors, receptor protein tyrosine kinases, g-protein coupled receptors, and integrins [67]. Src is frequently upregulated in GBM and the inhibition of SFK activity reduced glioma invasion [68,69,70]. Src increases EGFR-mediated pathway activity in many cancer types and can be activated via FAK-binding which initiates a crosstalk between integrin-FAK signaling and other pathways involved in glioma invasion [71,72,73].
3.2.1. EGFR in SFK Signaling
A recent study suggests and confirms a difference in EGFR and EGFRvIII downstream signaling. Eskilsson et al. found that EGFR was more associated with classical receptor tyrosine kinase signaling and involved in increased cell invasion. Moreover, they could provide evidence that SFK signaling was more specific for EGFRvIII as c-Src was upregulated and constitutively activated in EGFRvIII-expressing GBM cells. Surprisingly, they found those cells to be rather more aggressive in tumor growth and angiogenesis than invasive [74]. In contrast, other findings have demonstrated that Src is involved in enhancing tumor cell invasion and that the interaction is not only exclusive to EGFRvIII but includes interaction with EGFR [71,75]. Both Src and Flyn (both members of SFK) associate with and are phosphorylated by EGFR in glioma cells. Inhibition by the SFK inhibitor dasatinib blocked the EGFR-mediated cell motility in vitro and in vivo [75].
3.2.2. EGFRvIII in SFK Signaling
The kinases Src, Lyn, and Fyn are also expressed in EGFRvIII-expressing glioma cells and promote their tumorigenesis and invasion. As for EGFR-expressing cells, inhibition of these kinases led to decreased invasiveness [75,76]. SFKs also phosphorylate Dock180, a guanine nucleotide exchange factor that activates Rac1 and was found to be involved in the regulation of cell motility and invasion [77]. EGFRvIII stimulates Rac1 signaling through phosphorylation of Dock180 which is mediated by SFKs. Specific shRNA-induced downregulation of SFKs also attenuated EGFRvIII-induced Dock180 activation and decreased glioblastoma cell migration which suggests an EGFRvIII-SFK-Dock180-Rac1 pathway [78].
3.3. JAK/STAT Signaling
The Stat family of transcription factors acts as important signaling components and transfers the signal transduction of various cytokines and growth factors like EGF from the extracellular environment to the nucleus. Stat3 is upregulated in many cancer types and promotes immune responses and differentiation as well as cellular transformation, proliferation, and invasion. Upon activation of cell surface receptors through binding of its ligands, the receptor-associated Janus kinase (JAK) and the Src kinase act as connection links between the receptors and Stat3. Stat3 is phosphorylated and translocated into the nucleus where it induces the transcription of multiple genes involved in the regulation of cellular and tissue processes including those that function in cell motility [79].
3.3.1. EGFR in JAK/STAT Signaling
The interaction of EGFR and Stat3 is both physical and functional and described for cytoplasmic EGFR as well as for nuclear EGFR; the level of EGFR correlated significantly with activated Stat3 [80,81]. TWIST is a transcription factor which is required for epithelial-mesenchymal transition (EMT), and elevated levels of TWIST protein correlate with glioma invasion [82]. The binding of EGF to EGFR leads to an activation of Stat3 and subsequently a Stat3-induced increased expression of TWIST [83].
3.3.2. EGFRvIII in JAK/STAT Signaling
Even though EGFR levels show a strong correlation with p-Stat3, the correlation of EGFRvIII with activated Stat3 seems to be even stronger [81]. JAK2 and Stat3 form a complex with EGFRvIII attached to the FAK-Src complex to start an activation loop in EGFRvIII-overexpressing U87 glioma cells which promotes tumor cell invasion. JAK2 inhibitors disrupted this formation leading to a decrease in malignant tumor progression in vivo which was at least partly due to a reduced level of MMP-2 and MMP-9 [84]. Stat3 also associated with EGFRvIII in the nucleus and thereby transformed from a tumor-suppressive protein into an oncogenic protein with the largest effect in a small subset of cells both PTEN-deficient and EGFRvIII-overexpressing [85]. However, how EGFRvIII finds its way into the nucleus remains elusive. To complicate this already complex network of cross-talk between signaling pathways further, it seems that EGFRvIII also cooperates with the wild type receptor as suggested by Fan et al. They showed that even though EGFRvIII is unable to bind constitutive ligands, the binding of EGF does not only lead to a phosphorylation of EGFR but also of the mutant receptor. Phosphorylated EGFRvIII was translocated into the nucleus where it forms a complex and thus enhances the phosphorylation of Stat3 [86].
3.4. PI3K/Akt/mTOR Signaling
The PI3K/Akt pathway is known to control cell proliferation, growth, and apoptosis but changes in this pathway can also result in increased metastasis and cell invasion by inhibiting ECM detachment-induced cell death and stimulating MMP secretion [87]. PI3K mediates cell signaling by phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2) which generates the second messenger PIP3. This leads to the activation of Akt, also known as protein kinase B, which transfers the signaling to intracellular targets such as mammalian target of rapamycin (mTOR) and the S6 kinase. The PTEN gene mutation, which is a common occurrence in glioblastoma and which occurs in 30–40% of gliomas, leads to a dysfunctional inhibition of PI3K/Akt signaling by dephosphorylation of PIP3 [88].
3.4.1. EGFR in PI3K/Akt/mTOR Signaling
EGFR activates the PI3K/Akt pathway in both normal and cancer cells and the EGFR gene amplification in glioma cells induces an constitutive activation of PI3K [89]. Inhibition of EGFR through EGFR inhibitors PD153035 or AG1478 leads to the inactivation of the pathway [90]. As a large percentage of gliomas display gene amplification of EGFR or gene mutation of PTEN, the PI3K/Akt pathway is activated in most of them [91]. Evidence has shown that these two pathways not only crosstalk but are also influenced by the evolutionary conserved Notch pathway which is involved in cell fate determination, apoptosis, and cell proliferation. Studies have demonstrated that Akt also induces Notch1 expression in a mouse glioma model and EGFR, conversely, seems to be under the transcriptional control of Notch signaling [92,93,94].
3.4.2. EGFRvIII in PI3K/Akt/mTOR Signaling
In fibroblasts, EGFRvIII activates PI3K more excessively than the wild type receptor [95]. Expression of EGFRvIII increases the activation of Akt through downregulation of the cell cycle inhibitor p27 and enhances cell proliferation [96,97]. In vivo, EGFRvIII has a strong association with the phosphorylation of mTOR and it has been demonstrated that the mutant receptor might be an activator of PI3K in the absence of PTEN loss [98].
3.5. Wnt/β-Catenin Signaling
The canonical Wnt/β-catenin pathway has crucial roles in various cellular processes such as cell proliferation as well as cell migration and survival. In the absence of activating ligands, β-catenin, which is located in the cytoplasm, is degraded by a destruction complex consisting of several proteins including the glycogen synthase kinase 3 (GSK3). The binding of Wnt however, leads to a disruption of the destruction complex, followed by a translocation to the nucleus where it serves as a co-activator of transcription factor Tcf/Lef [99]. Aberrations and/or overexpression of key players of the Wnt/β-catenin pathway, like proteins related to the destruction complex or β-catenin itself, have frequently been found in brain tumor cells [100,101,102]. It has been shown that the knockdown of β-catenin inhibited cell proliferation and invasion in glioma cells but the mechanisms behind this are not fully determined [103].
3.5.1. EGFR in Wnt/β-Catenin Signaling
Binding of EGF to EGFR increases activation of Erk which leads to phosphorylation of α-catenin and transactivation of β-catenin [104]. Studies demonstrated further crosstalk between the Wnt/β-catenin pathway and the EGFR signaling pathway since the expression of multiple components of the EGFR pathway such as Stat3, Akt, MMP-2, and MMP-9 were downregulated after β-catenin knockdown [105]. Binding of EGF also induces the endocytosis of E-catherin, a cell adhesion transmembrane protein, resulting in disruption of cell-cell contacts and β-catenin-Tcf/Lef transactivation which contributes to tumor cell invasion [106].
3.5.2. EGFRvIII in Wnt/β-Catenin Signaling
A direct link of EGFRvIII to the Wnt/β-catenin pathway has not been discovered so far. But given the fact that crosstalk between different signaling pathways has been proposed, an indirect involvement of EGFRvIII via constitutive activation of the PI3K/Akt pathway can be assumed. Reports suggested that the activation of GSK-3β, which phosphorylates β-catenin, is regulated by the PI3K/Akt pathway and in glioma, LY294002-induced inhibition of PI3K decreased cell proliferation and cell invasiveness through the downregulation of members of the Wnt/β-catenin pathway, including GSK-3β and β-catenin [107,108].
4. Combined Therapy as an Effective Approach for Glioma Treatment
The application of a single target-specific agent as treatment for glioma patients quite often does not produce the desired results, especially in EGFR-or EGFRvIII-specific treatments. The agents on the market for EGFR and EGFRvIII are mostly antibodies binding the extracellular domain or small inhibitory molecules binding the intracellular domain of the protein which block EGFR or EGFRvIII signaling (Figure 2). Although a few of these agents are already approved by the Food and Drug Administration (FDA) for different cancer types, e.g., cetuximab for colorectal cancer and squamous cell carcinoma of the head and neck, gefitinib for non-small cell lung cancer, and erlotinib for non-small cell lung cancer and pancreatic cancer (see Table 1), unfortunately, none are approved for glioma treatment yet. The FDA has designated the monoclonal human antibody nimotuzumab as orphan status for glioma, which means the drug was originally developed to treat diseases affecting fewer than 200,000 people, but also in this case, approval has not yet been granted [109]. Still, efforts are made to develop an effective treatment. Various clinical trials are still ongoing or were recently completed, but in many cases, the desired outcome was not reached. The latest unfortunate examples are a failed phase III clinical trial for rindopepimut with no detected benefit of the vaccine for patient survival and a phase II study of erlotinib and bevacizumab (targeting VEGF) after radiation and temozolomide treatment, which also did not increase the survival of the treated glioma patients [21,110].
This lack of success can be due to different factors including cell resistance against the therapy or heterogeneity of the cells within the tumor. Combination gene therapy raises the possibility to target multiple molecules or pathways to diminish the cell’s ability to upregulate related pathways and circumvent the expected treatment effects. So far, the vast majority of combination studies included EGFR- or EGFRvIII-specific agents together with radiation or broad alkylating reagents like temozolomide. However those therapies might be even more effective when combined with more specific targets. Therefore, in addition to EGFR and EGFRvIII therapy, targeting the responsible matrix-degrading proteases or downstream signaling pathways might prevent the degradation of the ECM and subsequently decrease tumor-related angiogenesis and cell invasion. This could be achieved in more than one way: The direct blocking of the enzyme catalytic activity might prevent the activation of signaling cascades through specific inhibitors, for example tissue inhibitors of the MMPs, the TIMPs. Additionally, one could downregulate the enzymes themselves by downregulating the gene expression by either targeting the preceding signaling pathways, translational factors that activate the expression, or the protein’s mRNA directly. There are various approaches for the inhibition of proteases and members of the signaling pathways available and also already tested. Gene silencing of uPAR, uPA, and MMP-9 by the application of small hairpin RNA (hpRNA) significantly reduced glioma cell invasion and angiogenesis in vitro and direct intratumoral injections of plasmid DNA expressing the hpRNAs regressed pre-established intracranial tumors in nude mice [111]. Moreover, the combinational inhibition of cathepsin B expression and MMP-9 expression via RNA interference lead to reduced glioma tumor growth, invasion, and angiogenesis [112].
The combination gene therapies so far focused rather on the combination of EGFR therapies and targets of the downstream signaling pathways. One well known example is temsirolimus, a specific mTOR inhibitor, which was tested together with erlotinib or with the monoclonal antibody against EGFR, cetuximab, in phase I/II clinical trials [113]. Cetuximab, also called C225, is a chimeric antibody that binds specifically to EGFR consequently preventing the binding of the ligands to the receptor. It is a powerful tool, as it leads not only to an inhibition of the MAPK pathway, but it has also been shown that cetuximab decreases the expression and activity of MMPs like MMP-9, thereby decreasing tumor growth and tumor-related cell invasion in human head and neck squamous carcinoma cell lines and human transitional cell carcinoma [114,115,116,117]. The combination of EGFR inhibition and mTOR signaling pathway inhibition was also tested in GBM with different therapeutics: the application of AEE788, a tyrosine kinase inhibitor for EGFR and in high concentrations for VEGFR together with the application of RAD001, a mTOR inhibitor structurally related to rapamaycin, reduced proliferation and tumor growth in vitro and in vivo by blocking the phosphorylation of Erk and Akt [118]. A recent study has focused on a newly developed antibody against EGFRvIII, called CH12. When used in combination with rapamycin in EGFRvIII-positive and PTEN-negative glioma cells, it effectively inhibited the EGFRvIII downstream signals, including PI3K/Akt and Erk [119]. These studies all confirm that combination gene therapy is a promising approach to effectively increase the inhibitory effect on cell invasion and angiogenesis of mono-therapies against EGFR or EGFRvIII alone.
5. Conclusions
The strong ability of glioma cells to invade healthy tissue and to promote tumor-related angiogenesis may limit applicable therapies because of the high risk of tumor recurrence and the capacity of invasive cells to escape the therapy. EGFR and its mutant EGFRvIII have been extensively studied for decades as molecular markers in the context of GBM. In fact, it has been shown that the inhibition of wild type EGFR leads to a less invasive tumor type by switching to an angiogenic state, which shows that even shared mechanisms can be activated in an independent manner [120]. This flexibility of tumor cells, together with the heterogeneity of EGFR expression in general [121], the strong crosstalk and association of the receptor with multiple pathways and invasion- and angiogenic-regulating proteins, and the ability to develop resistance mechanisms are the reasons why mono-therapy of EGFR therapeutics have so far only been partially successful. It ultimately limits the success of therapies such as monoclonal antibody-bases therapies, EGFR- and EGFRvIII-specific small molecule inhibitors, or vaccines, because it is not possible to address all single glioma cells with only one specific treatment. Therefore, new therapies focusing on combination gene therapy with EGFR or EGFRvIII-specific treatments and agents targeting other molecular markers or molecules of collaborating pathways may offer therapeutic advantages. To cover more than one target or more than one mechanism which leads to cell invasion and angiogenesis could increase inhibitory effects. In this review, we focused on the impact of EGFR and EGFRvIII on the upregulation of ECM-degrading proteases and the activation of signaling pathways responsible for increased glioma cell invasion and angiogenesis and the subsequent possibilities to develop effective combined gene therapy. Unfortunately, there are still many unanswered questions and further genetic and molecular investigations are needed to fully reveal the mechanisms behind both glioma cell invasion and angiogenesis. However, early as well as recent studies have shown the promising effect of combination gene therapy, highlighting the potential of future novel therapies to successfully treat human glioma.
Acknowledgments
We thank Cheryl Ernest for proofreading the manuscript. This work was supported by the Frankfurt Initiative for Neurooncology Research (FIN) and a German Cancer Consortium (DKTK) grant to Mirko H. H. Schmidt.
Author Contributions
Stefanie Keller and Mirko H. H. Schmidt wrote and edited the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| EGFR | Epidermal growth factor receptor |
| GBM | Glioblastoma multiforme |
| ECM | Extracellular matrix |
| IDH | Isocitrate dehydrogenase |
| VEGF | Vascular endothelial growth factor receptor |
| EGF | Epidermal growth factor |
| FGF | Fibroblast growth factor |
| MMP | Matrix metalloprotease |
| MAPK | Mitogen-activated protein kinase |
| PI3K | Phosphoinositide 3-kinase |
| GSC | Glioma stem-like cells |
| ERK | Extracellular-signal regulated kinase |
| uPA | Urokinase-type plasminogen activator |
| tPA | Tissue-type plasminogen activator |
| PAI | Plasminogen activator inhibitors |
| uPAR | Urokinase-type plasminogen activator receptor |
| PLCγ | Phospholipase Cγ |
| TGFα | Transforming growth factor |
| EGFL7 | EGF-like domain-containing protein 7 |
| Grb2 | Grb2 Growth factor receptor-bound protein 2 |
| PTEN | Phosphatase and tensin homolog |
| SFK | Src family kinase |
| JAK | Receptor-associated Janus kinase |
| PIP2 | Phosphatidylinositol-4,5-bisphosphate |
| mTOR | Mammalian target of rapamycin |
| GSK3 | Glycogen synthase kinase 3 |
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.A.; Ye, X.; Piantadosi, S.; Desideri, S.; Nabors, L.B.; Rosenfeld, M.; Fisher, J. Survival of Patients with Newly Diagnosed Glioblastoma Treated with Radiation and Temozolomide in Research Studies in the United States. Clin. Cancer Res. 2010, 16, 2443–2449. [Google Scholar] [CrossRef] [PubMed]
- Scherer, H. Structural development in gliomas. Am. J. Cancer 1938, 34, 333–351. [Google Scholar]
- Rao, J.S. Molecular mechanisms of glioma invasiveness: The role of proteases. Nat. Rev. Cancer 2003, 3, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Fuller, G.N.; Bigner, S.H. Amplified cellular oncogenes in neoplasms of the human central nervous system. Mutat. Res. Rev. Genet. Toxicol. 1992, 276, 299–306. [Google Scholar] [CrossRef]
- Grandal, M.V.; Zandi, R.; Pedersen, M.W.; Willumsen, B.M.; van Deurs, B.; Poulsen, H.S. EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis 2007, 28, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.H.; Furnari, F.B.; Cavenee, W.K.; Bögler, O. Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proc. Natl. Acad. Sci. USA 2003, 100, 6505–6510. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-J.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.-D.; Huang, C.-M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Mukasa, A.; Bonavia, R.; Flynn, R.A.; Brewer, Z.E.; Cavenee, W.K.; Furnari, F.B.; White, F.M. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl. Acad. Sci. USA 2007, 104, 12867–12872. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Glazer, C.A.; Martinson, H.M.; Friedman, H.S.; Archer, G.E.; Sampson, J.H.; Riggins, G.J. Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res. 2002, 62, 3335–3339. [Google Scholar] [PubMed]
- Penar, P.L.; Khoshyomn, S.; Bhushan, A.; Tritton, T.R. Inhibition of epidermal growth factor receptor-associated tyrosine kinase blocks glioblastoma invasion of the brain. Neurosurgery 1997, 40, 141–151. [Google Scholar] [PubMed]
- Guillamo, J.-S.; de Boüard, S.; Valable, S.; Marteau, L.; Leuraud, P.; Marie, Y.; Poupon, M.-F.; Parienti, J.-J.; Raymond, E.; Peschanski, M. Molecular mechanisms underlying effects of epidermal growth factor receptor inhibition on invasion, proliferation, and angiogenesis in experimental glioma. Clin. Cancer Res. 2009, 15, 3697–3704. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, R.; Inda, M.; Vandenberg, S.; Cheng, S.; Nagane, M.; Hadwiger, P.; Tan, P.; Sah, D.; Cavenee, W.; Furnari, F. EGFRvIII promotes glioma angiogenesis and growth through the NF-κB, interleukin-8 pathway. Oncogene 2012, 31, 4054–4066. [Google Scholar] [CrossRef] [PubMed]
- Price, J.; Wilson, H.; Haites, N. Epidermal growth factor (EGF) increases the in vitro invasion, motility and adhesion interactions of the primary renal carcinoma cell line, A704. Eur. J. Cancer 1996, 32, 1977–1982. [Google Scholar] [CrossRef]
- Rosen, E.M.; Goldberg, I.D. Protein factors which regulate cell motility. Cell Dev. Biol. 1989, 25, 1079–1087. [Google Scholar] [CrossRef]
- Shibata, T.; Kawano, T.; Nagayasu, H.; Okumura, K.; Arisue, M.; Hamada, J.; Takeichi, N.; Hosokawa, M. Enhancing effects of epidermal growth factor on human squamous cell carcinoma motility and matrix degradation but not growth. Tumor Biol. 1996, 17, 168–175. [Google Scholar] [CrossRef]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front. Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Wikstrand, C.J.; Stanley, S.D.; Humphrey, P.A.; Pegram, C.N.; Archer, G.E.; Kurpad, S.; Shibuya, M.; Bigner, D.D. Investigation of a synthetic peptide as immunogen for a variant epidermal growth factor receptor associated with gliomas. J. Neuroimmunol. 1993, 46, 165–173. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors—Impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.E.; Furnari, F.B.; Cavenee, W.K. Targeting EGFR for treatment of glioblastoma: Molecular basis to overcome resistance. Curr. Cancer Drug Targets 2012, 12, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Malkki, H. Trial Watch: Glioblastoma vaccine therapy disappointment in Phase III trial. Nat. Rev. Neurol. 2016, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro Oncol. 2015, 17, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, P.; Wong, H.; Laing, T.; Rewcastle, N.; Morris, D.; Muzik, H.; Leco, K.; Johnston, R.; Brasher, P.; Sutherland, G.; et al. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br. J. Cancer 1999, 79, 1828. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Steck, P.A.; Mohanam, S.; Stetler-Stevenson, W.G.; Liotta, L.A.; Sawaya, R. Elevated levels of Mr 92,000 type IV collagenase in human brain tumors. Cancer Res. 1993, 53, 2208–2211. [Google Scholar] [PubMed]
- Anand, M.; Van Meter, T.; Fillmore, H. Epidermal growth factor induces matrix metalloproteinase-1 (MMP-1) expression and invasion in glioma cell lines via the MAPK pathway. J. Neuro Oncol. 2011, 104, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Kesanakurti, D.; Chetty, C.; Maddirela, D.R.; Gujrati, M.; Rao, J. Functional cooperativity by direct interaction between PAK4 and MMP-2 in the regulation of anoikis resistance, migration and invasion in glioma. Cell Death Dis. 2012, 3, e445. [Google Scholar] [CrossRef] [PubMed]
- Ellerbroek, S.M.; Halbleib, J.M.; Benavidez, M.; Warmka, J.K.; Wattenberg, E.V.; Stack, M.S.; Hudson, L.G. Phosphatidylinositol 3-kinase activity in epidermal growth factor-stimulated matrix metalloproteinase-9 production and cell surface association. Cancer Res. 2001, 61, 1855–1861. [Google Scholar] [PubMed]
- Choe, G.; Park, J.K.; Jouben-Steele, L.; Kremen, T.J.; Liau, L.M.; Vinters, H.V.; Cloughesy, T.F.; Mischel, P.S. Active matrix metalloproteinase 9 expression is associated with primary glioblastoma subtype. Clin. Cancer Res. 2002, 8, 2894–2901. [Google Scholar] [PubMed]
- Wang, F.; Xiao, W.; Sun, J.; Han, D.; Zhu, Y. MiRNA-181c inhibits EGFR-signaling-dependent MMP9 activation via suppressing Akt phosphorylation in glioblastoma. Tumor Biol. 2014, 35, 8653–8658. [Google Scholar] [CrossRef] [PubMed]
- Sangar, V.; Funk, C.C.; Kusebauch, U.; Campbell, D.S.; Moritz, R.L.; Price, N.D. Quantitative proteomic analysis reveals effects of epidermal growth factor receptor (EGFR) on invasion-promoting proteins secreted by glioblastoma cells. Mol. Cell. Proteom. 2014, 13, 2618–2631. [Google Scholar] [CrossRef] [PubMed]
- Lakka, S.S.; Jasti, S.L.; Kyritsis, A.P.; Yung, W.A.; Ali-Osman, F.; Nicolson, G.L.; Rao, J.S. Regulation of MMP-9 (type IV collagenase) production and invasiveness in gliomas by the extracellular signal-regulated kinase and jun amino-terminal kinase signaling cascades. Clin. Exp. Metastasis 2000, 18, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Lorimer, I.A.; Lavictoire, S.J. Activation of extracellular-regulated kinases by normal and mutant EGF receptors. Biochim. Biophy. Acta Mol. Cell Res. 2001, 1538, 1–9. [Google Scholar] [CrossRef]
- Bonavia, R.; Mukasa, A.; Narita, Y.; Sah, D.W.; Vandenberg, S.; Brennan, C.; Johns, T.G.; Bachoo, R.; Hadwiger, P.; Tan, P.; et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010, 24, 1731–1745. [Google Scholar]
- Jin, X.; Jin, X.; Sohn, Y.-W.; Yin, J.; Kim, S.-H.; Joshi, K.; Nam, D.-H.; Nakano, I.; Kim, H. Blockade of EGFR signaling promotes glioma stem-like cell invasiveness by abolishing ID3-mediated inhibition of p27 KIP1 and MMP3 expression. Cancer Lett. 2013, 328, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Salajegheh, M.; Rudnicki, A.; Smith, T.W. Expression of urokinase-type plasminogen activator receptor (uPAR) in primary central nervous system neoplasms. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Chandrasekar, N.; Kin, Y.; Lakka, S.S.; Mohanam, S.; Yanamandra, N.; Mohan, P.M.; Fuller, G.N.; Fang, B.; Fueyo, J.; et al. Suppression of glioma invasion and growth by adenovirus-mediated delivery of a bicistronic construct containing antisense uPAR and sense p16 gene sequences. Oncogene 2002, 21, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Czekay, R.-P.; Kuemmel, T.A.; Orlando, R.A.; Farquhar, M.G. Direct binding of occupied urokinase receptor (uPAR) to LDL receptor-related protein is required for endocytosis of uPAR and regulation of cell surface urokinase activity. Mol. Biol. Cell 2001, 12, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Muracciole, X.; Romain, S.; Dufour, H.; Palmari, J.; Chinot, O.; Ouafik, L.H.; Grisoli, F.; Figarella, D.; Martin, P.-M. PAI-1 and EGFR expression in adult glioma tumors: Toward a molecular prognostic classification. Int. J. Radiat. Oncol. Biol. Phys. 2002, 52, 592–598. [Google Scholar] [CrossRef]
- Kasza, A.; Kowanetz, M.; Poślednik, K.; Witek, B.; Kordula, T.; Koj, A. Epidermal growth factor and pro-inflammatory cytokines regulate the expression of components of plasminogen activation system in U373-MG astrocytoma cells. Cytokine 2001, 16, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Abe, T.; Wakabayashi, Y.; Hikawa, T.; Matsuo, K.-I.; Yamada, Y.; Kuwano, M.; Hori, S. Up-regulation of urokinase-type plasminogen activator and its receptor correlates with enhanced invasion activity of human glioma cells mediated by transforming growth factor-α or basic fibroblast growth factor. J. Neuro-Oncol. 2000, 46, 115–123. [Google Scholar] [CrossRef]
- Mamoune, A.; Kassis, J.; Kharait, S.; Kloeker, S.; Manos, E.; Jones, D.A.; Wells, A. DU145 human prostate carcinoma invasiveness is modulated by urokinase receptor (uPAR) downstream of epidermal growth factor receptor (EGFR) signaling. Exp. Cell Res. 2004, 299, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Paugh, B.S.; Paugh, S.W.; Bryan, L.; Kapitonov, D.; Wilczynska, K.M.; Gopalan, S.M.; Rokita, H.; Milstien, S.; Spiegel, S.; Kordula, T. EGF regulates plasminogen activator inhibitor-1 (PAI-1) by a pathway involving c-Src, PKCδ, and sphingosine kinase 1 in glioblastoma cells. FASEB J. 2008, 22, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Jo, M.; Cavenee, W.K.; Furnari, F.; VandenBerg, S.R.; Gonias, S.L. Crosstalk between the urokinase-type plasminogen activator receptor and EGF receptor variant III supports survival and growth of glioblastoma cells. Proc. Natl. Acade. Sci. USA 2011, 108, 15984–15989. [Google Scholar] [CrossRef] [PubMed]
- Rempel, S.A.; Rosenblum, M.L.; Mikkelsen, T.; Yan, P.-S.; Ellis, K.D.; Golembieski, W.A.; Sameni, M.; Rozhin, J.; Ziegler, G.; Sloane, B.F. Cathepsin B expression and localization in glioma progression and invasion. Cancer Res. 1994, 54, 6027–6031. [Google Scholar] [PubMed]
- Gole, B.; Huszthy, P.C.; Popović, M.; Jeruc, J.; Ardebili, Y.S.; Bjerkvig, R.; Lah, T.T. The regulation of cysteine cathepsins and cystatins in human gliomas. Int. J. Cancer 2012, 131, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, S.; Alapati, K.; Malla, R.R.; Gondi, C.S.; Mohanam, S.; Dinh, D.H.; Rao, J.S. Mechanism of p27 upregulation induced by downregulation of cathepsin B and uPAR in glioma. Mol. Oncol. 2011, 5, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR signaling networks in glioma. Sci. Signal 2009, 2, re6. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015, 15, 302. [Google Scholar] [CrossRef] [PubMed]
- Nikolić, I.; Stanković, N.D.; Bicker, F.; Meister, J.; Braun, H.; Awwad, K.; Baumgart, J.; Simon, K.; Thal, S.C.; Patra, C.; et al. EGFL7 ligates αvβ3 integrin to enhance vessel formation. Blood 2013, 121, 3041–3050. [Google Scholar] [CrossRef] [PubMed]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Sato, T.; D’Avirro, N.; Morimoto, C. Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. J. Exp. Med. 1995, 182, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Appeddu, P.A.; Parsons, J.T.; Hildebrand, J.D.; Schaller, M.D.; Guan, J.-L. Interaction of focal adhesion kinase with cytoskeletal protein talin. J. Biol. Chem. 1995, 270, 16995–16999. [Google Scholar] [CrossRef] [PubMed]
- Rutka, J.T.; Muller, M.; Hubbard, S.L.; Forsdike, J.; Dirks, P.B.; Jung, S.; Tsugu, A.; Ivanchuk, S.; Costello, P.; Mondal, S.; et al. Astrocytoma adhesion to extracellular matrix: Functional significance of integrin and focal adhesion kinase expression. J. Neuropathol. Exp. Neurol. 1999, 58, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.C.; Song, L.; Haura, E.B. Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol. 2009, 6, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Guan, J.-L. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc.Natl. Acad. Sci. USA 1994, 91, 10148–10152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chattopadhyay, A.; Ji, Q.-S.; Owen, J.D.; Ruest, P.J.; Carpenter, G.; Hanks, S.K. Focal adhesion kinase promotes phospholipase C-γ1 activity. Proc.Natl. Acad. Sci. USA 1999, 96, 9021–9026. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Hanks, S.K.; Hunter, T.; van der Geer, P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 1994, 372, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Teramoto, H.; Gutkind, J.S.; Yamada, K.M. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: Roles of integrin aggregation and occupancy of receptors. J. Cell Biol. 1996, 135, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Moro, L.; Venturino, M.; Bozzo, C.; Silengo, L.; Altruda, F.; Beguinot, L.; Tarone, G.; Defilippi, P. Integrins induce activation of EGF receptor: Role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 1998, 17, 6622–6632. [Google Scholar] [CrossRef] [PubMed]
- Sieg, D.J.; Hauck, C.R.; Ilic, D.; Klingbeil, C.K.; Schaefer, E.; Damsky, C.H.; Schlaepfer, D.D. FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2000, 2, 249–256. [Google Scholar] [PubMed]
- Lu, Z.; Jiang, G.; Blume-Jensen, P.; Hunter, T. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol. Cell. Biol. 2001, 21, 4016–4031. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.B.; Moscatello, D.K.; Wong, A.J.; Cooper, J.A.; Stahl, W.L. Differential modulation of mitogen-activated protein (MAP) kinase/extracellular signal-related kinase kinase and MAP kinase activities by a mutant epidermal growth factor receptor. J. Biol. Chem. 1995, 270, 30562–30566. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Tamura, M.; Pankov, R.; Danen, E.H.; Takino, T.; Matsumoto, K.; Yamada, K.M. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J. Cell Biol. 1999, 146, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.M.; Tao, B.B.; Wang, L.Y.; Liang, Y.L.; Jin, J.W.; Yang, Y.; Hu, Y.L.; Zha, X.L. Protein phosphatase activity of PTEN inhibited the invasion of glioma cells with epidermal growth factor receptor mutation type III expression. Int. J. Cancer 2005, 117, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, Y.; Wang, C.; Sun, L.; Mei, C.; Yao, W.; Liu, Y.; Shi, Y.; Qiu, S.; Fan, J.; et al. The effect of epidermal growth factor receptor variant III on glioma cell migration by stimulating ERK phosphorylation through the focal adhesion kinase signaling pathway. Arch. Biochem. Biophys. 2010, 502, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Bernasconi, P.; Clauser, K.R.; Mani, D.; Finn, S.P.; Beroukhim, R.; Burns, M.; Julian, B.; Peng, X.P.; Hieronymus, H.; et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat. Biotechnol. 2009, 27, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Angers-Loustau, A.; Hering, R.; Werbowetski, T.E.; Kaplan, D.R.; Del Maestro, R.F. Src Regulates Actin Dynamics and Invasion of Malignant Glial Cells in Three Dimensions. Mol. Cancer Res. 2004, 2, 595–605. [Google Scholar] [PubMed]
- Huveldt, D.; Lewis-Tuffin, L.J.; Carlson, B.L.; Schroeder, M.A.; Rodriguez, F.; Giannini, C.; Galanis, E.; Sarkaria, J.N.; Anastasiadis, P.Z. Targeting Src family kinases inhibits bevacizumab-induced glioma cell invasion. PLoS ONE 2013, 8, e56505. [Google Scholar] [CrossRef] [PubMed]
- Ishizawar, R.; Parsons, S.J. c-Src and cooperating partners in human cancer. Cancer Cell 2004, 6, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Thomas, S.M.; Xi, S.; Smithgall, T.E.; Siegfried, J.M.; Kamens, J.; Gooding, W.E.; Grandis, J.R. SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res. 2004, 64, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK–Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Eskilsson, E.; Rosland, G.V.; Talasila, K.M.; Knappskog, S.; Keunen, O.; Sottoriva, A.; Foerster, S.; Solecki, G.; Taxt, T.; Jirik, R.; et al. EGFRvIII mutations can emerge as late and heterogenous events in glioblastoma development and promote angiogenesis through Src activation. Neuro Oncol. 2016, 18, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Zhu, S.; Cvrljevic, A.; Huang, T.T.; Sarkaria, S.; Ahkavan, D.; Dang, J.; Dinca, E.B.; Plaisier, S.B.; Oderberg, I.; et al. Fyn and SRC are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Res. 2009, 69, 6889–6898. [Google Scholar] [CrossRef] [PubMed]
- Stettner, M.R.; Wang, W.; Nabors, L.B.; Bharara, S.; Flynn, D.C.; Grammer, J.R.; Gillespie, G.Y.; Gladson, C.L. Lyn kinase activity is the predominant cellular SRC kinase activity in glioblastoma tumor cells. Cancer Res. 2005, 65, 5535–5543. [Google Scholar] [CrossRef] [PubMed]
- Jarzynka, M.J.; Hu, B.; Hui, K.-M.; Bar-Joseph, I.; Gu, W.; Hirose, T.; Haney, L.B.; Ravichandran, K.S.; Nishikawa, R.; Cheng, S.-Y. ELMO1 and Dock180, a bipartite Rac1 guanine nucleotide exchange factor, promote human glioma cell invasion. Cancer Res. 2007, 67, 7203–7211. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Hu, B.; Jarzynka, M.J.; Li, Y.; Keezer, S.; Johns, T.G.; Tang, C.K.; Hamilton, R.L.; Vuori, K.; Nishikawa, R.; et al. Phosphorylation of dedicator of cytokinesis 1 (Dock180) at tyrosine residue Y722 by Src family kinases mediates EGFRvIII-driven glioblastoma tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 3018–3023. [Google Scholar] [CrossRef] [PubMed]
- Groner, B.; Lucks, P.; Borghouts, C. The Function of Stat3 in Tumor Cells and Their Microenvironment. Semin. Cell Dev. Biol. 2008, 19, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G.; Liao, H.-J. Trafficking of receptor tyrosine kinases to the nucleus. Exp. Cell Res. 2009, 315, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, M.; Betensky, R.A.; Batchelor, T.T.; Bernay, D.C.; Louis, D.N.; Nutt, C.L. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: Correlation with EGFR status, tumor grade, and survival. J. Neuropathol. Exp. Neurol. 2006, 65, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.C.; Tozer, K.R.; Silber, J.R.; Mikheeva, S.; Deng, M.; Morrison, R.S.; Manning, T.C.; Silbergeld, D.L.; Glackin, C.A.; Reh, T.A.; et al. TWIST is expressed in human gliomas, promotes invasion. Neoplasia 2005, 7, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.-W.; Hsu, S.-C.; Xia, W.; Cao, X.; Shih, J.-Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.-C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Han, L.; Dong, Y.; Tian, J.; Huang, W.; Liu, Z.; Jia, X.; Jiang, T.; Zhang, J.; Li, X.; et al. JAK2/STAT3 targeted therapy suppresses tumor invasion via disruption of the EGFRvIII/JAK2/STAT3 axis and associated focal adhesion in EGFRvIII-expressing glioblastoma. Neuro Oncol. 2014, 16, 1229–1243. [Google Scholar] [CrossRef] [PubMed]
- De la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; DePinho, R.A.; Bonni, A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.-W.; Cheng, C.K.; Gustafson, W.C.; Charron, E.; Zipper, P.; Wong, R.A.; Chen, J.; Lau, J.; Knobbe-Thomsen, C.; Weller, M.; et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 2013, 24, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Vara, J.Á.F.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.I.; Puc, J.; Li, J.; Bruce, J.N.; Cairns, P.; Sidransky, D.; Parsons, R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997, 57, 4183–4186. [Google Scholar] [PubMed]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.K.; Sharma, P.; Harbor, P.C.; Rahaman, S.O.; Haque, S.J. PI3K-AKT pathway negatively controls EGFR-dependent DNA-binding activity of Stat3 in glioblastoma multiforme cells. Oncogene 2005, 24, 7290–7300. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Tachibana, I.; Passe, S.M.; Huntley, B.K.; Borell, T.J.; Iturria, N.; O’fallon, J.R.; Schaefer, P.L.; Scheithauer, B.W.; James, C.D.; et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J. Natl. Cancer Inst. 2001, 93, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, V.K.; Viale, A.; Socci, N.D.; Wiedmann, M.; Hu, X.; Holland, E.C. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol. Cell 2003, 12, 889–901. [Google Scholar] [CrossRef]
- Purow, B.W.; Sundaresan, T.K.; Burdick, M.J.; Kefas, B.A.; Comeau, L.D.; Hawkinson, M.P.; Su, Q.; Kotliarov, Y.; Lee, J.; Zhang, W.; et al. Notch-1 regulates transcription of the epidermal growth factor receptor through p53. Carcinogenesis 2008, 29, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Teodorczyk, M.; Schmidt, M.H. Notching on cancer’s door: Notch signaling in brain tumors. Front. Oncol. 2015, 4, 341. [Google Scholar] [CrossRef] [PubMed]
- Moscatello, D.K.; Holgado-Madruga, M.; Emlet, D.R.; Montgomery, R.B.; Wong, A.J. Constitutive activation of phosphatidylinositol 3-kinase by a naturally occurring mutant epidermal growth factor receptor. J. Biol. Chem. 1998, 273, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yuan, M.; Kim, I.-A.; Chang, C.-M.; Bernhard, E.J.; Shu, H.-K.G. Mutant epidermal growth factor receptor displays increased signaling through the phosphatidylinositol-3 kinase/AKT pathway and promotes radioresistance in cells of astrocytic origin. Oncogene 2004, 23, 4594–4602. [Google Scholar] [CrossRef] [PubMed]
- Narita, Y.; Nagane, M.; Mishima, K.; Huang, H.S.; Furnari, F.B.; Cavenee, W.K. Mutant epidermal growth factor receptor signaling down-regulates p27 through activation of the phosphatidylinositol 3-kinase/Akt pathway in glioblastomas. Cancer Res. 2002, 62, 6764–6769. [Google Scholar] [PubMed]
- Choe, G.; Horvath, S.; Cloughesy, T.F.; Crosby, K.; Seligson, D.; Palotie, A.; Inge, L.; Smith, B.L.; Sawyers, C.L.; Mischel, P.S. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003, 63, 2742–2746. [Google Scholar] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Nagase, H.; Horii, A.; Miyoshi, Y.; Nakatsuru, S.; Aoki, T.; Arakawa, H.; Nakamura, Y.; Shimano, T.; Yanagisawa, A.; et al. Germ-line and somatic mutations of the APC gene in patients with turcot syndrome and analysis of APC mutations in brain tumors. Genes Chromosom. Cancer 1994, 9, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Dahmen, R.; Koch, A.; Denkhaus, D.; Tonn, J.; Sörensen, N.; Berthold, F.; Behrens, J.; Birchmeier, W.; Wiestler, O.; Pietsch, T. Deletions of AXIN1, a component of the WNT/wingless pathway, in sporadic medulloblastomas. Cancer Res. 2001, 61, 7039–7043. [Google Scholar] [PubMed]
- Koch, A.; Hrychyk, A.; Hartmann, W.; Waha, A.; Mikeska, T.; Waha, A.; Schüller, U.; Sörensen, N.; Berthold, F.; Goodyer, C.G.; et al. Mutations of the Wnt antagonist AXIN2 (Conductin) result in TCF-dependent transcription in medulloblastomas. Int. J. Cancer 2007, 121, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Pu, P.; Zhang, Z.; Kang, C.; Jiang, R.; Jia, Z.; Wang, G.; Jiang, H. Downregulation of Wnt2 and β-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Ther. 2009, 16, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Wang, J.; Nika, H.; Hawke, D.; Keezer, S.; Ge, Q.; Fang, B.; Fang, X.; Fang, D.; Litchfield, D.W.; et al. EGF-induced ERK activation promotes CK2-mediated disassociation of α-catenin from β-catenin and transactivation of β-catenin. Mol. Cell 2009, 36, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Lan, F.; Yang, W.; Yang, Y.; Han, L.; Zhang, A.; Liu, J.; Zeng, H.; Jiang, T.; Pu, P.; et al. Interruption of β-catenin suppresses the EGFR pathway by blocking multiple oncogenic targets in human glioma cells. Brain Res. 2010, 1366, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Ghosh, S.; Wang, Z.; Hunter, T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of β-catenin, and enhanced tumor cell invasion. Cancer Cell 2003, 4, 499–515. [Google Scholar] [CrossRef]
- Han, L.; Yang, Y.; Yue, X.; Huang, K.; Liu, X.; Pu, P.; Jiang, H.; Yan, W.; Jiang, T.; Kang, C. Inactivation of PI3K/AKT signaling inhibits glioma cell growth through modulation of β-catenin-mediated transcription. Brain Res. 2010, 1366, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.S.; Eswaraiah, A.; Crombet, T.; Piedra, P.; Saurez, G.; Iyer, H.; Arvind, A. Nimotuzumab, a promising therapeutic monoclonal for treatment of tumors of epithelial origin. MAbs 2009, 1, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.; Giglio, P.; Hu, J.; Groves, M.; Merrell, R.; Conrad, C.; Phuphanich, S.; Puduvalli, V.K.; Loghin, M.; Paleologos, N.; et al. A phase II study of bevacizumab and erlotinib after radiation and temozolomide in MGMT unmethylated GBM patients. J. Neuro Oncol. 2016, 126, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Gondi, C.S.; Lakka, S.S.; Dinh, D.H.; Olivero, W.C.; Gujrati, M.; Rao, J.S. Downregulation of uPA, uPAR and MMP-9 using small, interfering, hairpin RNA (siRNA) inhibits glioma cell invasion, angiogenesis and tumor growth. Neuron Glia Biol. 2004, 1, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Lakka, S.S.; Gondi, C.S.; Yanamandra, N.; Olivero, W.C.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene 2004, 23, 4681–4689. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Norden, A.D.; Cloughesy, T.F.; Robins, H.I.; Lieberman, F.S.; Gilbert, M.R.; Mehta, M.P.; et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro Oncol. 2014, 16, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Canadas, M.; Codony, J.; Hueto, J.; Arcas, A.; LLadó, A.; Puig, X.; Guix, M.; Raspall, G.; Albanell, J. Activated epidermal growth factor receptor: Studies in head and neck tumors and tumor cell lines after exposure to ligand and receptor tyrosine-kinase inhibitors. In Proceedings of the 35th Annual Meeting of the American Society of Clinical Oncology, Atlanta, GA, USA, 15–18 May 1999; p. 2392. [Google Scholar]
- Rhys-Evans, P.; Modjtahedi, H.; Court, W.; Box, G.; Eccles, S. Overexpression of epidermal growth factor receptor in human head and neck squamous carcinoma cell lines correlates with matrix metalloproteinase-9 expression and in vitro invasion. Int. J. Cancer 2000, 86, 307–317. [Google Scholar]
- Pornchai, O.; Rhys-Evans, P.; Box, G.M.; Eccles, S.A. Differential modulation of proliferation, matrix metalloproteinase expression and invasion of human head and neck squamous carcinoma cells by c-erbB ligands. Clin. Exp. Metastasis 1999, 17, 631–639. [Google Scholar]
- Matsumoto, T.; Perrotte, P.; Bar-Eli, M.; Inoue, K.; Kuniyasu, H.; Eve, B. Blockade of EGF-R signaling with anti-EGFR monoclonal antibody (Mab) C225 inhibits matrix metalloproteinase-9 (MMP-9) expression and invasion of human transitional cell carcinoma (TCC) in vitro and in vivo. Proc. Am. Assoc. Cancer Res. 1998, 39, 83. [Google Scholar]
- Goudar, R.K.; Shi, Q.; Hjelmeland, M.D.; Keir, S.T.; McLendon, R.E.; Wikstrand, C.J.; Reese, E.D.; Conrad, C.A.; Traxler, P.; Lane, H.A.; et al. Combination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibition. Mol. Cancer Ther. 2005, 4, 101–112. [Google Scholar] [PubMed]
- Xu, W.; Bi, Y.; Kong, J.; Zhang, J.; Wang, B.; Li, K.; Tian, M.; Pan, X.; Shi, B.; Gu, J.; et al. Combination of an anti-EGFRvIII antibody CH12 with Rapamycin synergistically inhibits the growth of EGFRvIII+PTEN− glioblastoma in vivo. Oncotarget 2016, 7, 24752. [Google Scholar] [PubMed]
- Talasila, K.M.; Soentgerath, A.; Euskirchen, P.; Rosland, G.V.; Wang, J.; Huszthy, P.C.; Prestegarden, L.; Skaftnesmo, K.O.; Sakariassen, P.Ø.; Eskilsson, E.; et al. EGFR wild-type amplification and activation promote invasion and development of glioblastoma independent of angiogenesis. Acta Neuropathol. 2013, 125, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, R.; Cavenee, W.K.; Furnari, F.B. Heterogeneity maintenance in glioblastoma: A social network. Cancer Res. 2011, 71, 4055–4060. [Google Scholar] [CrossRef] [PubMed]
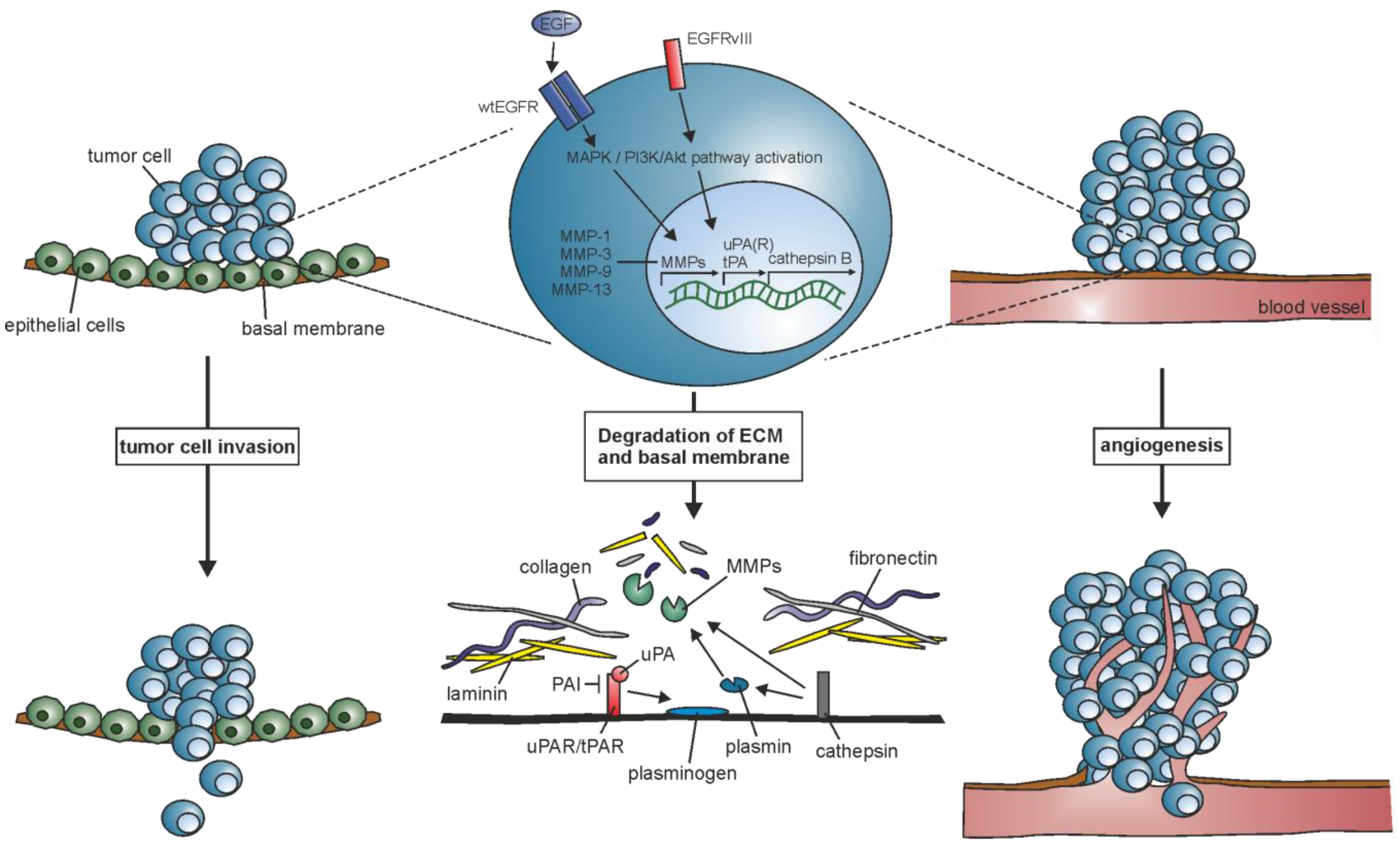
Figure 1.
EGFR and EGFRvIII contribute to the upregulation of the extracellular matrix (ECM)-degrading proteases matrix metalloproteases (MMPs), urokinase-type plasminogen activator (uPA), tissue-type plasminogen activator (tPA), and cathepsin B, which leads to degradation of basal membrane components like collagen, laminin, and fibronectin, and facilitate the invasion of tumor cells into the surrounding tissue and blood vessels.
Figure 1.
EGFR and EGFRvIII contribute to the upregulation of the extracellular matrix (ECM)-degrading proteases matrix metalloproteases (MMPs), urokinase-type plasminogen activator (uPA), tissue-type plasminogen activator (tPA), and cathepsin B, which leads to degradation of basal membrane components like collagen, laminin, and fibronectin, and facilitate the invasion of tumor cells into the surrounding tissue and blood vessels.

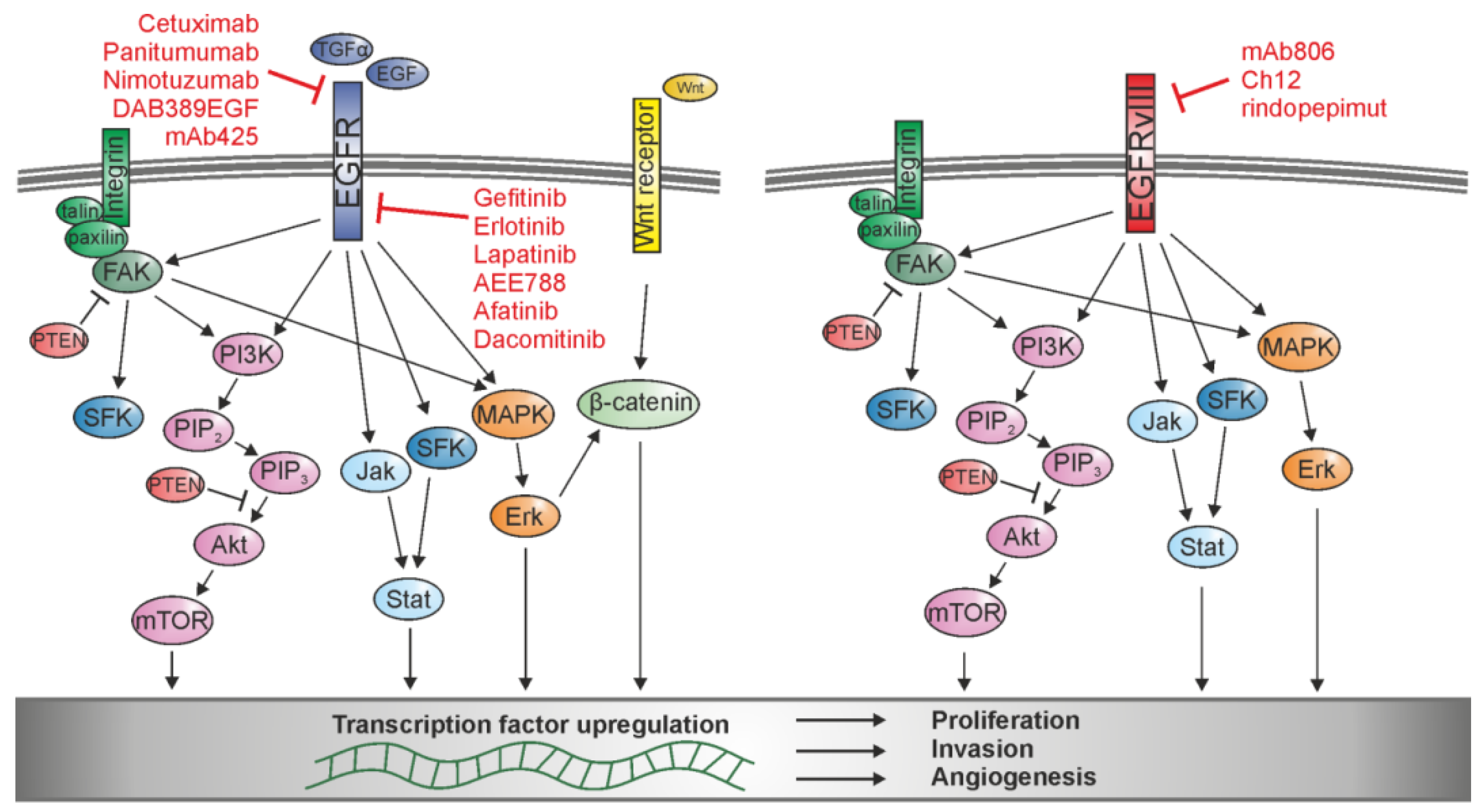
Figure 2.
Schematic of signaling pathways activated by EGFR and EGFRvIII and their interaction with integrin and Wnt signaling. Activation upregulates different transcription factors involved in tumor cell proliferation, invasion, and angiogenesis, which can be blocked by various EGFR- and EGFRvIII-specific agents.
Figure 2.
Schematic of signaling pathways activated by EGFR and EGFRvIII and their interaction with integrin and Wnt signaling. Activation upregulates different transcription factors involved in tumor cell proliferation, invasion, and angiogenesis, which can be blocked by various EGFR- and EGFRvIII-specific agents.


{kind=link}
{kind=link}
Table 1.
Selection of EGFR and EGFRvIII therapeutics.
| Target | Therapy | Class | Targeting also | FDA Approval |
|---|---|---|---|---|
| EGFR 1 | Monoclonal antibodies | |||
| Cetuximab | Mouse-human chimeric antibody | HER1 | Colorectal cancer Squamous cell carcinoma of the head and neck | |
| Nimotuzumab | Human antibody | HER1 | Orphan status for glioma Squamous cell carcinoma of the head and neck | |
| Panitumumab | Human antibody | HER1 | Metastatic colorectal cancer | |
| 125 I-Mab 425 | Radiolabeled murine antibody | - | N/A 3 | |
| Immunotoxins | ||||
| DAB389EGF | EGFR-toxin fusion protein | - | N/A | |
| Small molecule inhibitors | ||||
| Gefitinib | Anilinoquinazoline-based reversible inhibitor | HER1 | Non-small cell lung cancer | |
| Erlotinib | Anilinoquinazoline-based reversible inhibitor | HER1 | Non-small cell lung cancer Pancreatic cancer | |
| Lapatinib | Thiazolylquinazoline-based reversible inhibitor | HER1/2 | HER2+ breast cancer | |
| Afatinib | Anilinoquinazoline-based reversible inhibitor | HER1/2/4 | Metastasized non-small cell lung cancer | |
| Dacomitinib | Anilinoquinazoline-based reversible inhibitor | HER1/2/4 | N/A | |
| AEE788 | Tyrosine kinase inhibitor | VEGFR 2, HER1/2, ErbB2 | N/A | |
| EGFRvIII | Monoclonal antibodies | |||
| mAb806 | Human antibody | - | N/A | |
| CH12 | Human antibody | - | N/A | |
| Vaccines | ||||
| Rindopepimut | Peptide vaccination | - | N/A |
1 EGFR: epidermal growth factor receptor; 2 VEGFR: vascular endothelial growth factor receptor; 3 N/A: not available.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Keller, S.; Schmidt, M.H.H. EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment. Int. J. Mol. Sci. 2017, 18, 1295. https://doi.org/10.3390/ijms18061295
AMA Style
Keller S, Schmidt MHH. EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment. International Journal of Molecular Sciences. 2017; 18(6):1295. https://doi.org/10.3390/ijms18061295
Chicago/Turabian StyleKeller, Stefanie, and Mirko H. H. Schmidt. 2017. "EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment" International Journal of Molecular Sciences 18, no. 6: 1295. https://doi.org/10.3390/ijms18061295
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.