Genotoxic Anti-Cancer Agents and Their Relationship to DNA Damage, Mitosis, and Checkpoint Adaptation in Proliferating Cancer Cells


Abstract
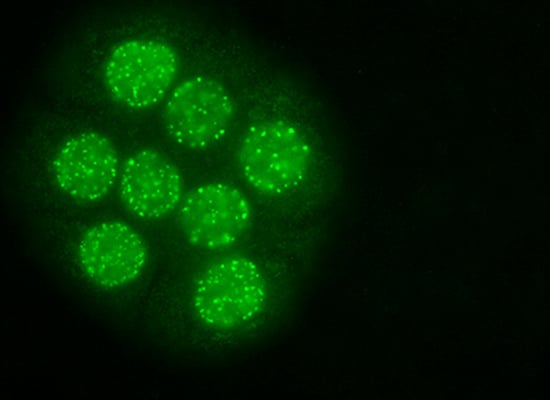
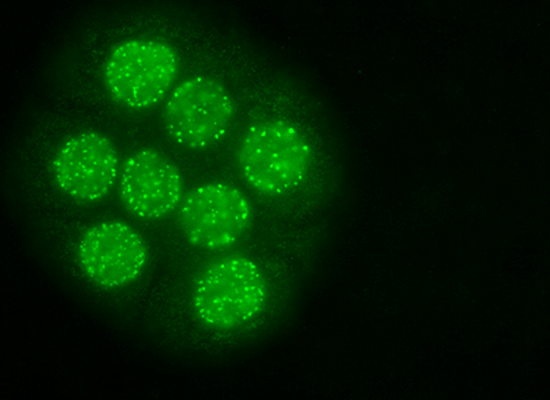
:
1. Introduction
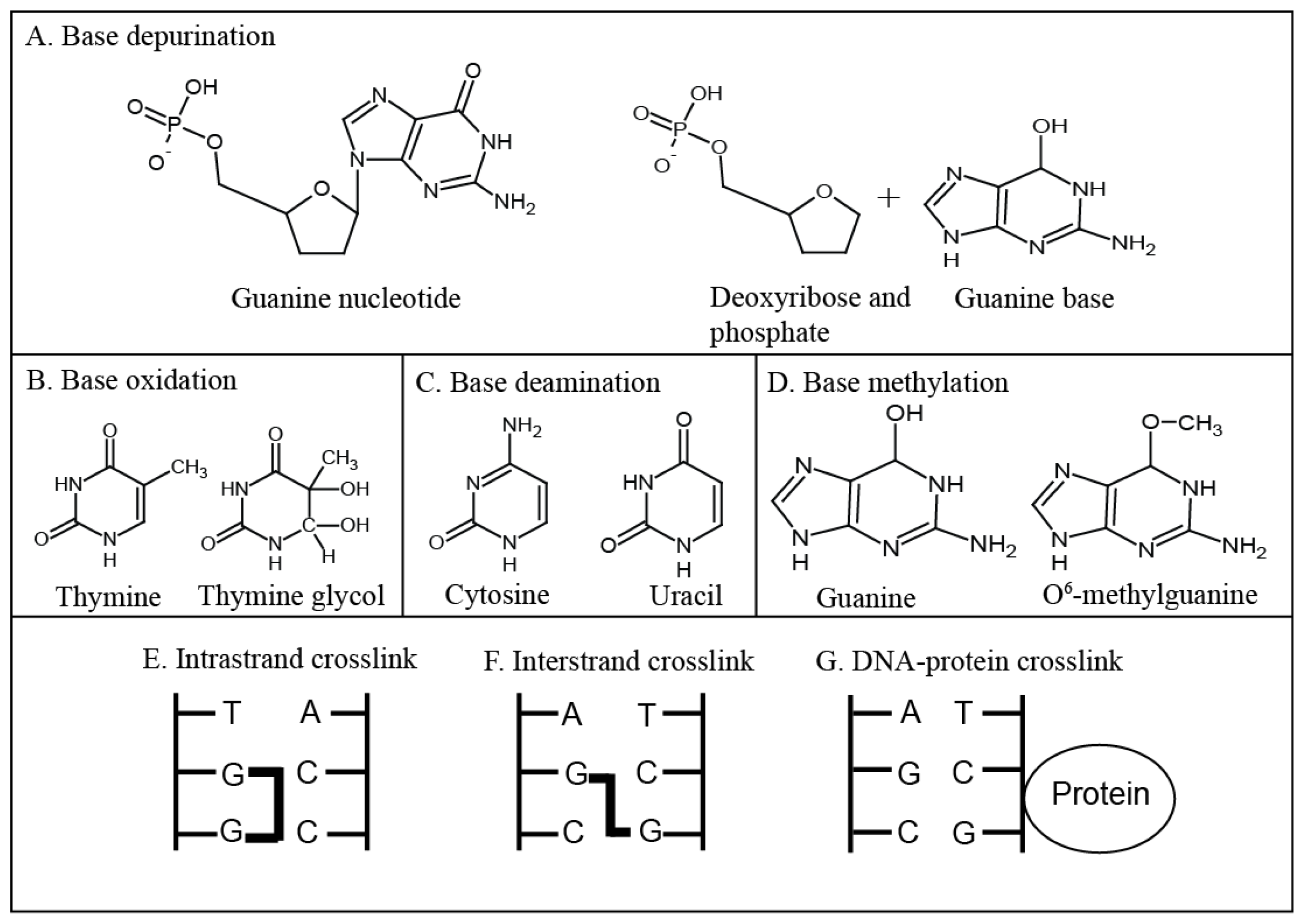
2. Types of DNA Damage
3. DNA Damaging Agents as Anti-Cancer Drugs
3.1. Alkylating Agents
3.2. Platinum Drugs
3.3. Antimetabolites
3.4. Topoisomerase Inhibitors
3.5. Ionising Radiation
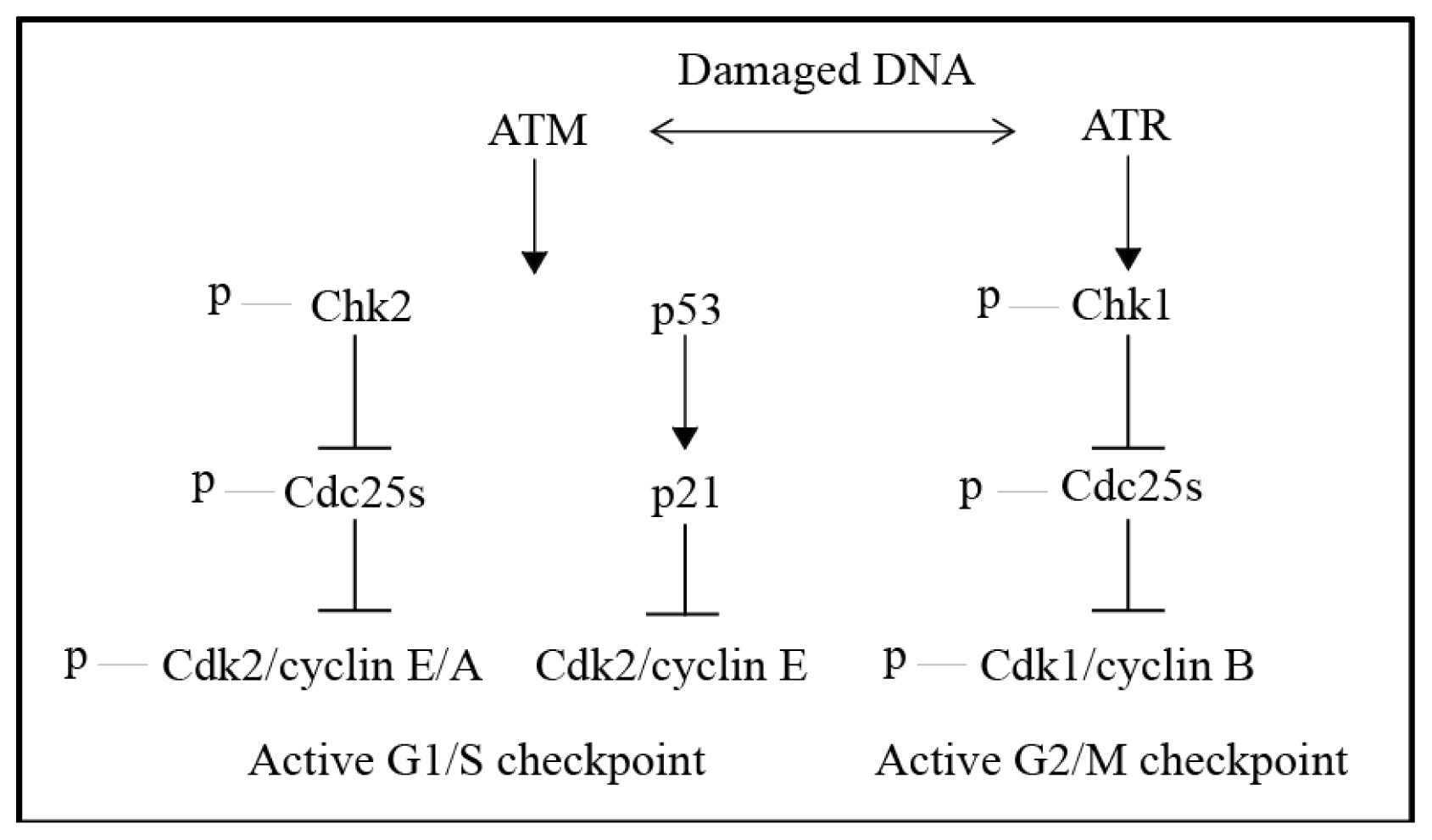
4. The DNA Damage Response (DDR)—DNA Damage Signalling and Cell Cycle Checkpoints
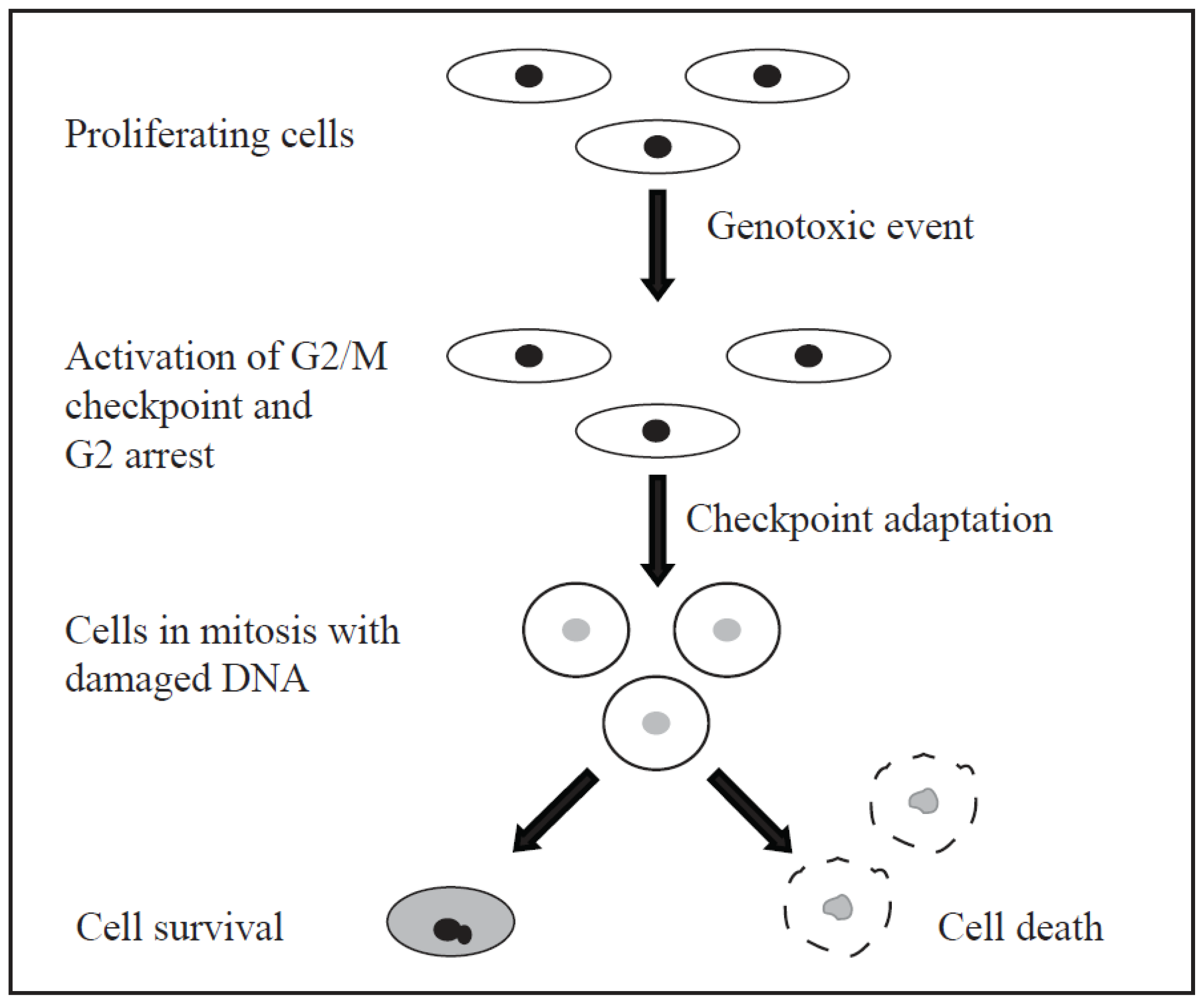
5. Cell Cycle Checkpoints and Checkpoint Adaptation
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ATM | Ataxia telangiectasia mutated |
| ATR | ATM and Rad3-related |
| ATRIP | ATR-interacting protein |
| Cdk1 | Cyclin-dependent kinase 1 |
| Cdk2 | Cyclin-dependent kinase 2 |
| Chk1 | Checkpoint kinase 1 |
| Chk2 | Checkpoint kinase 2 |
| CPT | Camptothecin |
| DDR | DNA damage response |
| DNA | Deoxyribonucleic acid |
| dNTPs | Deoxynucleotide triphosphates |
| DSBs | Double-strand breaks |
| hmdUrd | 5-hydroxymethyl-2′-deoxyuridine |
| MGMT | O6-methylguanine-DNA methyltransferase |
| MRN | Mre11-Rad50-Nbs1 |
| RFC | Replication factor C |
| ROS | Reactive oxygen species |
| RPA | Replication protein A |
| SSBs | Single-strand breaks |
| ssDNA | Single-stranded DNA |
| TMZ | Temozolomide |
| TopB1 | DNA topoisomerase II binding protein 1 |
| Top1 | Topoisomerase I |
| Top2 | Topoisomerase II |
| UV | Ultraviolet |
| 5-FU | 5-fluorouracil |
| 9-1-1 | Rad9-Rad1-Hus1 |
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar]
- Cavalieri, E.; Saeed, M.; Zahid, M.; Cassada, D.; Snow, D.; Miljkovic, M.; Rogan, E. Mechanism of DNA depurination by carcinogens in relation to cancer initiation. IUBMB Life 2012, 64, 169–179. [Google Scholar]
- Chakravarti, D.; Pelling, J.C.; Cavalieri, E.L.; Rogan, E.G. Relating aromatic hydrocarbon-induced DNA adducts and c-H-ras mutations in mouse skin papillomas: The role of apurinic sites. Proc. Natl. Acad. Sci. USA 1995, 92, 10422–10426. [Google Scholar]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012, 46, 382–419. [Google Scholar]
- Chaung, W.; Boorstein, R.J. Molecular spectrum of mutations induced by 5-hydroxymethyl-2′-deoxyuridine in (CHO)-PL61 cells. Mutat. Res. 1997, 373, 125–137. [Google Scholar]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms mutation and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar]
- Yoon, J.H.; Bhatia, G.; Prakash, S.; Prakash, L. Error-free replicative bypass of thymine glycol by the combined action of DNA polymerases kappa and zeta in human cells. Proc. Natl. Acad. Sci. USA 2010, 107, 14116–14121. [Google Scholar]
- Kow, Y.W. Repair of deaminated bases in DNA. Free Radic. Biol. Med. 2002, 33, 886–893. [Google Scholar]
- Kreutzer, D.A.; Essigmann, J.M. Oxidized deaminated cytosines are a source of C→T transitions in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 3578–3582. [Google Scholar]
- Drablos, F.; Feyzi, E.; Aas, P.A.; Vaagbo, C.B.; Kavli, B.; Bratlie, M.S.; Pena-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA—Repair mechanisms and medical significance. DNA Repair (Amst.) 2004, 3, 1389–1407. [Google Scholar]
- Warren, J.J.; Forsberg, L.J.; Beese, L.S. The structural basis for the mutagenicity of O(6)-methylguanine lesions. Proc. Natl. Acad. Sci. USA 2006, 103, 19701–19706. [Google Scholar]
- Mojas, N.; Lopes, M.; Jiricny, J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes Dev. 2007, 21, 3342–3355. [Google Scholar]
- Yarosh, D.B.; Foote, R.S.; Mitra, S.; Day, R.S. Repair of O6-methylguanine in DNA by demethylation is lacking in Mer- human tumor cell strains. Carcinogenesis 1983, 4, 199–205. [Google Scholar]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar]
- Hemminki, K. Cancer IAfRo. In DNA Adducts: Identification and Biological Significance; IARC Scientific Publication: Lyon, France, 1994. [Google Scholar]
- Swenberg, J.A.; Lu, K.; Moeller, B.C.; Gao, L.; Upton, P.B.; Nakamura, J.; Starr, T.B. Endogenous versus exogenous DNA adducts: Their role in carcinogenesis epidemiology and risk assessment. Toxicol. Sci. 2011, 120, S130–S145. [Google Scholar]
- Schorr, S.; Schneider, S.; Lammens, K.; Hopfner, K.P.; Carell, T. Mechanism of replication blocking and bypass of Y-family polymerase {eta} by bulky acetylaminofluorene DNA adducts. Proc. Natl. Acad. Sci. USA 2010, 107, 20720–20725. [Google Scholar]
- Kondo, N.; Takahashi, A.; Ono, K.; Ohnishi, T. DNA damage induced by alkylating agents and repair pathways. J. Nucleic Acids 2010, 2010, 543531. [Google Scholar]
- Noll, D.M.; Mason, T.M.; Miller, P.S. Formation and repair of interstrand cross-links in DNA. Chem. Rev. 2006, 106, 277–301. [Google Scholar]
- Kowalczyk, A.; Carmical, J.R.; Zou, Y.; van Houten, B.; Lloyd, R.S.; Harris, C.M.; Harris, T.M. Intrastrand DNA cross-links as tools for studying DNA replication and repair: Two- three- and four-carbon tethers between the N(2) positions of adjacent guanines. Biochemistry 2002, 41, 3109–3118. [Google Scholar]
- Yaghi, B.M.; Turner, P.M.; Denny, W.A.; Turner, P.R.; O’Connor, C.J.; Ferguson, L.R. Comparative mutational spectra of the nitrogen mustard chlorambucil and its half-mustard analogue in Chinese hamster AS52 cells. Mutat. Res. 1998, 401, 153–164. [Google Scholar]
- Barker, S.; Weinfeld, M.; Murray, D. DNA-protein crosslinks: Their induction repair and biological consequences. Mutat. Res. 2005, 589, 111–135. [Google Scholar]
- Alexander, P.; Moroson, H. Cross-linking of deoxyribonucleic acid to protein following ultra-violet irradiation different cells. Nature 1962, 194, 882–883. [Google Scholar]
- Shoulkamy, M.I.; Nakano, T.; Ohshima, M.; Hirayama, R.; Uzawa, A.; Furusawa, Y.; Ide, H. Detection of DNA-protein crosslinks (DPCs) by novel direct fluorescence labeling methods: Distinct stabilities of aldehyde and radiation-induced DPCs. Nucleic Acids Res. 2012, 40, e143. [Google Scholar]
- Connelly, J.C.; Leach, D.R. Repair of DNA covalently linked to protein. Mol. Cell 2004, 13, 307–316. [Google Scholar]
- Biedermann, K.A.; Sun, J.R.; Giaccia, A.J.; Tosto, L.M.; Brown, J.M. Scid mutation in mice confers hypersensitivity to ionizing radiation and a deficiency in DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 1991, 88, 1394–1397. [Google Scholar]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar]
- Rastogi, R.P.; Richa Kumar, A.; Tyagi, M.B.; Sinha, R.P. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J. Nucleic Acids 2010, 2010, 592980. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar]
- Holland, J.F.; Kufe, D.W.; Weichselbaum, R.R.; Pollock, R.E.; Frei, E.; Gansler, T.S.; Bast, R.C., Jr. Holland-Frei Cancer Medicine, 6th Ed. ed; BC Decker: Hamilton, ON, Canada, 2003. [Google Scholar]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar]
- Woods, D.; Turchi, J.J. Chemotherapy induced DNA damage response: Convergence of drugs and pathways. Cancer Biol. Ther. 2013, 14, 379–389. [Google Scholar]
- Chabner, B.A.; Roberts, T.G., Jr. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar]
- Krumbhaar, E.B.; Krumbhaar, H.D. The blood and bone marrow in yellow cross gas (mustard gas) poisoning: Changes produced in the bone marrow of fatal cases. J. Med. Res. 1919, 40, 497–508.3. [Google Scholar]
- Gilman, A. The initial clinical trial of nitrogen mustard. Am. J. Surg 1963, 105, 574–578. [Google Scholar]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar]
- Tong, W.P.; Ludlum, D.B. Crosslinking of DNA by busulfan Formation of diguanyl derivatives. Biochim. Biophys. Acta 1980, 608, 174–181. [Google Scholar]
- Newlands, E.S.; Stevens, M.F.; Wedge, S.R.; Wheelhouse, R.T.; Brock, C. Temozolomide: A review of its discovery chemical properties pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar]
- Stupp, R.; van den Bent, M.J.; Hegi, M.E. Optimal role of temozolomide in the treatment of malignant gliomas. Curr. Neurol. Neurosci. Rep. 2005, 5, 198–206. [Google Scholar]
- Payne, M.J.; Pratap, S.E.; Middleton, M.R. Temozolomide in the treatment of solid tumours: Current results and rationale for dosing/scheduling. Crit. Rev. Oncol. Hematol. 2005, 53, 241–252. [Google Scholar]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neurooncol. 2012, 107, 359–364. [Google Scholar]
- Hammond, L.A.; Eckardt, J.R.; Baker, S.D.; Eckhardt, S.G.; Dugan, M.; Forral, K.; Reidenberg, P.; Statkevich, P.; Weiss, G.R.; Rinaldi, D.A.; et al. Phase I and pharmacokinetic study of temozolomide on a daily-for-5-days schedule in patients with advanced solid malignancies. J. Clin. Oncol. 1999, 17, 2604–2613. [Google Scholar]
- Cahuzac, N.; Studeny, A.; Marshall, K.; Versteege, I.; Wetenhall, K.; Pfeiffer, B.; Leonce, S.; Hickman, J.A.; Pierre, A.; Golsteyn, R.M. An unusual DNA binding compound S23906 induces mitotic catastrophe in cultured human cells. Cancer Lett. 2010, 289, 178–187. [Google Scholar]
- Tillequin, F. Sarcomelicope alkaloids as leads for the discovery of new antitumor acronycine derivatives. Phytochem. Rev. 2002, 1, 355–368. [Google Scholar]
- David-Cordonnier, M.H.; Laine, W.; Lansiaux, A.; Kouach, M.; Briand, G.; Pierre, A.; Hickman, J.A.; Bailly, C. Alkylation of guanine in DNA by S23906–1 a novel potent antitumor compound derived from the plant alkaloid acronycine. Biochemistry 2002, 41, 9911–9920. [Google Scholar]
- Leonce, S.; Perez, V.; Lambel, S.; Peyroulan, D.; Tillequin, F.; Michel, S.; Koch, M.; Pfeiffer, B.; Atassi, G.; Hickman, J.A.; et al. Induction of cyclin E and inhibition of DNA synthesis by the novel acronycine derivative S23906–1 precede the irreversible arrest of tumor cells in S phase leading to apoptosis. Mol. Pharmacol. 2001, 60, 1383–1391. [Google Scholar]
- Charlier, C.; Kintz, P.; Dubois, N.; Plomteux, G. Fatal overdosage with cisplatin. J. Anal. Toxicol. 2004, 28, 138–140. [Google Scholar]
- Oldfield, E.H.; Dedrick, R.L.; Yeager, R.L.; Clark, W.C.; DeVroom, H.L.; Chatterji, D.C.; Doppman, J.L. Reduced systemic drug exposure by combining intra-arterial chemotherapy with hemoperfusion of regional venous drainage. J. Neurosurg. 1985, 63, 726–732. [Google Scholar]
- Vermorken, J.B.; van der Vijgh, W.J.; Klein, I.; Hart, A.A.; Gall, H.E.; Pinedo, H.M. Pharmacokinetics of free and total platinum species after short-term infusion of cisplatin. Cancer Treat. Rep. 1984, 68, 505–513. [Google Scholar]
- Harland, S.J.; Newell, D.R.; Siddik, Z.H.; Chadwick, R.; Calvert, A.H.; Harrap, K.R. Pharmacokinetics of cis-diammine-1 1-cyclobutane dicarboxylate platinum(II) in patients with normal and impaired renal-function. Cancer Res. 1984, 44, 1693–1697. [Google Scholar]
- Elferink, F.; van der Vijgh, W.J.; Klein, I.; Vermorken, J.B.; Gall, H.E.; Pinedo, H.M. Pharmacokinetics of carboplatin after iv administration. Cancer Treat. Rep. 1987, 71, 1231–1237. [Google Scholar]
- Graham, M.A.; Lockwood, G.F.; Greenslade, D.; Brienza, S.; Bayssas, M.; Gamelin, E. Clinical pharmacokinetics of oxaliplatin: A critical review. Clin. Cancer Res. 2000, 6, 1205–1218. [Google Scholar]
- Takimoto, C.H.; Yee, L.K.; Venzon, D.J.; Schuler, B.; Grollman, F.; Chabuk, C.; Hamilton, J.M.; Chen, A.P.; Allegra, C.J.; Grem, J.L. High inter- and intrapatient variation in 5-fluorouracil plasma concentrations during a prolonged drug infusion. Clin. Cancer Res. 1999, 5, 1347–1352. [Google Scholar]
- Raymond, E.; Campone, M.; Stupp, R.; Menten, J.; Chollet, P.; Lesimple, T.; Fety-Deporte, R.; Lacombe, D.; Paoletti, X.; Fumoleau, P. Multicentre phase II and pharmacokinetic study of RFS2000 (9-nitro-camptothecin) administered orally 5 days a week in patients with glioblastoma multiforme. Eur J. Cancer 2002, 38, 1348–1350. [Google Scholar]
- Millward, M.J.; Newell, D.R.; Yuen, K.; Matthews, J.P.; Balmanno, K.; Charlton, C.J.; Gumbrell, L.; Lind, M.J.; Chapman, F.; Proctor, M.; et al. Pharmacokinetics and pharmacodynamics of prolonged oral etoposide in women with metastatic breast cancer. Cancer Chemother. Pharmacol. 1995, 37, 161–167. [Google Scholar]
- Hande, K.R.; Wedlund, P.J.; Noone, R.M.; Wilkinson, G.R.; Greco, F.A.; Wolff, S.N. Pharmacokinetics of high-dose etoposide (VP-16–213) administered to cancer patients. Cancer Res. 1984, 44, 379–382. [Google Scholar]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar]
- Rosenberg, B.; VanCamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar]
- Zamble, D.B.; Lippard, S.J. Cisplatin and DNA repair in cancer chemotherapy. Trends Biochem. Sci. 1995, 20, 435–439. [Google Scholar]
- Go, R.S.; Adjei, A.A. Review of the comparative pharmacology and clinical activity of cisplatin and carboplatin. J. Clin. Oncol. 1999, 17, 409–422. [Google Scholar]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar]
- Yoshioka, A.; Tanaka, S.; Hiraoka, O.; Koyama, Y.; Hirota, Y.; Ayusawa, D.; Seno, T.; Garrett, C.; Wataya, Y. Deoxyribonucleoside triphosphate imbalance 5-Fluorodeoxyuridineinduced DNA double strand breaks in mouse FM3A cells and the mechanism of cell death. J. Biol. Chem. 1987, 262, 8235–8241. [Google Scholar]Int. J. Mol. Sci. 2014, 15, 3425.
- Houghton, J.A.; Tillman, D.M.; Harwood, F.G. Ratio of 2′-deoxyadenosine-5′-triphosphate/thymidine-5′-triphosphate influences the commitment of human colon carcinoma cells to thymineless death. Clin. Cancer Res. 1995, 1, 723–730. [Google Scholar]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar]
- Froelich-Ammon, S.J.; Osheroff, N. Topoisomerase poisons: Harnessing the dark side of enzyme mechanism. J. Biol. Chem. 1995, 270, 21429–21432. [Google Scholar]
- Hsiang, Y.H.; Lihou, M.G.; Liu, L.F. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989, 49, 5077–5082. [Google Scholar]
- Weinfeld, M.; Mani, R.S.; Abdou, I.; Aceytuno, R.D.; Glover, J.N. Tidying up loose ends: The role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends Biochem. Sci. 2011, 36, 262–271. [Google Scholar]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 260, 14873–14878. [Google Scholar]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar]
- Chen, G.L.; Yang, L.; Rowe, T.C.; Halligan, B.D.; Tewey, K.M.; Liu, L.F. Nonintercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J. Biol. Chem. 1984, 259, 13560–13566. [Google Scholar]
- Baldwin, E.L.; Osheroff, N. Etoposide topoisomerase II and cancer. Curr. Med. Chem. Anticancer Agents 2005, 5, 363–372. [Google Scholar]
- Nakada, S.; Katsuki, Y.; Imoto, I.; Yokoyama, T.; Nagasawa, M.; Inazawa, J.; Mizutani, S. Early G2/M checkpoint failure as a molecular mechanism underlying etoposide-induced chromosomal aberrations. J. Clin. Investig. 2006, 116, 80–89. [Google Scholar]
- Smith, M.A.; Rubinstein, L.; Anderson, J.R.; Arthur, D.; Catalano, P.J.; Freidlin, B.; Heyn, R.; Khayat, A.; Krailo, M.; Land, V.J.; et al. Secondary leukemia or myelodysplastic syndrome after treatment with epipodophyllotoxins. J. Clin. Oncol. 1999, 17, 569–577. [Google Scholar]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci 2012, 9, 193–199. [Google Scholar]
- Bernier, J.; Hall, E.J.; Giaccia, A. Radiation oncology: A century of achievements. Nat. Rev. Cancer 2004, 4, 737–747. [Google Scholar]
- Bucci, M.K.; Bevan, A.; Roach, M., 3rd. Advances in radiation therapy: Conventional to 3D to IMRT to 4D and beyond. CA Cancer J. Clin. 2005, 55, 117–134. [Google Scholar]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar]
- Emami, B.; Lyman, J.; Brown, A.; Coia, L.; Goitein, M.; Munzenrider, J.E.; Shank, B.; Solin, L.J.; Wesson, M. Tolerance of normal tissue to therapeutic irradiation. Int. J. Radiat. Oncol. Biol. Phys. 1991, 21, 109–122. [Google Scholar]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 7th ed.; Lippincott Williams and Wilkins: Riverwoods, IL, USA, 2012. [Google Scholar]Int. J. Mol. Sci. 2014, 15, 3426.
- Dewey, W.C.; Ling, C.C.; Meyn, R.E. Radiation-induced apoptosis: Relevance to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 1995, 33, 781–796. [Google Scholar]
- Jonathan, E.C.; Bernhard, E.J.; McKenna, W.G. How does radiation kill cells? Curr. Opin. Chem. Biol. 1999, 3, 77–83. [Google Scholar]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar]
- Bartek, J.; Lukas, J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar]
- Rhind, N.; Russell, P. Chk1 and Cds1: Linchpins of the DNA damage and replication checkpoint pathways. J. Cell Sci. 2000, 113(Pt. 22), 3889–3896. [Google Scholar]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar]
- Korwek, Z.; Sewastianik, T.; Bielak-Zmijewska, A.; Mosieniak, G.; Alster, O.; Moreno-Villanueva, M.; Burkle, A.; Sikora, E. Inhibition of ATM blocks the etoposide-induced DNA damage response and apoptosis of resting human T cells. DNA Repair (Amst.) 2012, 11, 864–873. [Google Scholar]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar]
- Lukas, C.; Melander, F.; Stucki, M.; Falck, J.; Bekker-Jensen, S.; Goldberg, M.; Lerenthal, Y.; Jackson, S.P.; Bartek, J.; Lukas, J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004, 23, 2674–2683. [Google Scholar]
- Kurz, E.U.; Lees-Miller, S.P. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst.) 2004, 3, 889–900. [Google Scholar]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar]
- Chen, L.; Gilkes, D.M.; Pan, Y.; Lane, W.S.; Chen, J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005, 24, 3411–3422. [Google Scholar]
- Flynn, R.L.; Zou, L. ATR: A master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 2011, 36, 133–140. [Google Scholar]
- Wilsker, D.; Bunz, F. Loss of ataxia telangiectasia mutated- and Rad3-related function potentiates the effects of chemotherapeutic drugs on cancer cell survival. Mol. Cancer Ther. 2007, 6, 1406–1413. [Google Scholar]
- Yoshioka, K.; Yoshioka, Y.; Hsieh, P. ATR kinase activation mediated by MutS alpha and MutL alpha in response to cytotoxic O(6)-methylguanine adducts. Mol. Cell 2006, 22, 501–510. [Google Scholar]Int. J. Mol. Sci. 2014, 15, 3427.
- Wagner, J.M.; Karnitz, L.M. Cisplatin-induced DNA damage activates replication checkpoint signaling components that differentially affect tumor cell survival. Mol. Pharmacol. 2009, 76, 208–214. [Google Scholar]
- Myers, J.S.; Cortez, D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J. Biol. Chem. 2006, 281, 9346–9350. [Google Scholar]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar]
- Delacroix, S.; Wagner, J.M.; Kobayashi, M.; Yamamoto, K.; Karnitz, L.M. The Rad9-Hus1-Rad1 (9–1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007, 21, 1472–1477. [Google Scholar]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol. 2001, 21, 4129–4139. [Google Scholar]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by ATR and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar]
- Chen, P.; Luo, C.; Deng, Y.; Ryan, K.; Register, J.; Margosiak, S.; Tempczyk-Russell, A.; Nguyen, B.; Myers, P.; Lundgren, K.; et al. The 17 A crystal structure of human cell cycle checkpoint kinase Chk1: Implications for Chk1 regulation. Cell 2000, 100, 681–692. [Google Scholar]
- Chini, C.C.; Chen, J. Human claspin is required for replication checkpoint control. J. Biol. Chem. 2003, 278, 30057–30062. [Google Scholar]
- Weiss, R.S.; Matsuoka, S.; Elledge, S.J.; Leder, P. Hus1 acts upstream of chk1 in a mammalian DNA damage response pathway. Curr. Biol. 2002, 12, 73–77. [Google Scholar]
- Zou, L.; Cortez, D.; Elledge, S.J. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002, 16, 198–208. [Google Scholar]
- Zou, L.; Liu, D.; Elledge, S.J. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 13827–13832. [Google Scholar]
- Kumagai, A.; Dunphy, W.G. Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1. Nat. Cell Biol. 2003, 5, 161–165. [Google Scholar]
- Jin, J.; Ang, X.L.; Ye, X.; Livingstone, M.; Harper, J.W. Differential roles for checkpoint kinases in DNA damage-dependent degradation of the Cdc25A protein phosphatase. J. Biol. Chem. 2008, 283, 19322–19328. [Google Scholar]
- Ferguson, A.M.; White, L.S.; Donovan, P.J.; Piwnica-Worms, H. Normal cell cycle and checkpoint responses in mice and cells lacking Cdc25B and Cdc25C protein phosphatases. Mol. Cell Biol. 2005, 25, 2853–2860. [Google Scholar]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Kroemer, G. Cyclin-dependent kinase-1: Linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002, 9, 1287–1293. [Google Scholar]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337.
- Chen, T.; Stephens, P.A.; Middleton, F.K.; Curtin, N.J. Targeting the S and G2 checkpoint to treat cancer. Drug Discov. Today 2012, 17, 194–202. [Google Scholar]
- Bucher, N.; Britten, C.D. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br. J. Cancer 2008, 98, 523–528. [Google Scholar]
- Bettayeb, K.; Oumata, N.; Echalier, A.; Ferandin, Y.; Endicott, J.A.; Galons, H.; Meijer, L. CR8 a potent and selective roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene 2008, 27, 5797–5807. [Google Scholar]
- Rizzolio, F.; Tuccinardi, T.; Caligiuri, I.; Lucchetti, C.; Giordano, A. CDK inhibitors: From the bench to clinical trials. Curr. Drug Targets 2010, 11, 279–290. [Google Scholar]
- Clemenson, C.; Marsolier-Kergoat, M.C. DNA damage checkpoint inactivation: Adaptation and recovery. DNA Repair (Amst.) 2009, 8, 1101–1109. [Google Scholar]
- Toczyski, D.P.; Galgoczy, D.J.; Hartwell, L.H. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 1997, 90, 1097–1106. [Google Scholar]
- Tansley, K.; Spear, F.G.; Glucksmann, A. The effect of gamma rays on cell division in the developing rat retina. Br. J. Ophthalmol. 1937, 21, 273–298. [Google Scholar]
- Kubara, P.M.; Kerneis-Golsteyn, S.; Studeny, A.; Lanser, B.B.; Meijer, L.; Golsteyn, R.M. Human cells enter mitosis with damaged DNA after treatment with pharmacological concentrations of genotoxic agents. Biochem. J. 2012, 446, 373–381. [Google Scholar]
- Sandell, L.L.; Zakian, V.A. Loss of a yeast telomere: Arrest recovery and chromosome loss. Cell 1993, 75, 729–739. [Google Scholar]
- Yoo, H.Y.; Kumagai, A.; Shevchenko, A.; Dunphy, W.G. Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell 2004, 117, 575–588. [Google Scholar]
- Pellicioli, A.; Lee, S.E.; Lucca, C.; Foiani, M.; Haber, J.E. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol. Cell 2001, 7, 293–300. [Google Scholar]
- Vaze, M.B.; Pellicioli, A.; Lee, S.E.; Ira, G.; Liberi, G.; Arbel-Eden, A.; Foiani, M.; Haber, J.E. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol. Cell 2002, 10, 373–385. [Google Scholar]
- Leroy, C.; Lee, S.E.; Vaze, M.B.; Ochsenbein, F.; Guerois, R.; Haber, J.E.; Marsolier-Kergoat, M.C. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol. Cell 2003, 11, 827–835. [Google Scholar]
- Lupardus, P.J.; Cimprich, K.A. Checkpoint adaptation; molecular mechanisms uncovered. Cell 2004, 117, 555–556. [Google Scholar]
- Syljuasen, R.G.; Jensen, S.; Bartek, J.; Lukas, J. Adaptation to the ionizing radiation-induced G2 checkpoint occurs in human cells and depends on checkpoint kinase 1 and Polo-like kinase 1 kinases. Cancer Res. 2006, 66, 10253–10257. [Google Scholar]
- Rezacova, M.; Rudolfova, G.; Tichy, A.; Bacikova, A.; Mutna, D.; Havelek, R.; Vavrova, J.; Odrazka, K.; Lukasova, E.; Kozubek, S. Accumulation of DNA damage and cell death after fractionated irradiation. Radiat. Res. 2011, 175, 708–718. [Google Scholar]
- Roninson, I.B.; Broude, E.V.; Chang, B.D. If not apoptosis then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist. Updat. 2001, 4, 303–313. [Google Scholar]
- Golsteyn, R.M. Cdk1 and Cdk2 complexes (cyclin dependent kinases) in apoptosis: A role beyond the cell cycle. Cancer Lett. 2005, 217, 129–138. [Google Scholar]
- Borgne, A.; Golsteyn, R.M. The role of cyclin-dependent kinases in apoptosis. Prog. Cell Cycle Res. 2003, 5, 453–459. [Google Scholar]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar]
- Huang, L.C.; Clarkin, K.C.; Wahl, G.M. Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proc. Natl. Acad. Sci. USA 1996, 93, 4827–4832. [Google Scholar]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Curman, D.; Cinel, B.; Williams, D.E.; Rundle, N.; Block, W.D.; Goodarzi, A.A.; Hutchins, J.R.; Clarke, P.R.; Zhou, B.B.; Lees-Miller, S.P.; et al. Inhibition of the G2 DNA damage checkpoint and of protein kinases Chk1 and Chk2 by the marine sponge alkaloid debromohymenialdisine. J. Biol. Chem. 2001, 276, 17914–17919. [Google Scholar]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. Abrogation of the Chk1-mediated G(2) checkpoint pathway potentiates temozolomide-induced toxicity in a p53-independent manner in human glioblastoma cells. Cancer Res. 2001, 61, 5843–5849. [Google Scholar]
- Syljuasen, R.G.; Sorensen, C.S.; Nylandsted, J.; Lukas, C.; Lukas, J.; Bartek, J. Inhibition of Chk1 by CEP-3891 accelerates mitotic nuclear fragmentation in response to ionizing Radiation. Cancer Res. 2004, 64, 9035–9040. [Google Scholar]
- Tse, A.N.; Schwartz, G.K. Potentiation of cytotoxicity of topoisomerase I poison by concurrent and sequential treatment with the checkpoint inhibitor UCN-01 involves disparate mechanisms resulting in either p53-independent clonogenic suppression or p53-dependent mitotic catastrophe. Cancer Res. 2004, 64, 6635–6644. [Google Scholar]
- Ferry, G.; Studeny, A.; Bossard, C.; Kubara, P.M.; Zeyer, D.; Renaud, J.P.; Casara, P.; de Nanteuil, G.; Wierzbicki, M.; Pfeiffer, B.; et al. Characterization of novel checkpoint kinase 1 inhibitors by in vitro assays and in human cancer cells treated with topoisomerase inhibitors. Life Sci. 2011, 89, 259–268. [Google Scholar]
- Ianzini, F.; Mackey, M.A. Spontaneous premature chromosome condensation and mitotic catastrophe following irradiation of HeLa S3 cells. Int. J. Radiat. Biol. 1997, 72, 409–421. [Google Scholar]
- Tounekti, O.; Pron, G.; Belehradek, J., Jr; Mir, L.M. Bleomycin an apoptosis-mimetic drug that induces two types of cell death depending on the number of molecules internalized. Cancer Res. 1993, 53, 5462–5469. [Google Scholar]
- Holgersson, A.; Heiden, T.; Castro, J.; Edgren, M.R.; Lewensohn, R.; Meijer, A.E. Different G2/M accumulation in M059J and M059K cells after exposure to DNA double-strand break-inducing agents. Int. J. Radiat. Oncol. Biol. Phys. 2005, 61, 915–921. [Google Scholar]
- Lock, R.B.; Stribinskiene, L. Dual modes of death induced by etoposide in human epithelial tumor cells allow Bcl-2 to inhibit apoptosis without affecting clonogenic survival. Cancer Res. 1996, 56, 4006–4012. [Google Scholar]
- Demarcq, C.; Bunch, R.T.; Creswell, D.; Eastman, A. The role of cell cycle progression in cisplatin-induced apoptosis in Chinese hamster ovary cells. Cell Growth Differ. 1994, 5, 983–993. [Google Scholar]
- Vakifahmetoglu, H.; Olsson, M.; Tamm, C.; Heidari, N.; Orrenius, S.; Zhivotovsky, B. DNA damage induces two distinct modes of cell death in ovarian carcinomas. Cell Death Differ. 2008, 15, 555–566. [Google Scholar]
- Chang, B.D.; Broude, E.V.; Dokmanovic, M.; Zhu, H.; Ruth, A.; Xuan, Y.; Kandel, E.S.; Lausch, E.; Christov, K.; Roninson, I.B. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999, 59, 3761–3767. [Google Scholar]
- Yoshikawa, R.; Kusunoki, M.; Yanagi, H.; Noda, M.; Furuyama, J.I.; Yamamura, T.; Hashimoto-Tamaoki, T. Dual antitumor effects of 5-fluorouracil on the cell cycle in colorectal carcinoma cells: A novel target mechanism concept for pharmacokinetic modulating chemotherapy. Cancer Res. 2001, 61, 1029–1037. [Google Scholar]
- Eom, Y.W.; Kim, M.A.; Park, S.S.; Goo, M.J.; Kwon, H.J.; Sohn, S.; Kim, W.H.; Yoon, G.; Choi, K.S. Two distinct modes of cell death induced by doxorubicin: Apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 2005, 24, 4765–4777. [Google Scholar]
- Bhattathiri, N.V.; Bharathykkutty, C.; Prathapan, R.; Chirayathmanjiyil, D.A.; Nair, K.M. Prediction of radiosensitivity of oral cancers by serial cytological assay of nuclear changes. Radiother. Oncol. 1998, 49, 61–65. [Google Scholar]
- Kumari, R.; Chaugule, A.; Goyal, P.K. Karyoanomalic frequency during radiation therapy. J. Cancer Res. Ther 2005, 1, 187–190. [Google Scholar]
- Bhattathiri, V.N. Amitotic cell divisions and tumour growth: An alternative model for cell kinetic compartments in solid tumours. Oral. Oncol. 2001, 37, 288–295. [Google Scholar]
- Widel, M.; Jedrus, S.; Owczarek, S.; Konopacka, M.; Lubecka, B.; Kolosza, Z. The increment of micronucleus frequency in cervical carcinoma during irradiation in vivo and its prognostic value for tumour radiocurability. Br. J. Cancer 1999, 80, 1599–1607. [Google Scholar]
- Zolzer, F.; Alberti, W.; Pelzer, T.; Lamberti, G.; Hulskamp, F.H.; Streffer, C. Changes in S-phase fraction and micronucleus frequency as prognostic factors in radiotherapy of cervical carcinoma. Radiother. Oncol. 1995, 36, 128–132. [Google Scholar]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar]
- Chan, T.A.; Hermeking, H.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. 14–3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature 1999, 401, 616–620. [Google Scholar]
- de Bruin, E.C.; Medema, J.P. Apoptosis and non-apoptotic deaths in cancer development and treatment response. Cancer Treat. Rev. 2008, 34, 737–749. [Google Scholar]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar]
- Hayashi, M.T.; Karlseder, J. DNA damage associated with mitosis and cytokinesis failure. Oncogene 2013, 32, 4593–4601. [Google Scholar]
- Gascoigne, K.E.; Taylor, S.S. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar]
- Olive, P.L.; Banath, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar]
- Hendzel, M.J.; Wei, Y.; Mancini, M.A.; van Hooser, A.; Ranalli, T.; Brinkley, B.R.; Bazett-Jones, D.P.; Allis, C.D. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 1997, 106, 348–360. [Google Scholar]
- Lewis, C.W.; Taylor, R.G.; Kubara, P.M.; Marshall, K.; Meijer, L.; Golsteyn, R.M. A western blot assay to measure cyclin dependent kinase activity in cells or in vitro without the use of radioisotopes. FEBS Lett. 2013, 587, 3089–3095. [Google Scholar]
- Swift, L.H.; Golsteyn, R.M. Checkpoint adaptation induced by the cancer drug cisplatin in human colon cancer cells: Analyzing the role of mitosis. 2014, unpublished work. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
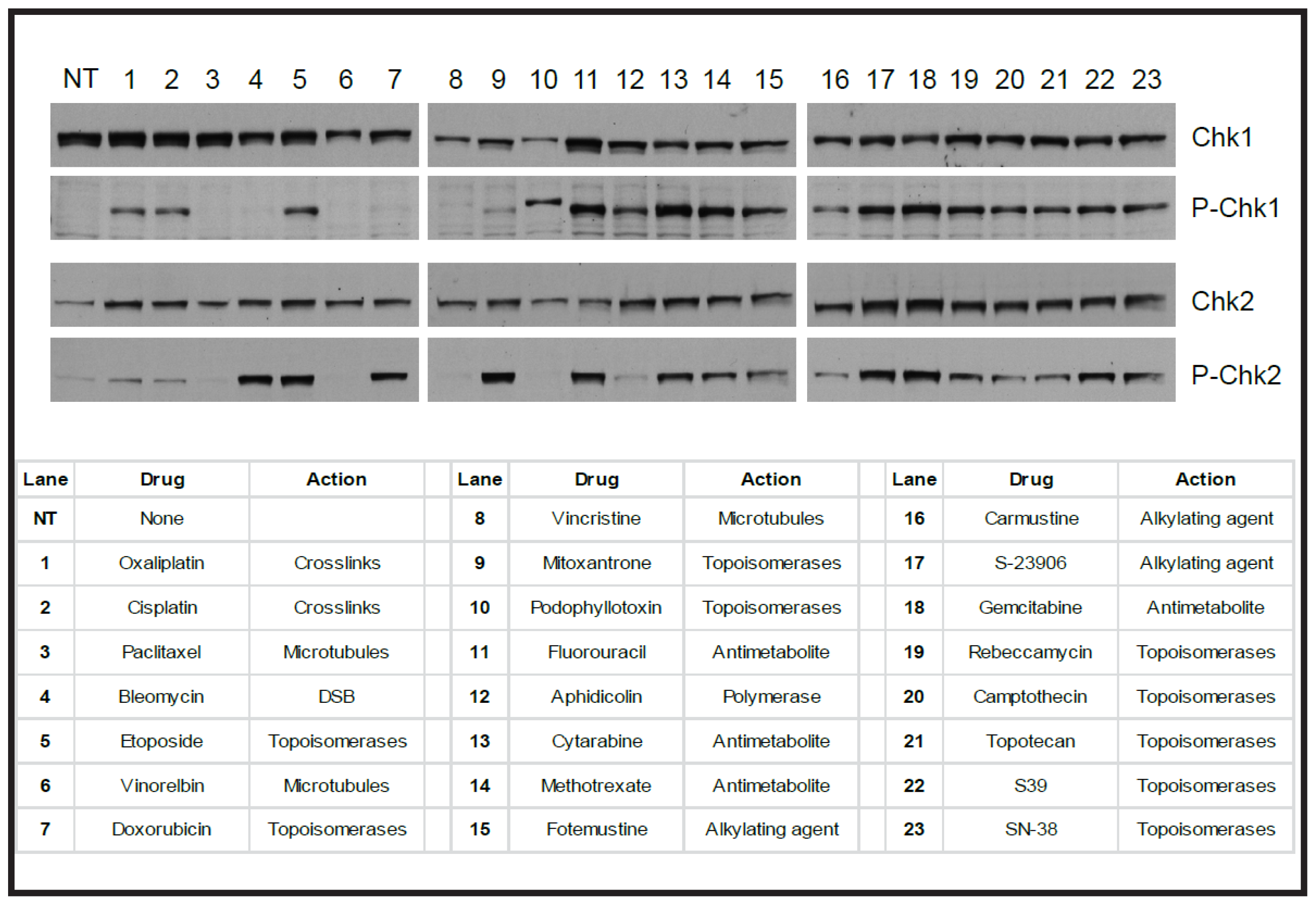
| Agent Type | Examples of Drugs | Mechanism of Action | Type of DNA Damage |
|---|---|---|---|
| Alkylating agents | Nitrogen mustards Nitrosoureas Temozolomide S23906 | Base alkylation-Monofunctional DNA adducts | Block the replication machinery leading to strand breaks |
| Inter, intra and DNA-protein crosslinks | Block the replication machinery leading to strand breaks | ||
| Platinum drugs | Cisplatin Carboplatin Oxaliplatin | Monofunctional DNA adducts | Block the replication machinery leading to strand breaks |
| Inter, intra and DNA-protein crosslinks | Block the replication machinery leading to strand breaks | ||
| Antimetabolites | 5-Fluorouracil | Misincorporates into DNA | Blocks the replication machinery leading to strand breaks |
| Depletes dNTPs | Blocks the replication machinery leading to strand breaks | ||
| Topoisomerase poisons | Camptothecin Etoposide | Inhibit topoisomerase enzymes in complex with DNA | SSBs and DSBs |
| Ionising radiation | Direct | SSBs and DSBs | |
| Indirect production of ROS | DNA adducts, base oxidation, SSBs, DSBs, base deamination, DNA-protein crosslinks | ||
| Drug | Administration | Dosage | Peak Plasma Concentration |
|---|---|---|---|
| Temozolomide | Oral capsule | 200 mg/m2/day for 5 days | 104 μM [45] |
| 150 mg/m2/day for 5 days | 66 μM [45] | ||
| 100 mg/m2/day for 5 days | 44 μM [45] | ||
| Cisplatin (Total platinum) | Toxic concentration in blood | 16 μM [50] | |
| 1 h intravenous | 100 mg/m2 | 12 μM [51] | |
| Rapid (4–15 min) intravenous | 100 mg/m2 | 40 μM [52] | |
| Carboplatin (Total platinum) | 1 h intravenous | 400 mg/m2 | 70 μM [53] |
| 37 min intravenous | 350 mg/m2 | 130 μM [54] | |
| Oxaliplatin (Total platinum) | 5 cycles, 2 h intravenous | 130 mg/m2 every 3 weeks | 10 μM [55] |
| 3 cycles, 2 h intravenous | 85 mg/m2 every 2 weeks | 6 μM [55] | |
| 5-Fluorouracil | 72 h intravenous | 1750 mg/m2/day | 10 μM [56] |
| Camptothecin | Oral | 1.5 mg/m2/day for 5 days | 75 nM [57] |
| Etoposide | Oral | 100 mg/day-8–15 days (typical dose) | 14 μM [58] |
| Intravenous, 500 mg/h | 400–800 mg/m2/day for 3 days (high dose study) | 45–194 μM [59] | |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Swift, L.H.; Golsteyn, R.M. Genotoxic Anti-Cancer Agents and Their Relationship to DNA Damage, Mitosis, and Checkpoint Adaptation in Proliferating Cancer Cells. Int. J. Mol. Sci. 2014, 15, 3403-3431. https://doi.org/10.3390/ijms15033403
Swift LH, Golsteyn RM. Genotoxic Anti-Cancer Agents and Their Relationship to DNA Damage, Mitosis, and Checkpoint Adaptation in Proliferating Cancer Cells. International Journal of Molecular Sciences. 2014; 15(3):3403-3431. https://doi.org/10.3390/ijms15033403
Chicago/Turabian StyleSwift, Lucy H., and Roy M. Golsteyn. 2014. "Genotoxic Anti-Cancer Agents and Their Relationship to DNA Damage, Mitosis, and Checkpoint Adaptation in Proliferating Cancer Cells" International Journal of Molecular Sciences 15, no. 3: 3403-3431. https://doi.org/10.3390/ijms15033403