High Sensitivity of Circulating Tumor Cells Derived from a Colorectal Cancer Patient for Dual Inhibition with AKT and mTOR Inhibitors

 , ,
, ,  and
and
Abstract
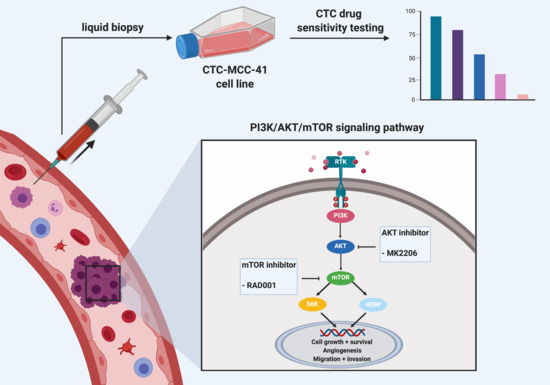
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
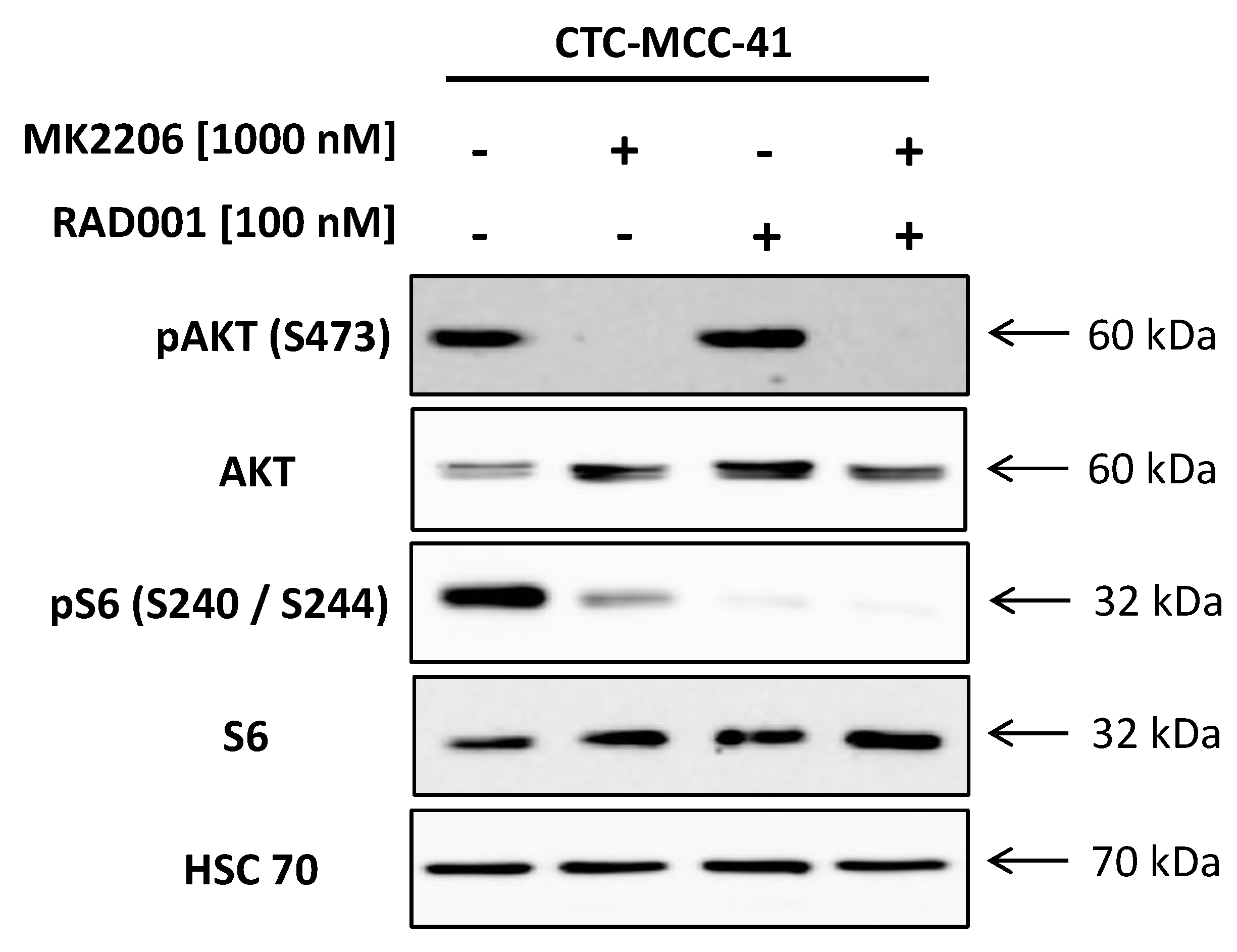
2.1. Strong Activation of the PI3K/AKT/mTOR Signaling Pathway in CTC-MCC-41
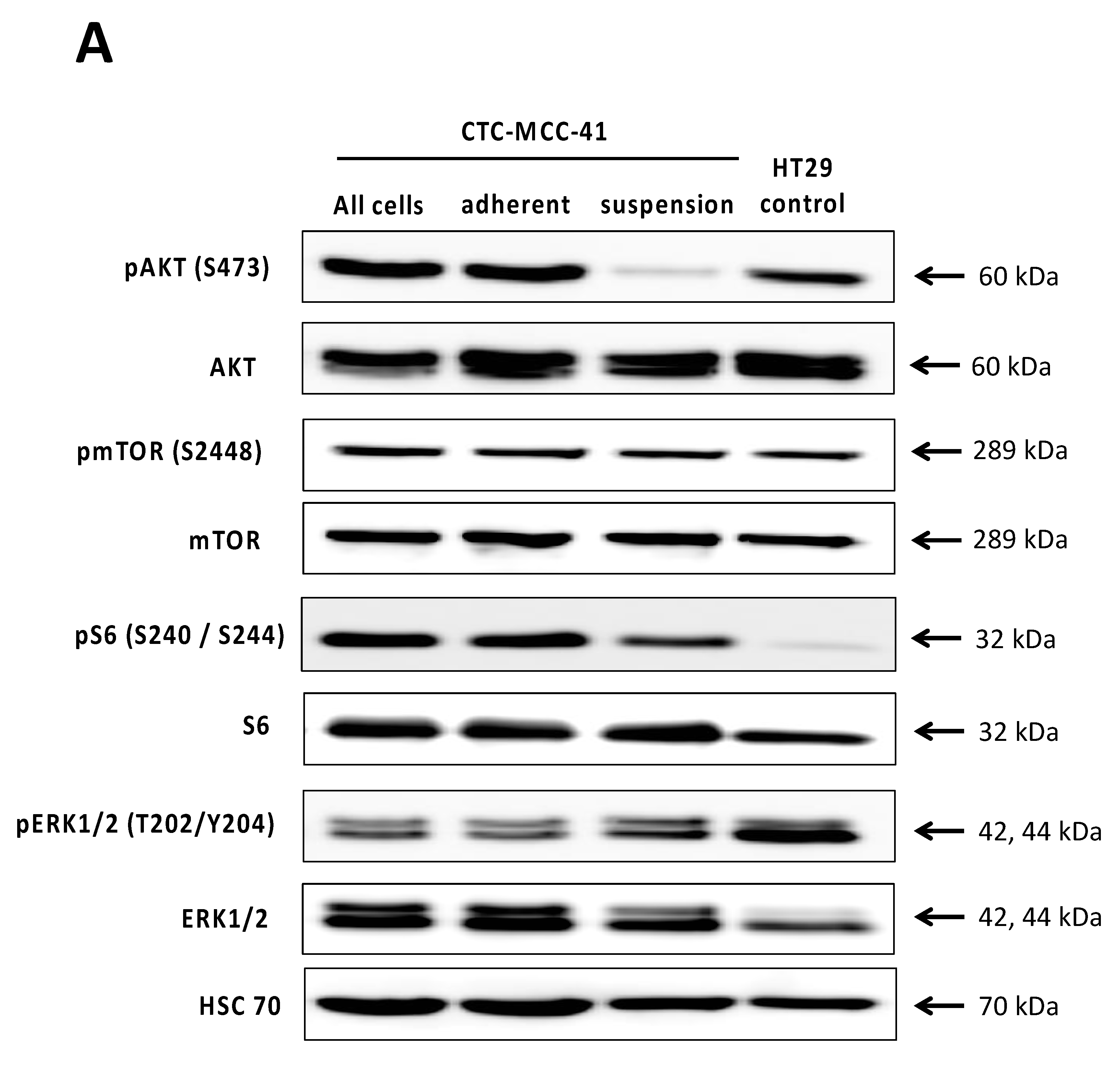
2.2. Differential PI3K/AKT/mTOR Signaling in Suspension and Adherent Phenotype of CTC-MCC-41 Cells
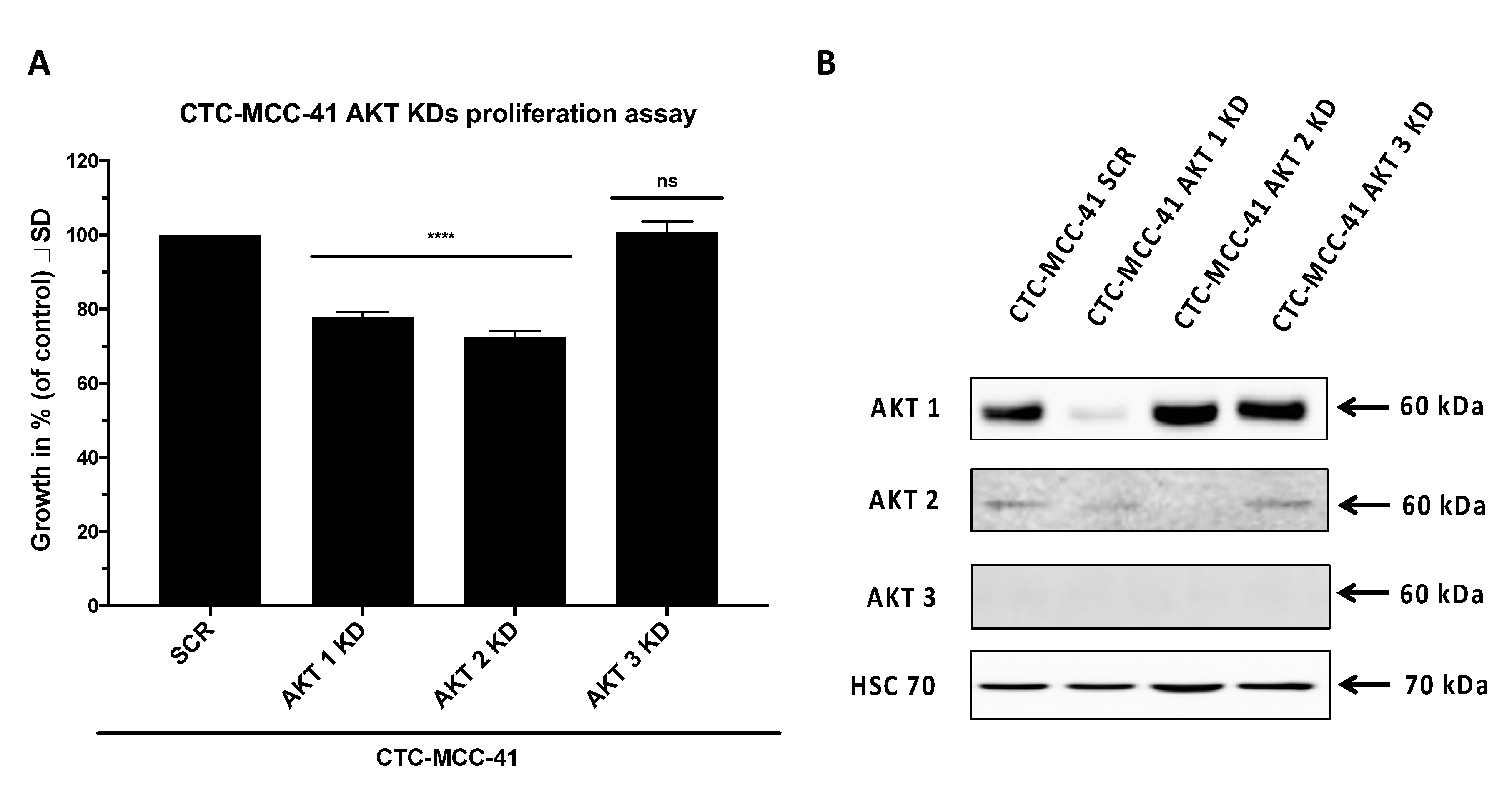
2.3. AKT Isoform Specific Signaling in CTC-MCC-41 Cells
3. Discussion
4. Materials and Methods
4.1. Standard Cell Culture
4.2. Stable AKT Isoform Specific Knockdown
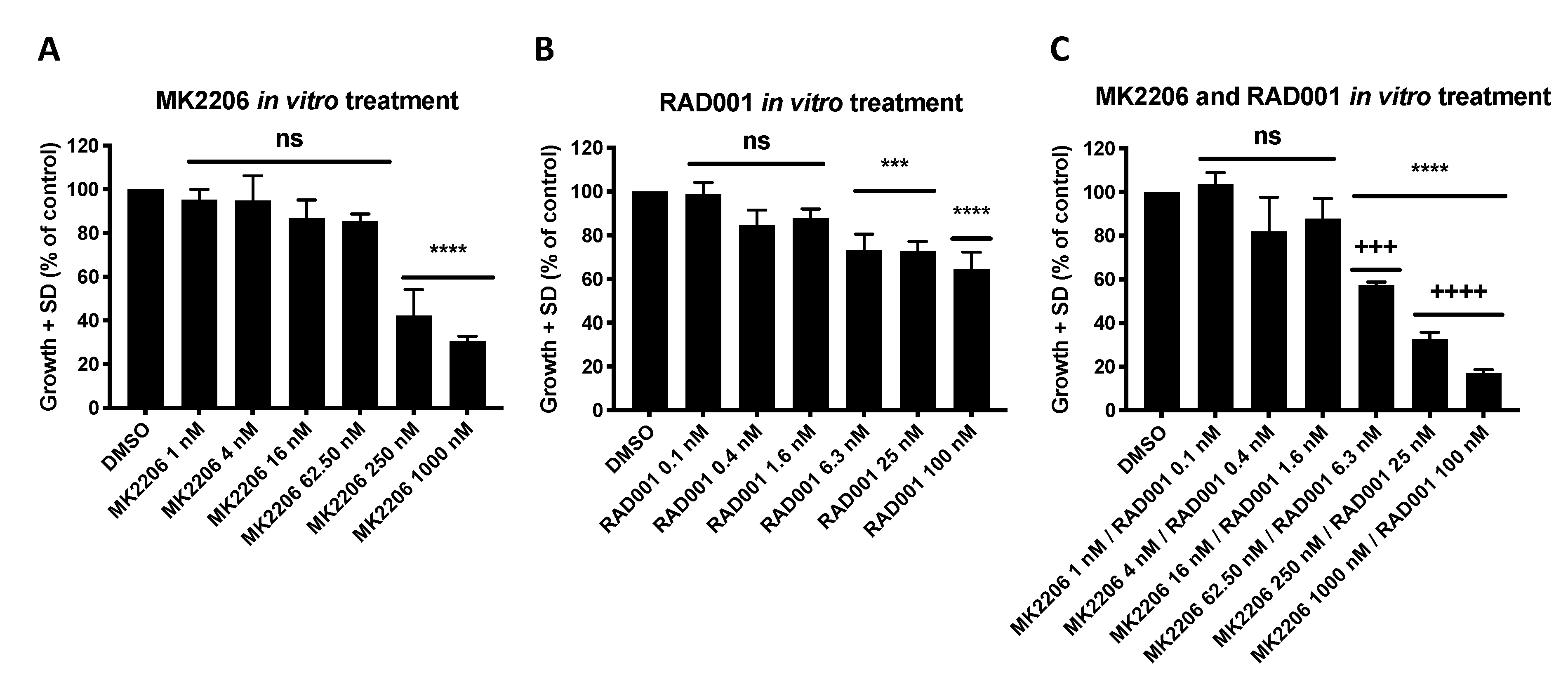
4.3. Proliferation Assay and Therapeutic Drug Sensitivity Testing
4.4. Apoptosis Assay
4.5. Western Blot Analysis and Densitometric Quantification
4.6. Calculation of Combination Indices
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krakhmal, N.V.; Zavyalova, M.V.; Denisov, E.V.; Vtorushin, S.V.; Perelmuter, V.M. Cancer Invasion: Patterns and Mechanisms. Acta Nat. 2015, 7, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Cayrefourcq, L.; Mazard, T.; Joosse, S.; Solassol, J.; Ramos, J.; Assenat, E.; Schumacher, U.; Costes, V.; Maudelonde, T.; Pantel, K.; et al. Establishment and characterization of a cell line from human circulating colon cancer cells. Cancer Res. 2015, 75, 892–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés-Hernández, L.E.; Eslami, S.Z.; Alix-Panabières, C. Circulating tumor cell as the functional aspect of liquid biopsy to understand the metastatic cascade in solid cancer. Mol. Asp. Med. 2020, 72, 100816. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabieres, C.; Pantel, K. Circulating tumor cells: Liquid biopsy of cancer. Clin. Chem. 2013, 59, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabieres, C. Cell lines from circulating tumor cells. Oncoscience 2015, 2, 815–816. [Google Scholar] [CrossRef]
- Burz, C.; Pop, V.V.; Buiga, R.; Daniel, S.; Samasca, G.; Aldea, C.; Lupan, I. Circulating tumor cells in clinical research and monitoring patients with colorectal cancer. Oncotarget 2018, 9, 24561–24571. [Google Scholar] [CrossRef]
- Hartkopf, A.D.; Wagner, P.; Wallwiener, D.; Fehm, T.; Rothmund, R. Changing levels of circulating tumor cells in monitoring chemotherapy response in patients with metastatic breast cancer. Anticancer Res. 2011, 31, 979–984. [Google Scholar]
- Bielcikova, Z.; Jakabova, A.; Pinkas, M.; Zemanova, M.; Kolostova, K.; Bobek, V. Circulating tumor cells: What we know, what do we want to know about them and are they ready to be used in clinics? Am. J. Transl. Res. 2017, 9, 2807–2823. [Google Scholar]
- Pailler, E.; Mezquita, L.; Faugeroux, V.; Oulhen, M.; Queffelec, P.; Remon, J.; Honore, A.; Laporte, M.; Forcato, C.; Lecluse, Y.; et al. Analysis of single circulating tumor cells (CTCs) to identify resistance mutations to ALK-inhibitors in both ALK-gene and bypass oncogenic pathways. J. Clin. Oncol. 2018, 36, 12038. [Google Scholar] [CrossRef]
- Que, Z.; Luo, B.; Zhou, Z.; Dong, C.; Jiang, Y.; Wang, L.; Shi, Q.; Tian, J. Establishment and characterization of a patient-derived circulating lung tumor cell line in vitro and in vivo. Cancer Cell Int. 2019, 19, 21. [Google Scholar] [CrossRef] [Green Version]
- Abdallah, E.A.; Fanelli, M.F.; Souza, E.S.V.; Machado Netto, M.C.; Gasparini Junior, J.L.; Araujo, D.V.; Ocea, L.M.; Buim, M.E.; Tariki, M.S.; Alves Vda, S.; et al. MRP1 expression in CTCs confers resistance to irinotecan-based chemotherapy in metastatic colorectal cancer. Int. J. Cancer 2016, 139, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Effenberger, K.E.; Schroeder, C.; Hanssen, A.; Wolter, S.; Eulenburg, C.; Tachezy, M.; Gebauer, F.; Izbicki, J.R.; Pantel, K.; Bockhorn, M. Improved Risk Stratification by Circulating Tumor Cell Counts in Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 2844–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeh, M.; Effenberger, K.E.; Koenig, A.M.; Riethdorf, S.; Eichstadt, D.; Vettorazzi, E.; Uzunoglu, F.G.; Vashist, Y.K.; Izbicki, J.R.; Pantel, K.; et al. Circulating Tumor Cells as a Biomarker for Preoperative Prognostic Staging in Patients With Esophageal Cancer. Ann. Surg. 2015, 261, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Garrigos, N.; Gallego, J.; Guillen-Ponce, C.; Guaraz, P.; Garcia-Bautista, M.; Castillejo, A.; Gomez-Martinez, A.; Carrato, A.; Rodriguez-Lescure, A.; Soto, J.L. Circulating tumour cell analysis as an early marker for relapse in stage II and III colorectal cancer patients: A pilot study. Clin. Transl. Oncol. 2010, 12, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabieres, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Alix-Panabieres, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.J.; Punt, C.J.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Geest, L.G.; Lam-Boer, J.; Koopman, M.; Verhoef, C.; Elferink, M.A.; de Wilt, J.H. Nationwide trends in incidence, treatment and survival of colorectal cancer patients with synchronous metastases. Clin. Exp. Metastasis 2015, 32, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, S.; Liu, Y.; Zhang, C.; Li, H.; Lai, B. Metastatic patterns and survival outcomes in patients with stage IV colon cancer: A population-based analysis. Cancer Med. 2020, 9, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahbari, N.N.; Bork, U.; Kircher, A.; Nimitz, T.; Scholch, S.; Kahlert, C.; Schmidt, T.; Steinert, G.; Ulrich, A.B.; Reissfelder, C.; et al. Compartmental differences of circulating tumor cells in colorectal cancer. Ann. Surg. Oncol. 2012, 19, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wu, H. The significant prognostic value of circulating tumor cells in colorectal cancer: A systematic review and meta-analysis. Curr. Probl. Cancer 2018, 42, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Keller, L.; Pantel, K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat. Rev. Cancer 2019, 19, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Koch, C.; Kuske, A.; Joosse, S.A.; Yigit, G.; Sflomos, G.; Thaler, S.; Smit, D.J.; Werner, S.; Borgmann, K.; Gartner, S.; et al. Characterization of circulating breast cancer cells with tumorigenic and metastatic capacity. EMBO Mol. Med. 2020, e11908. [Google Scholar] [CrossRef]
- Soler, A.; Cayrefourcq, L.; Mazard, T.; Babayan, A.; Lamy, P.J.; Assou, S.; Assenat, E.; Pantel, K.; Alix-Panabieres, C. Autologous cell lines from circulating colon cancer cells captured from sequential liquid biopsies as model to study therapy-driven tumor changes. Sci. Rep. 2018, 8, 15931. [Google Scholar] [CrossRef]
- Rodrigues, P.; Vanharanta, S. Circulating Tumor Cells: Come Together, Right Now, Over Metastasis. Cancer Discov. 2019, 9, 22–24. [Google Scholar] [CrossRef] [Green Version]
- Alix-Panabieres, C.; Cayrefourcq, L.; Mazard, T.; Maudelonde, T.; Assenat, E.; Assou, S. Molecular Portrait of Metastasis-Competent Circulating Tumor Cells in Colon Cancer Reveals the Crucial Role of Genes Regulating Energy Metabolism and DNA Repair. Clin. Chem. 2017, 63, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Hu, Y.Z. PI3K/Akt/mTOR pathway inhibitors in cancer: A perspective on clinical progress. Curr. Med. Chem. 2010, 17, 4326–4341. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Gao, J.; Du, Z.; Zhang, X.; Yang, F.; Gao, W. Expression of factors and key components associated with the PI3K signaling pathway in colon cancer. Oncol. Lett. 2018, 15, 5465–5472. [Google Scholar] [CrossRef] [Green Version]
- Itoh, N.; Semba, S.; Ito, M.; Takeda, H.; Kawata, S.; Yamakawa, M. Phosphorylation of Akt/PKB is required for suppression of cancer cell apoptosis and tumor progression in human colorectal carcinoma. Cancer 2002, 94, 3127–3134. [Google Scholar] [CrossRef]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, W.; Sedrani, R.; Cottens, S.; Haberlin, B.; Schulz, M.; Schuurman, H.J.; Zenke, G.; Zerwes, H.G.; Schreier, M.H. SDZ RAD, a new rapamycin derivative: Pharmacological properties in vitro and in vivo. Transplantation 1997, 64, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Ewald, F.; Grabinski, N.; Grottke, A.; Windhorst, S.; Norz, D.; Carstensen, L.; Staufer, K.; Hofmann, B.T.; Diehl, F.; David, K.; et al. Combined targeting of AKT and mTOR using MK-2206 and RAD001 is synergistic in the treatment of cholangiocarcinoma. Int. J. Cancer 2013, 133, 2065–2076. [Google Scholar] [CrossRef] [PubMed]
- Ewald, F.; Norz, D.; Grottke, A.; Bach, J.; Herzberger, C.; Hofmann, B.T.; Nashan, B.; Jucker, M. Vertical Targeting of AKT and mTOR as Well as Dual Targeting of AKT and MEK Signaling Is Synergistic in Hepatocellular Carcinoma. J. Cancer 2015, 6, 1195–1205. [Google Scholar] [CrossRef] [Green Version]
- Ewald, F.; Norz, D.; Grottke, A.; Hofmann, B.T.; Nashan, B.; Jucker, M. Dual Inhibition of PI3K-AKT-mTOR- and RAF-MEK-ERK-signaling is synergistic in cholangiocarcinoma and reverses acquired resistance to MEK-inhibitors. Investig. New Drugs 2014, 32, 1144–1154. [Google Scholar] [CrossRef]
- Schulte, A.; Ewald, F.; Spyra, M.; Smit, D.J.; Jiang, W.; Salamon, J.; Jücker, M.; Mautner, V.-F. Combined Targeting of AKT and mTOR Inhibits Proliferation of Human NF1-Associated Malignant Peripheral Nerve Sheath Tumour Cells In Vitro but not in a Xenograft Mouse Model In Vivo. Int. J. Mol. Sci. 2020, 21, 1548. [Google Scholar] [CrossRef] [Green Version]
- Francipane, M.G.; Lagasse, E. mTOR pathway in colorectal cancer: An update. Oncotarget 2014, 5, 49–66. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.M.; Gulhati, P.; Rampy, B.A.; Han, Y.; Rychahou, P.G.; Doan, H.Q.; Weiss, H.L.; Evers, B.M. Novel expression patterns of PI3K/Akt/mTOR signaling pathway components in colorectal cancer. J. Am. Coll. Surg. 2010, 210, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yue, P.; Kim, Y.A.; Fu, H.; Khuri, F.R.; Sun, S.Y. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res. 2008, 68, 7409–7418. [Google Scholar] [CrossRef] [Green Version]
- Ng, K.; Tabernero, J.; Hwang, J.; Bajetta, E.; Sharma, S.; Del Prete, S.A.; Arrowsmith, E.R.; Ryan, D.P.; Sedova, M.; Jin, J.; et al. Phase II study of everolimus in patients with metastatic colorectal adenocarcinoma previously treated with bevacizumab-, fluoropyrimidine-, oxaliplatin-, and irinotecan-based regimens. Clin. Cancer Res. 2013, 19, 3987–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearl, M.L.; Dong, H.; Zhao, Q.; Tulley, S.; Dombroff, M.K.; Chen, W.T. iCTC drug resistance (CDR) Testing ex vivo for evaluation of available therapies to treat patients with epithelial ovarian cancer. Gynecol. Oncol. 2017, 147, 426–432. [Google Scholar] [CrossRef]
- Hinz, N.; Jucker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A.; Testa, J.R.; Moore, R.; Larue, L. A portrait of AKT kinases: Human cancer and animal models depict a family with strong individualities. Cancer Biol. Ther. 2004, 3, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef]
- Linnerth-Petrik, N.M.; Santry, L.A.; Moorehead, R.; Jucker, M.; Wootton, S.K.; Petrik, J. Akt isoform specific effects in ovarian cancer progression. Oncotarget 2016, 7, 74820–74833. [Google Scholar] [CrossRef] [Green Version]
- Grabinski, N.; Bartkowiak, K.; Grupp, K.; Brandt, B.; Pantel, K.; Jucker, M. Distinct functional roles of Akt isoforms for proliferation, survival, migration and EGF-mediated signalling in lung cancer derived disseminated tumor cells. Cell. Signal. 2011, 23, 1952–1960. [Google Scholar] [CrossRef]
- Soung, Y.H.; Lee, J.W.; Nam, S.W.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutational analysis of AKT1, AKT2 and AKT3 genes in common human carcinomas. Oncology 2006, 70, 285–289. [Google Scholar] [CrossRef]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Aktas, B.; Tewes, M.; Fehm, T.; Hauch, S.; Kimmig, R.; Kasimir-Bauer, S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009, 11, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heron-Milhavet, L.; Franckhauser, C.; Rana, V.; Berthenet, C.; Fisher, D.; Hemmings, B.A.; Fernandez, A.; Lamb, N.J. Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol. Cell. Biol. 2006, 26, 8267–8280. [Google Scholar] [CrossRef] [Green Version]
- Riggio, M.; Perrone, M.C.; Polo, M.L.; Rodriguez, M.J.; May, M.; Abba, M.; Lanari, C.; Novaro, V. AKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteins. Sci. Rep. 2017, 7, 44244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, E.; Robb, C.M.; Smith, L.M.; Brattain, M.G.; Wang, J.; Black, J.D.; Chowdhury, S. Role of Akt2 in regulation of metastasis suppressor 1 expression and colorectal cancer metastasis. Oncogene 2017, 36, 3104–3118. [Google Scholar] [CrossRef] [PubMed]
- Haggblad Sahlberg, S.; Mortensen, A.C.; Haglof, J.; Engskog, M.K.; Arvidsson, T.; Pettersson, C.; Glimelius, B.; Stenerlow, B.; Nestor, M. Different functions of AKT1 and AKT2 in molecular pathways, cell migration and metabolism in colon cancer cells. Int. J. Oncol. 2017, 50, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santi, S.A.; Lee, H. Ablation of Akt2 induces autophagy through cell cycle arrest, the downregulation of p70S6K, and the deregulation of mitochondria in MDA-MB231 cells. PLoS ONE 2011, 6, e14614. [Google Scholar] [CrossRef]
- Koseoglu, S.; Lu, Z.; Kumar, C.; Kirschmeier, P.; Zou, J. AKT1, AKT2 and AKT3-dependent cell survival is cell line-specific and knockdown of all three isoforms selectively induces apoptosis in 20 human tumor cell lines. Cancer Biol. Ther. 2007, 6, 755–762. [Google Scholar] [CrossRef]
- Grottke, A.; Ewald, F.; Lange, T.; Norz, D.; Herzberger, C.; Bach, J.; Grabinski, N.; Graser, L.; Hoppner, F.; Nashan, B.; et al. Downregulation of AKT3 Increases Migration and Metastasis in Triple Negative Breast Cancer Cells by Upregulating S100A4. PLoS ONE 2016, 11, e0146370. [Google Scholar] [CrossRef] [Green Version]
- Moreb, J.S. Aldehyde dehydrogenase as a marker for stem cells. Curr. Stem Cell Res. Ther. 2008, 3, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Salt, M.B.; Bandyopadhyay, S.; McCormick, F. Epithelial-to-mesenchymal transition rewires the molecular path to PI3K-dependent proliferation. Cancer Discov. 2014, 4, 186–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alix-Panabieres, C.; Mader, S.; Pantel, K. Epithelial-mesenchymal plasticity in circulating tumor cells. J. Mol. Med. 2017, 95, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Paterlini-Brechot, P.; Benali, N.L. Circulating tumor cells (CTC) detection: Clinical impact and future directions. Cancer Lett. 2007, 253, 180–204. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smit, D.J.; Cayrefourcq, L.; Haider, M.-T.; Hinz, N.; Pantel, K.; Alix-Panabières, C.; Jücker, M. High Sensitivity of Circulating Tumor Cells Derived from a Colorectal Cancer Patient for Dual Inhibition with AKT and mTOR Inhibitors. Cells 2020, 9, 2129. https://doi.org/10.3390/cells9092129
Smit DJ, Cayrefourcq L, Haider M-T, Hinz N, Pantel K, Alix-Panabières C, Jücker M. High Sensitivity of Circulating Tumor Cells Derived from a Colorectal Cancer Patient for Dual Inhibition with AKT and mTOR Inhibitors. Cells. 2020; 9(9):2129. https://doi.org/10.3390/cells9092129
Chicago/Turabian StyleSmit, Daniel J., Laure Cayrefourcq, Marie-Therese Haider, Nico Hinz, Klaus Pantel, Catherine Alix-Panabières, and Manfred Jücker. 2020. "High Sensitivity of Circulating Tumor Cells Derived from a Colorectal Cancer Patient for Dual Inhibition with AKT and mTOR Inhibitors" Cells 9, no. 9: 2129. https://doi.org/10.3390/cells9092129