Protein Kinase B Inactivation Is Associated with Magnolol-Enhanced Therapeutic Efficacy of Sorafenib in Hepatocellular Carcinoma In Vitro and In Vivo



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
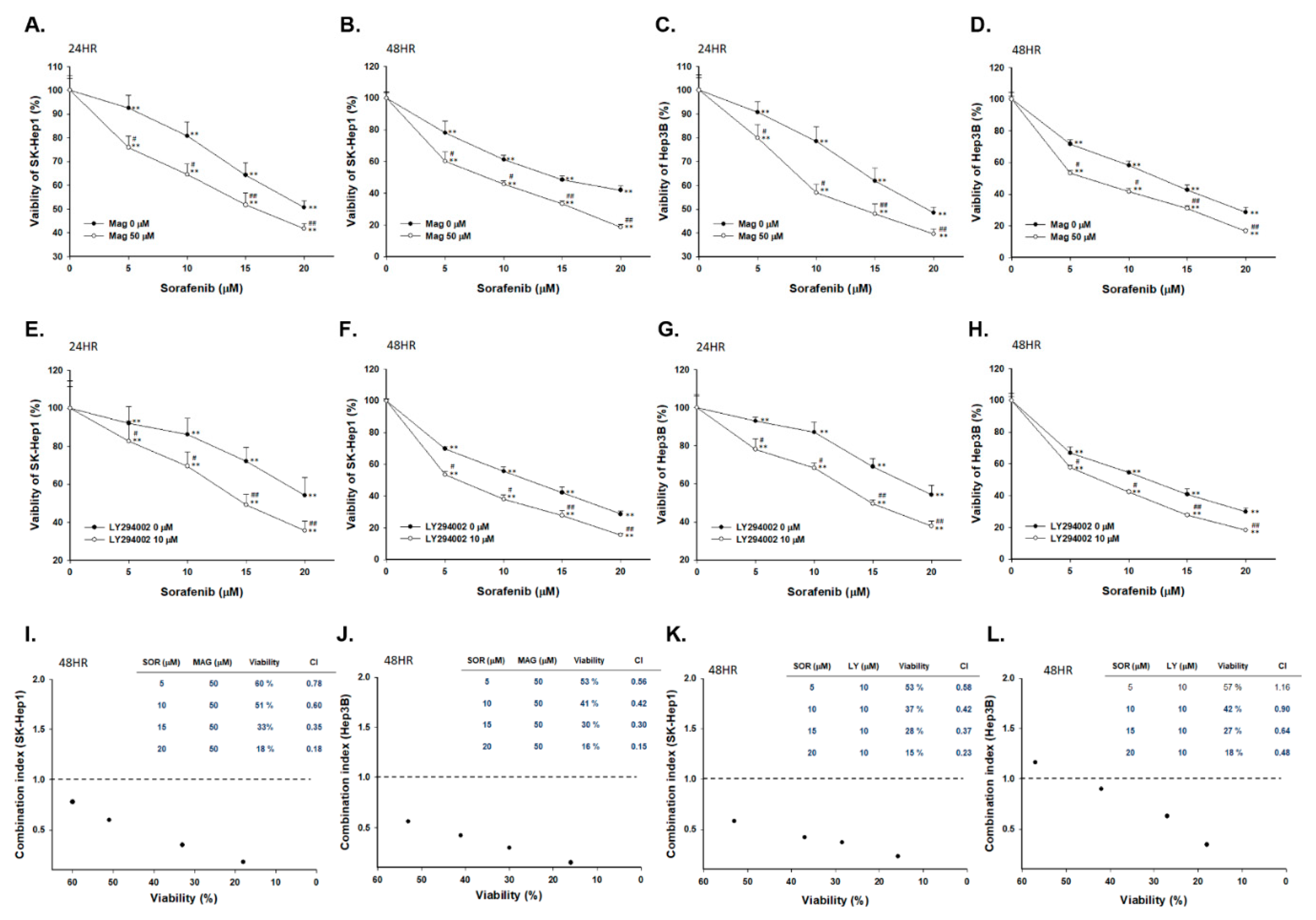
2.1. Both Magnolol and LY294002 Induced Cytotoxicity of Sorafenib on SK-Hep1 and Hep3B Cells
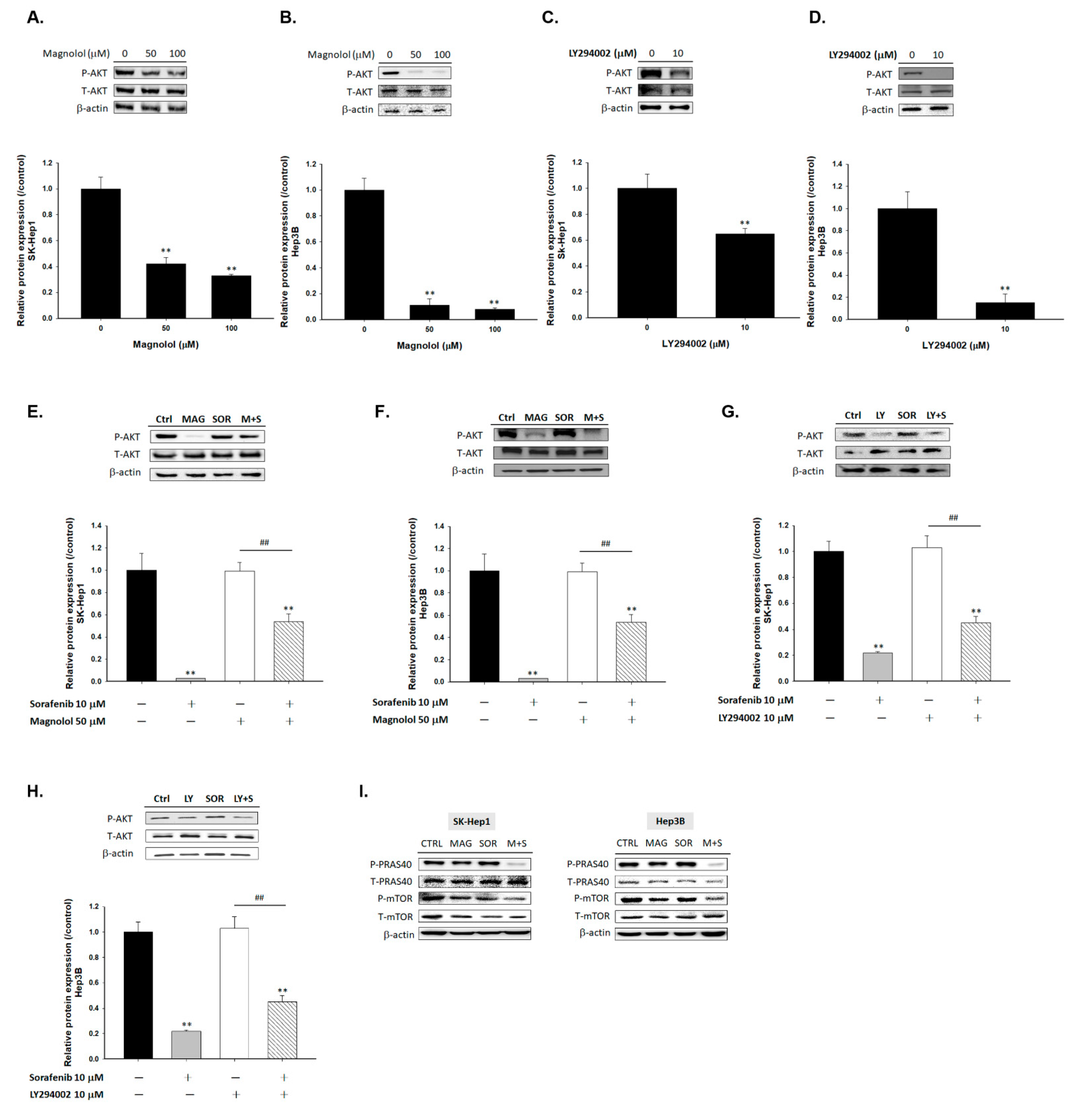
2.2. Magnolol Triggered the Dephosphorylation of AKT/mTOR/PRAS40 in Combined with Sorafenib
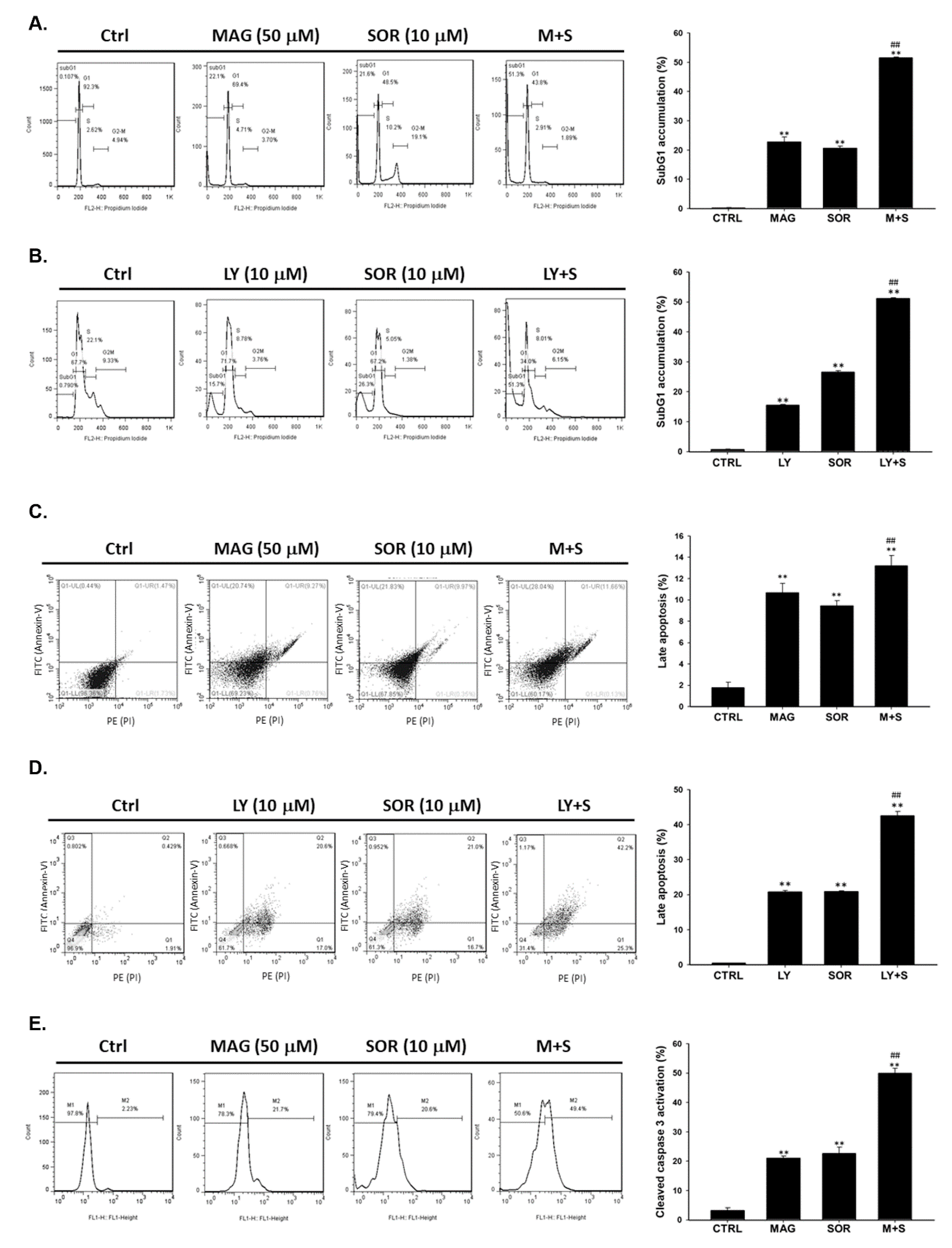
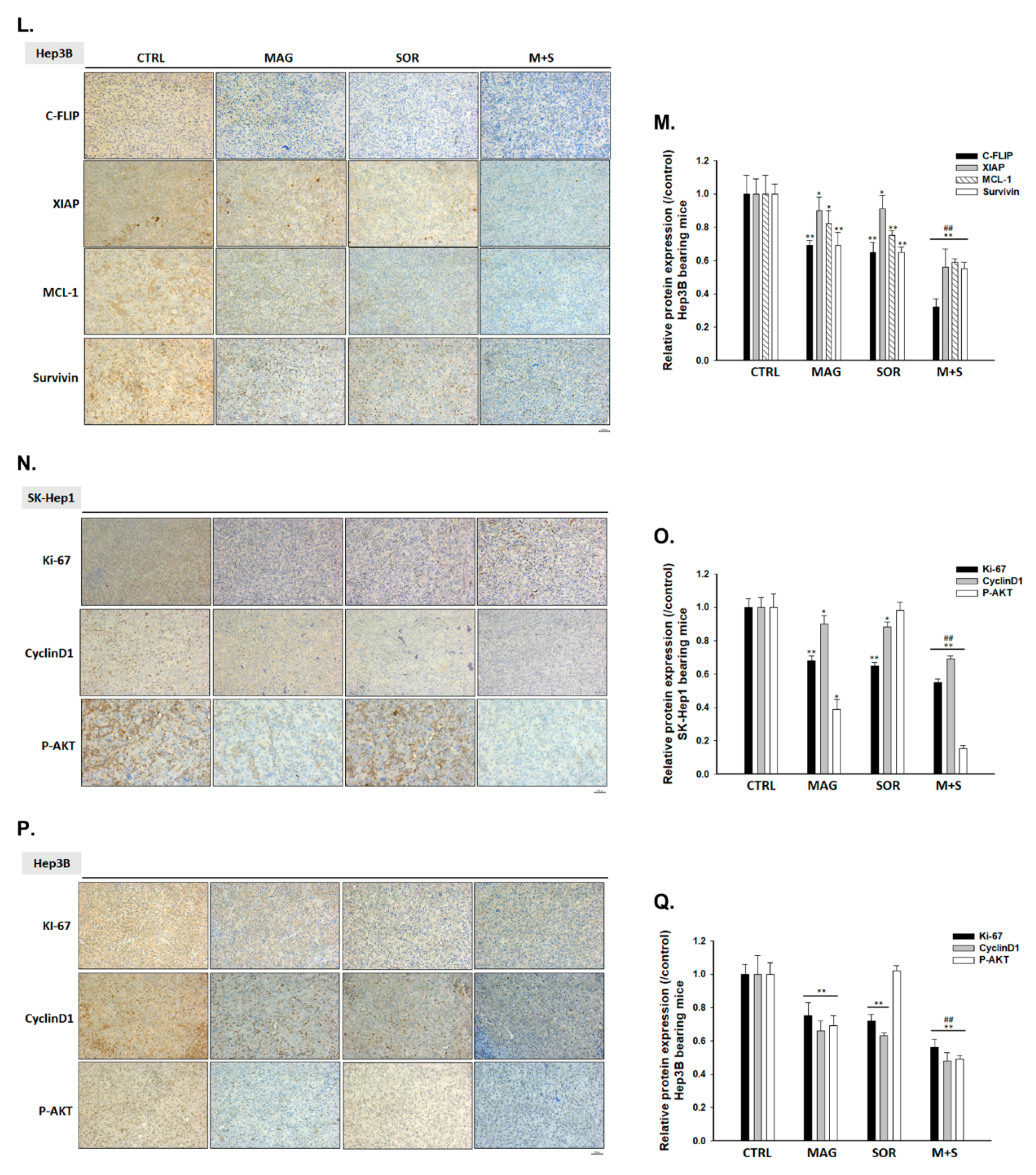
2.3. Both Magnolol and LY294002 Enhanced Sorafenib-Induced Apoptotic Cell Death and Reduced Anti-Apoptosis Proteins Expression of HCC Cells
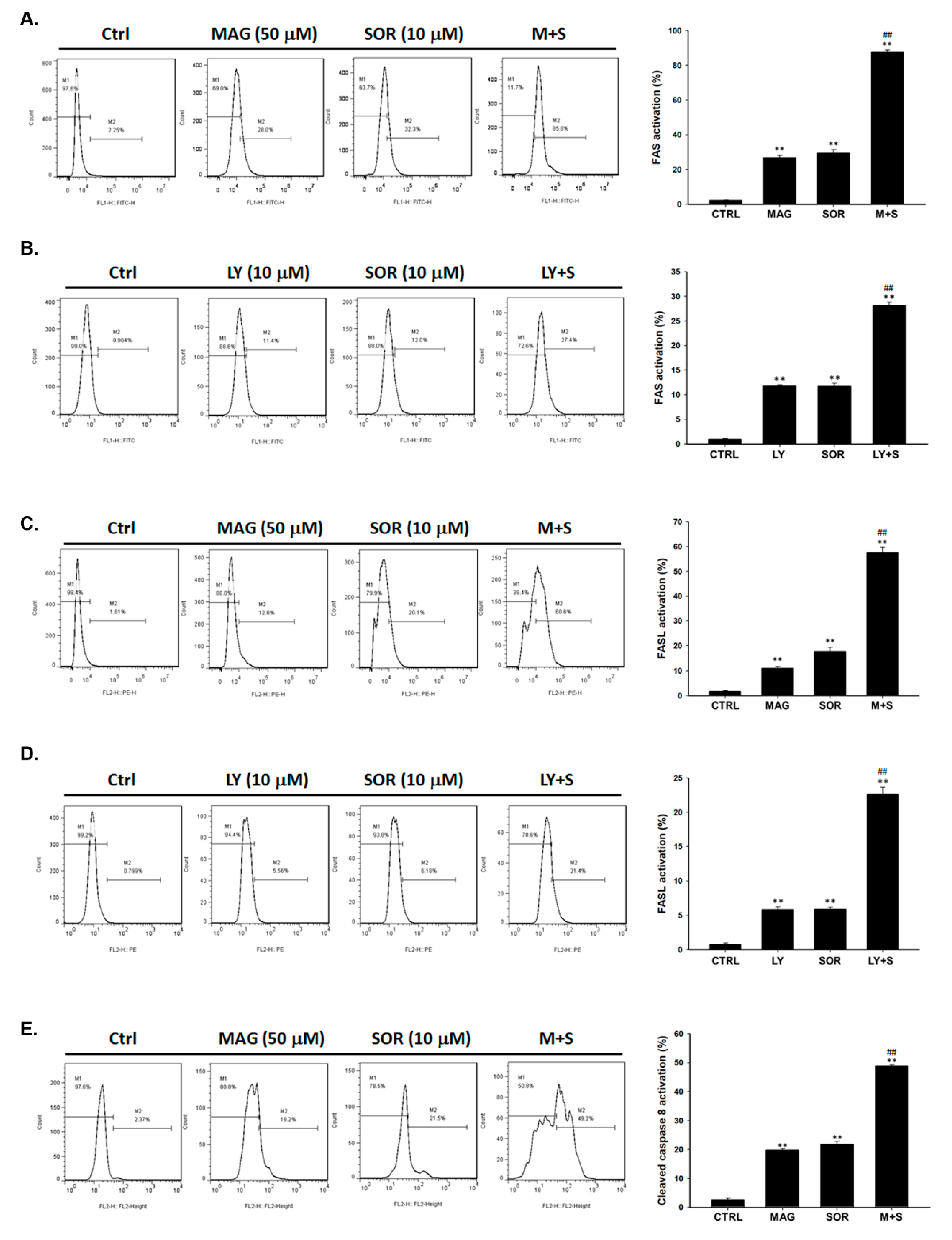
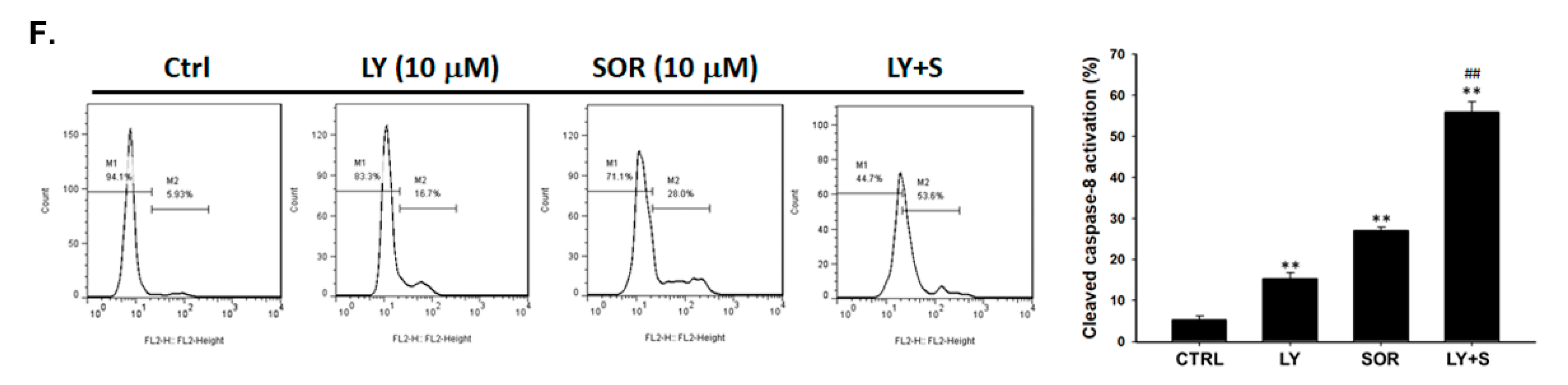
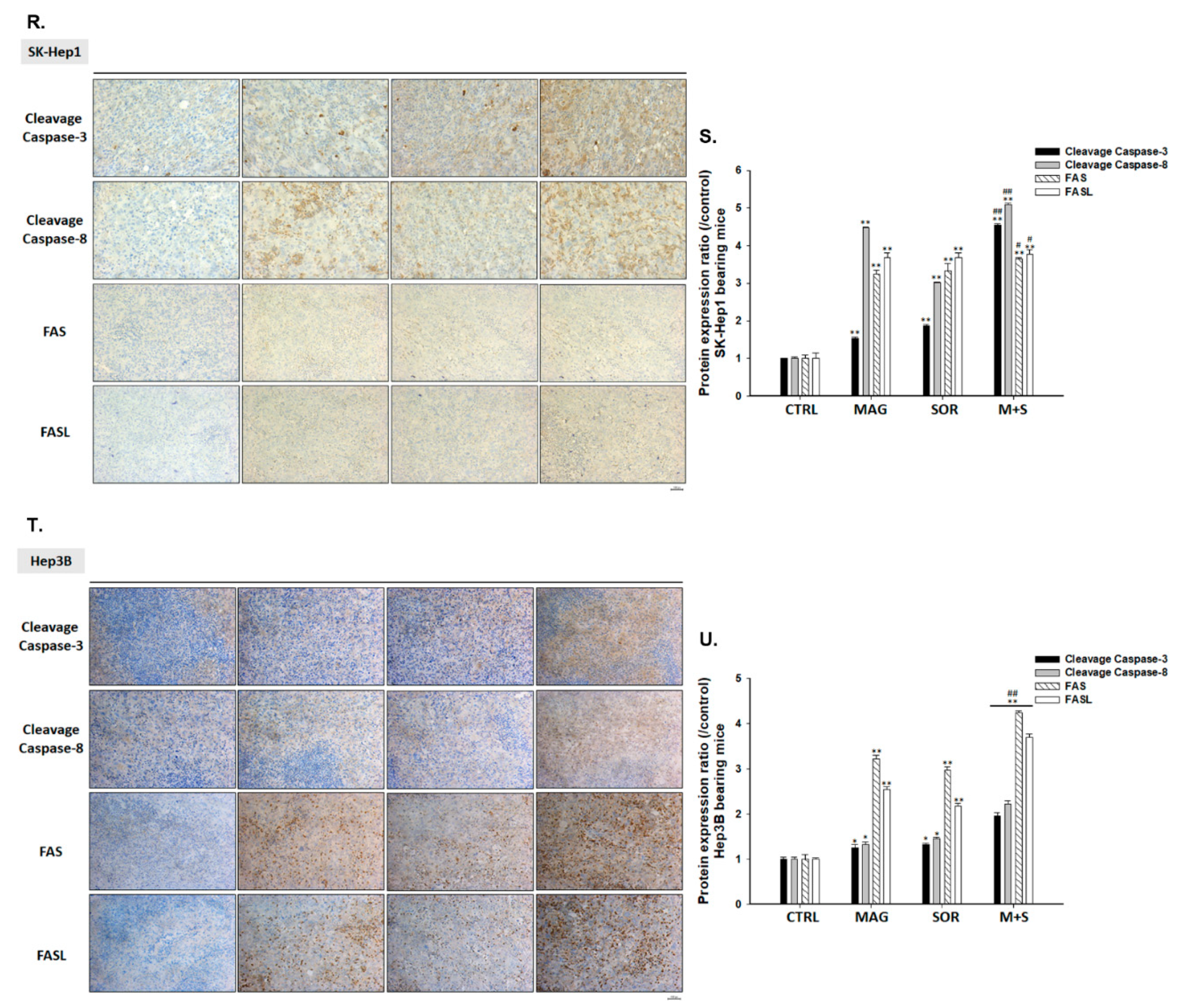
2.4. Both Magnolol and LY294002 Promoted Sorafenib-Induced Activation of Death Receptor Dependent Extrinsic Apoptosis in SK-Hep1 Cells
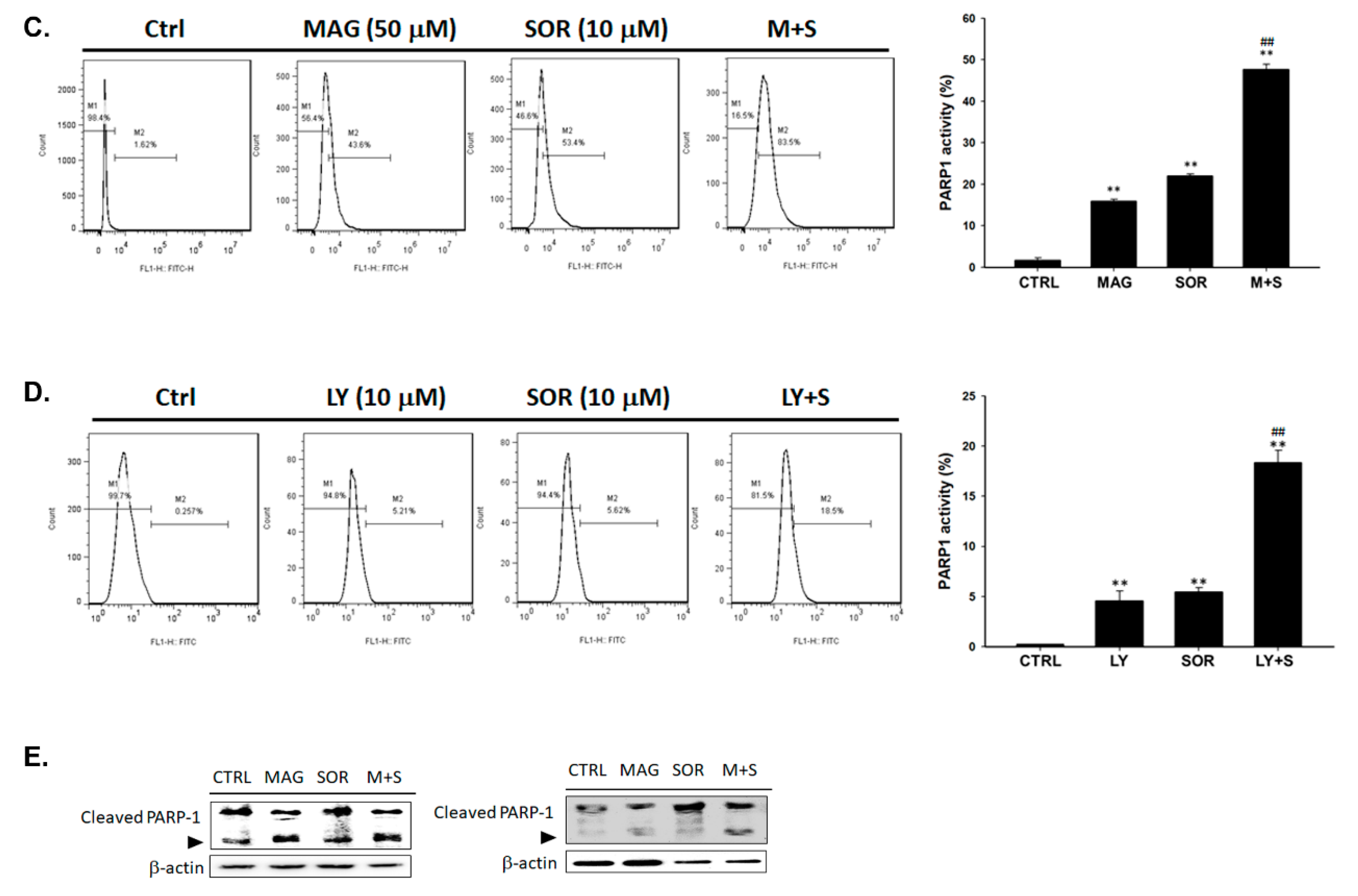
2.5. Both Magnolol and LY294002 Triggered Sorafenib-Initiated Intrinsic Apoptosis and Cleavage of PARP-1 in HCC Cells
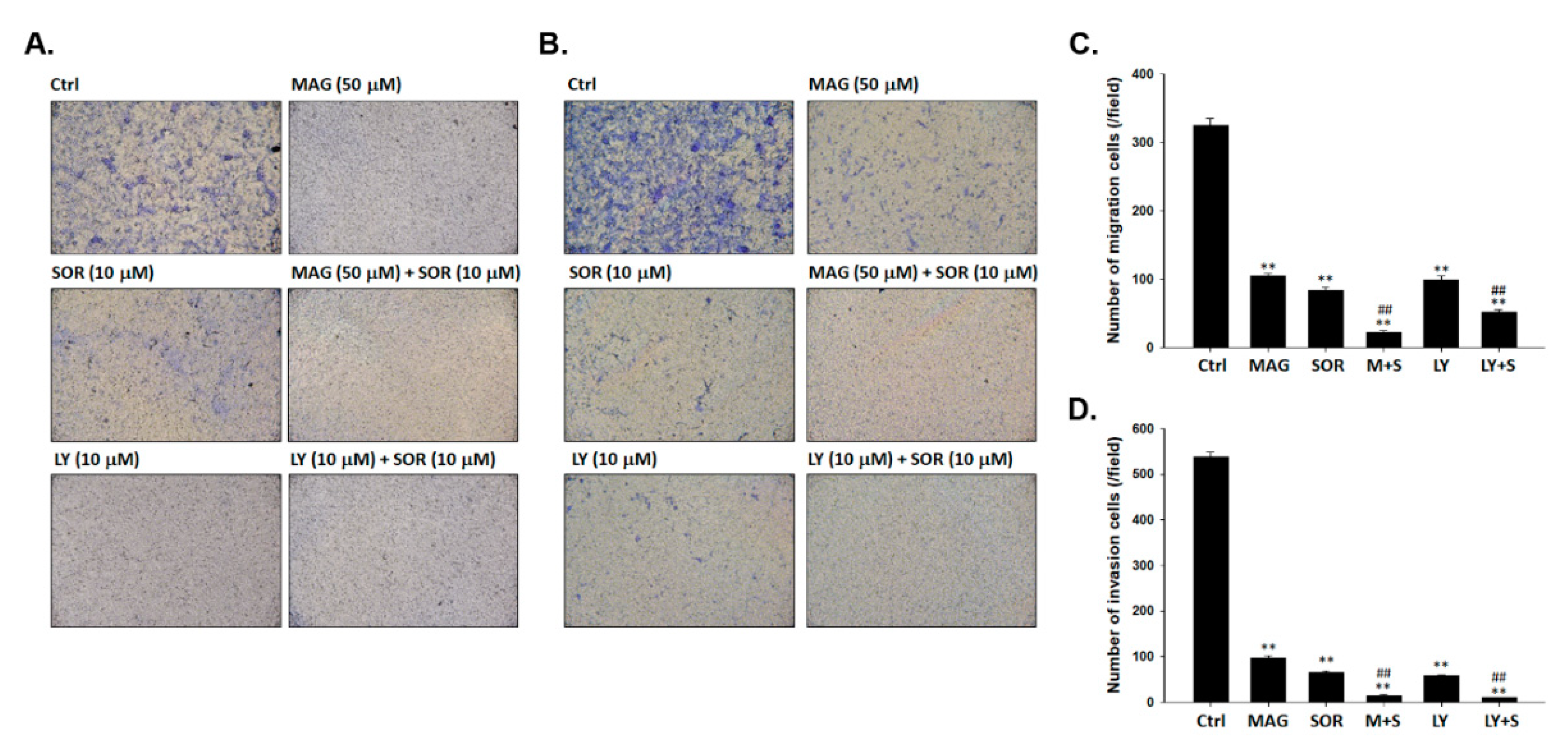
2.6. Both Magnolol and LY294002 Boosted Sorafenib-Reduced Migration and Invasion Ability of SK-Hep1 Cells
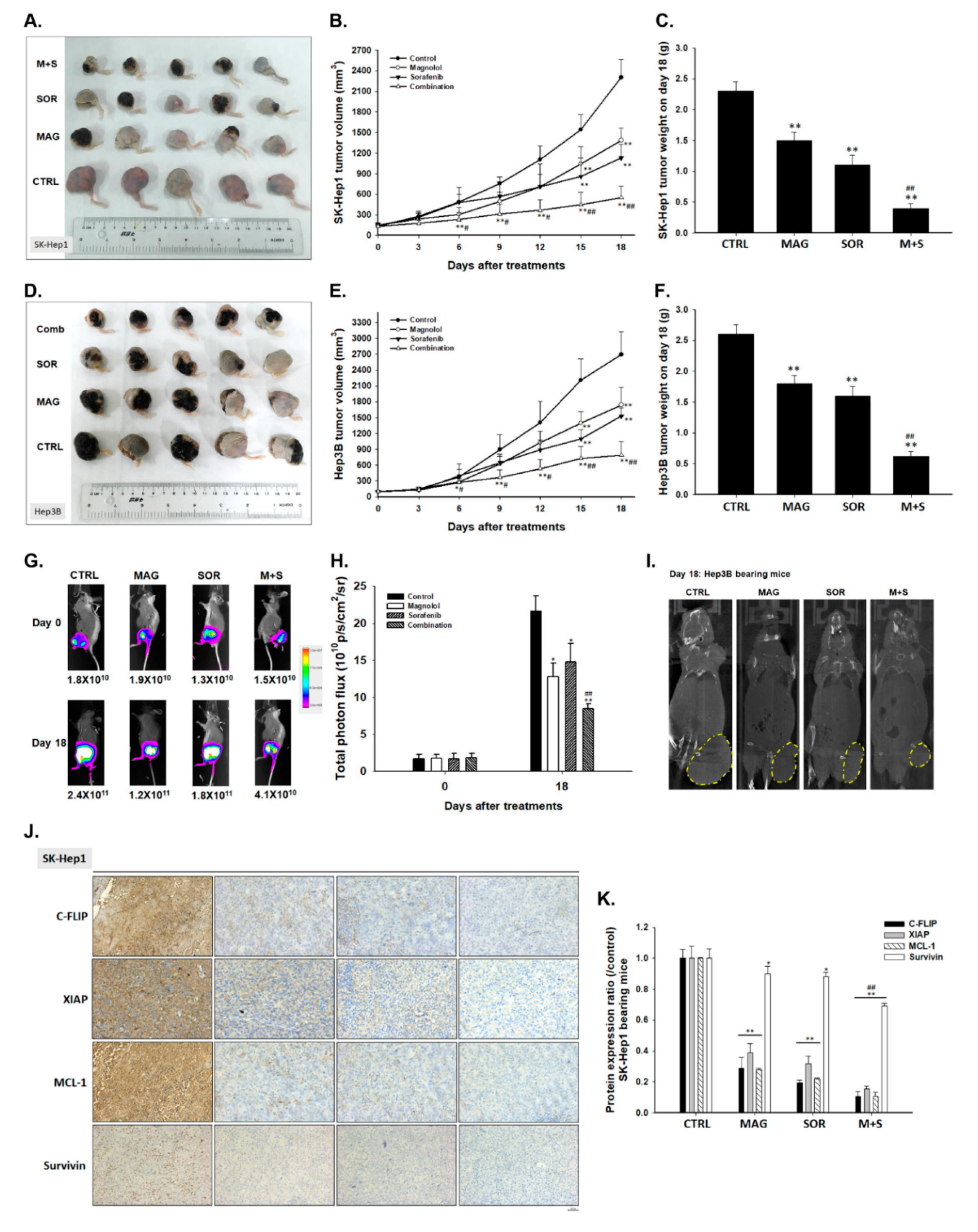
2.7. Magnolol and Sorafenib Co-Treatment Markedly Suppressed Tumor Growth, Anti-Apoptotic Proteins Expression and Induced Apoptosis Mechanism in Sk-Hep1/luc2 and Hep3B Bearing Mice
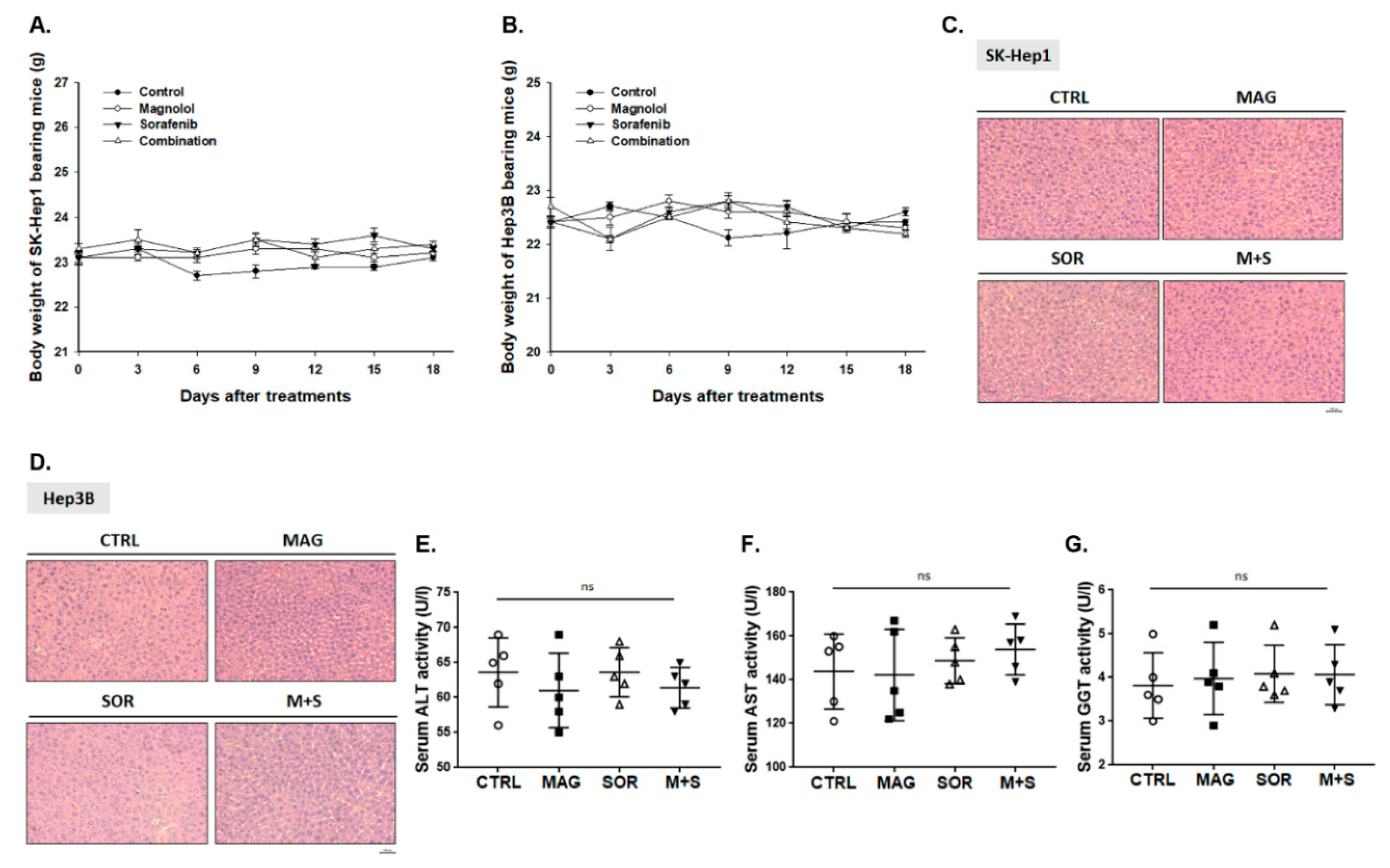
2.8. Magnolol and Sorafenib Co-Treatment may not Trigger Liver and General Toxicity of Sk-Hep1/luc2 and Hep3B Bearing Mice
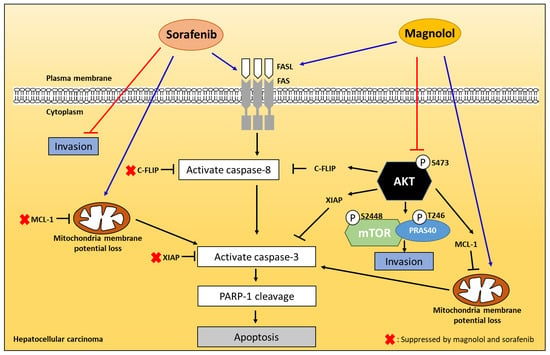
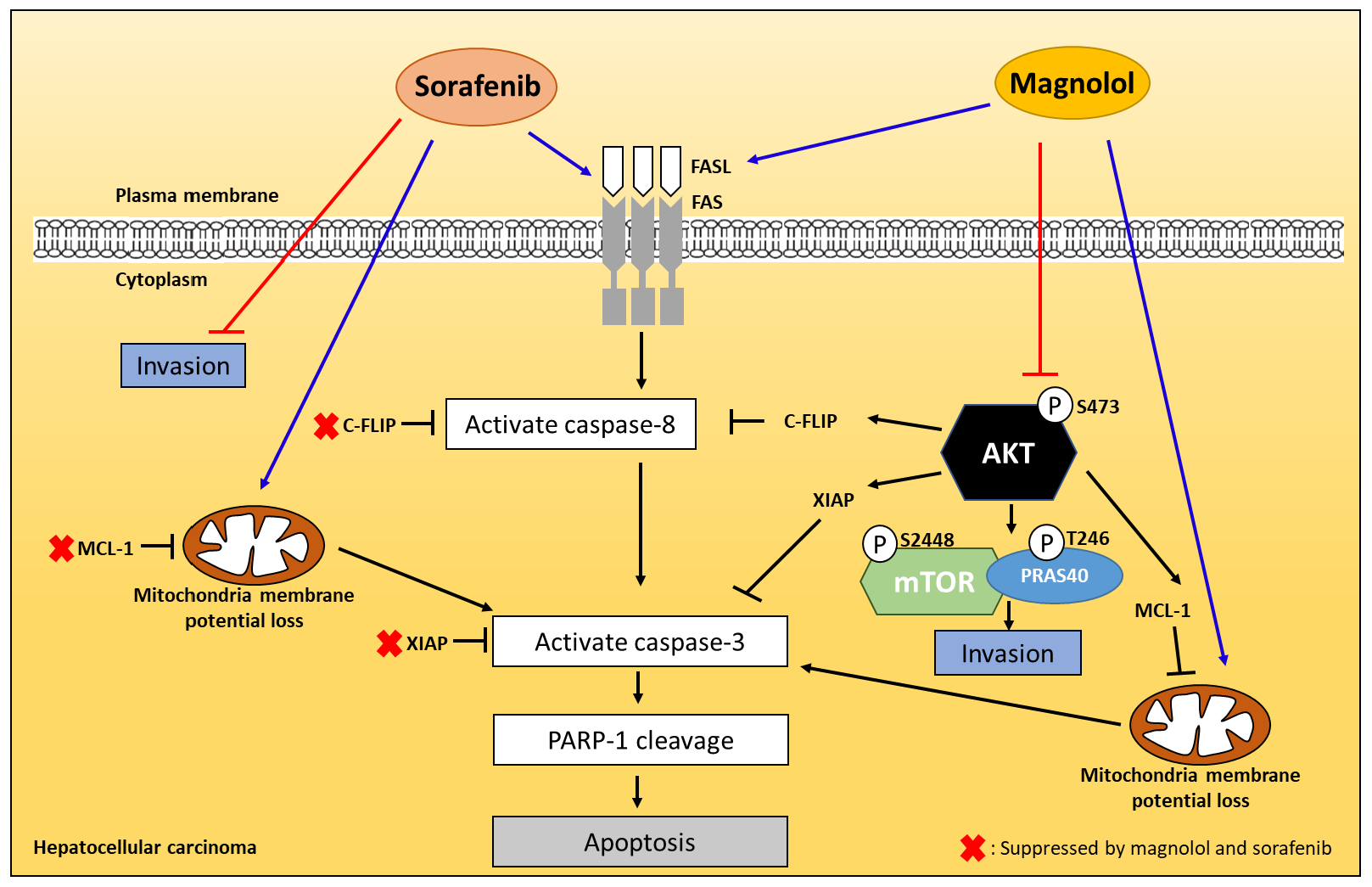
3. Discussion
4. Methods
4.1. Drugs and Chemical Reagents
4.2. Cell Culture
4.3. Transfection and Stable Clone Selection
4.4. Determinations for Viable Cells
4.5. Measurements of Apoptotic Cell Death
4.6. Measurements of Mitochondrial Membrane Potential (MMP, ΔΨm) in SK-Hep1 Cells
4.7. Measurements of Caspase-3 and -8 Activities
4.8. Measurements of FAS and FAS-L Activities
4.9. Measurements of Cleaved PARP-1 Activities
4.10. Measurements of Protein Expression by Western Blotting Analysis
4.11. Measurements of Migration Ability by Transwell Assay
4.12. Measurements of Invasion Ability by Transwell Assay
4.13. Measurements of Tumor Growth In Vivo by Caliper, Computer Tomography and Bioluminescent Imaging (BLI)
4.14. Measurements of Tumor Anti-Apoptosis and Apoptosis Protein Expression
4.15. Measurements of General Toxicity
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stickel, F.; Schuppan, D. Herbal medicine in the treatment of liver diseases. Dig. Liver Dis. 2007, 39, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I. Sho-saiko-to: Japanese herbal medicine for protection against hepatic fibrosis and carcinoma. J. Gastroenterol. Hepatol. 2000, 15, D84–D90. [Google Scholar] [CrossRef] [PubMed]
- Arase, Y.; Ikeda, K.; Murashima, N.; Chayama, K.; Tsubota, A.; Koida, I.; Suzuki, Y.; Saitoh, S.; Kobayashi, M.; Kumada, H. The long term efficacy of glycyrrhizin in chronic hepatitis C patients. Cancer 1997, 79, 1494–1500. [Google Scholar] [CrossRef]
- Ting, C.T.; Li, W.C.; Chen, C.Y.; Tsai, T.H. Preventive and therapeutic role of traditional Chinese herbal medicine in hepatocellular carcinoma. J. Chin. Med Assoc. 2015, 78, 139–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Martin, R.C., 2nd. Herbal medicine and hepatocellular carcinoma: Applications and challenges. Evid. Based Complemen. Altern. Med. 2011, 2011, 541209. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.H.; Chiang, I.T.; Ding, K.; Chung, J.G.; Lin, W.J.; Lin, S.S.; Hwang, J.J. Curcumin-induced apoptosis in human hepatocellular carcinoma j5 cells: Critical role of ca(+2)-dependent pathway. Evid Based Complem. Altern. Med. 2012, 2012, 512907. [Google Scholar] [CrossRef]
- Pan, Z.; Zhuang, J.; Ji, C.; Cai, Z.; Liao, W.; Huang, Z. Curcumin inhibits hepatocellular carcinoma growth by targeting VEGF expression. Oncol. Lett. 2018, 15, 4821–4826. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.H.; Zhang, Y.; Cheng, Y.N.; Gong, F.L.; Cao, Z.Q.; Yu, L.G.; Guo, X.L. Metformin incombination with curcumin inhibits the growth, metastasis, and angiogenesis of hepatocellular carcinoma in vitro and in vivo. Mol. Carcinog. 2018, 57, 44–56. [Google Scholar] [CrossRef]
- Yano, H.; Mizoguchi, A.; Fukuda, K.; Haramaki, M.; Ogasawara, S.; Momosaki, S.; Kojiro, M. The herbal medicine sho-saiko-to inhibits proliferation of cancer cell lines by inducing apoptosis and arrest at the G0/G1 phase. Cancer Res. 1994, 54, 448–454. [Google Scholar]
- Tsuchiya, M.; Kono, H.; Matsuda, M.; Fujii, H.; Rusyn, I. Protective effect of Juzen-taiho-to on hepatocarcinogenesis is mediated through the inhibition of Kupffer cell-induced oxidative stress. Int. J. Cancer 2008, 123, 2503–2511. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.J.; Jin, C.N.; Zheng, M.L.; Ouyang, X.N.; Zeng, J.X.; Dai, X.H. Clinical study on treatment of primary hepatocellular carcinoma by Shenqi mixture combined with microwave coagulation. Chin. J. Integr. Med. 2005, 11, 104–110. [Google Scholar] [PubMed]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.L.; Hsieh, C.L.; Chen, J.H.; Huang, C.S.; Chen, W.T.; Kuo, Y.C.; Chen, C.Y.; Hsu, F.T. Amentoflavone enhances sorafenib-induced apoptosis through extrinsic and intrinsic pathways in sorafenib-resistant hepatocellular carcinoma SK-Hep1 cells in vitro. Oncol. Lett. 2017, 14, 3229–3234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.J.; Hsu, F.T.; Pan, P.J.; Chen, C.W.; Kuo, Y.C. Amentoflavone Enhances the Therapeutic Efficacy of Sorafenib by Inhibiting Anti-Apoptotic Potential and Potentiating Apoptosis in Hepatocellular Carcinoma in vivo. Anticancer Res. 2018, 38, 2119–2125. [Google Scholar] [PubMed]
- Lam, W.; Jiang, Z.; Guan, F.; Huang, X.; Hu, R.; Wang, J.; Bussom, S.; Liu, S.H.; Zhao, H.; Yen, Y.; et al. PHY906(KD018), an adjuvant based on a 1800-year-old Chinese medicine, enhanced the anti-tumor activity of Sorafenib by changing the tumor microenvironment. Sci. Rep. 2015, 5, 9384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuan, L.Y.; Chen, W.L.; Chen, J.H.; Hsu, F.T.; Liu, T.T.; Chen, W.T.; Wang, K.L.; Chen, W.C.; Liu, Y.C.; Wang, W.S. Magnolol Induces Apoptosis and Inhibits ERK-modulated Metastatic Potential in Hepatocellular Carcinoma Cells. In Vivo 2018, 32, 1361–1368. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, Y.; Ichida, T.; Sugitani, S.; Genda, T.; Inayoshi, J.; Takamura, M.; Matsuda, Y.; Nomoto, M.; Aoyagi, Y. Overexpression of extracellular signal-regulated protein kinase and its correlation with proliferation in human hepatocellular carcinoma. Liver Int. 2004, 24, 432–436. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chen, P.M.; Ming, Y.Z.; Lin, P.Y.; Chu, C.P.; Chu, P.Y. Phosphorylated AKT expression in tumor-adjacent normal tissue is associated with poor prognosis in patients with hepatocellular carcinoma. Oncol. Lett. 2017, 14, 7461–7466. [Google Scholar] [CrossRef]
- Pinyol, R.; Montal, R.; Bassaganyas, L.; Sia, D.; Takayama, T.; Chau, G.Y.; Mazzaferro, V.; Roayaie, S.; Lee, H.C.; Kokudo, N.; et al. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut 2019, 68, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: Size matters. Oncogene 2003, 22, 8983–8998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, K.J.; Wohlschlaeger, J.; Lang, H.; Sotiropoulos, G.C.; Malago, M.; Steveling, K.; Reis, H.; Cicinnati, V.R.; Schmid, K.W.; Baba, H.A. Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection. J. Hepatol. 2008, 48, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.F.; Chen, H.L.; Tai, W.T.; Feng, W.C.; Hsu, C.H.; Chen, P.J.; Cheng, A.L. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J. Pharmacol. Exp. Ther. 2011, 337, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef] [Green Version]
- Jilkova, Z.M.; Kuyucu, A.Z.; Kurma, K.; Ahmad Pour, S.T.; Roth, G.S.; Abbadessa, G.; Yu, Y.; Schwartz, B.; Sturm, N.; Marche, P.N.; et al. Combination of AKT inhibitor ARQ 092 and sorafenib potentiates inhibition of tumor progression in cirrhotic rat model of hepatocellular carcinoma. Oncotarget 2018, 9, 11145–11158. [Google Scholar] [CrossRef]
- Llerena, S.; García-Díaz, N.; Curiel-Olm, S.; Agraz-Doblas, A.; García-Blanco, A.; Pisonero, H.; Varela, M.; Santibáñez, M.; Almaraz, C.; Cereceda, L.; et al. Applied diagnostics in liver cancer. Efficient combinations of sorafenib with targeted inhibitors blocking AKT/mTOR. Oncotarget 2018, 9, 30869–30882. [Google Scholar] [CrossRef] [Green Version]
- Plati, J.; Bucur, O.; Khosravi-Far, R. Dysregulation of apoptotic signaling in cancer: Molecular mechanisms and therapeutic opportunities. J. Cell. Biochem. 2008, 104, 1124–1149. [Google Scholar] [CrossRef] [Green Version]
- Panka, D.J.; Mano, T.; Suhara, T.; Walsh, K.; Mier, J.W. Phosphatidylinositol 3-kinase/Akt activity regulates c-FLIP expression in tumor cells. J. Biol. Chem. 2001, 276, 6893–6896. [Google Scholar] [CrossRef] [Green Version]
- Merlo, P.; Cecconi, F. XIAP: Inhibitor of two worlds. EMBO J. 2013, 32, 2187–2188. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.L.; Chuang, S.E.; Lin, M.T.; Yang, S.Y. The involvement of PI 3-K/Akt-dependent up-regulation of Mcl-1 in the prevention of apoptosis of Hep3B cells by interleukin-6. Oncogene 2001, 20, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, I.T.; Chen, W.T.; Tseng, C.W.; Chen, Y.C.; Kuo, Y.C.; Chen, B.J.; Weng, M.C.; Lin, H.J.; Wang, W.S. Hyperforin Inhibits Cell Growth by Inducing Intrinsic and Extrinsic Apoptotic Pathways in Hepatocellular Carcinoma Cells. Anticancer Res. 2017, 37, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Fan, Y.; Li, J.; Cheng, B.; Lin, W.; Li, X.; Du, J.; Ling, C. Inhibition of cFLIP overcomes acquired resistance to sorafenib via reducing ER stressrelated autophagy in hepatocellular carcinoma. Oncol. Rep. 2018, 40, 2206–2214. [Google Scholar] [PubMed]
- Hsu, C.; Lin, L.I.; Cheng, Y.C.; Feng, Z.R.; Shao, Y.Y.; Cheng, A.L.; Ou, D.L. Cyclin E1 Inhibition can Overcome Sorafenib Resistance in Hepatocellular Carcinoma Cells Through Mcl-1 Suppression. Clin. Cancer Res. 2016, 22, 2555–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.; Liu, T.; Mei, L.; Li, J.; Gong, K.; Yu, C.; Li, W. Synergistic antitumour activity of sorafenib in combination with tetrandrine is mediated by reactive oxygen species (ROS)/Akt signaling. Br. J. Cancer 2013, 109, 342–350. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, C.; Ning, Z.; Xu, L.; Zhu, X.; Meng, Z. Bufalin enhances anti-angiogenic effect of sorafenib via AKT/VEGF signaling. Int. J. Oncol. 2016, 48, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Matter, M.S.; Decaens, T.; Andersen, J.B.; Thorgeirsson, S.S. Targeting the mTOR pathway in hepatocellular carcinoma: Current state and future trends. J. Hepatol. 2014, 60, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Lv, D.; Guo, L.; Zhang, T.; Huang, L. PRAS40 signaling in tumor. Oncotarget 2017, 8, 69076–69085. [Google Scholar] [CrossRef] [Green Version]
- Roth, G.S.; Macek, J.Z.; Zeybek, K.A.; Kurma, K.; Ahmad Pour, S.T.; Abbadessa, G.; Yu, Y.; Busser, B.; Marche, P.N.; Leroy, V.; et al. Efficacy of AKT Inhibitor ARQ 092 Compared with Sorafenib in a Cirrhotic Rat Model with Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 2157–2165. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Zhang, R.; Zhang, Y.; Liu, X.; Wang, R.; Liu, J.; Liu, X.; Xie, Y.; Cao, W.; Xu, R.; et al. BEZ235 increases sorafenib inhibition of hepatocellular carcinoma cells by suppressing the PI3K/AKT/mTOR pathway. Am. J. Transl. Res. 2019, 11, 5573–5585. [Google Scholar]
- Tsai, J.J.; Pan, P.J.; Hsu, F.T. Regorafenib induces extrinsic and intrinsic apoptosis through inhibition of ERK/NF-kappaB activation in hepatocellular carcinoma cells. Oncol. Rep. 2017, 37, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.C.; Wang, M.H.; Tsai, J.J.; Kuo, Y.C.; Liu, Y.C.; Hsu, F.T.; Wang, H.E. Regorafenib inhibits tumor progression through suppression of ERK/NF-κB activation in hepatocellular carcinoma bearing mice. Biosci. Rep. 2018, 38, BSR20171264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, I.T.; Liu, Y.C.; Wang, W.H.; Hsu, F.T.; Chen, H.W.; Lin, W.J.; Chang, W.Y.; Hwang, J.J. Sorafenib inhibits TPA-induced MMP-9 and VEGF expression via suppression of ERK/NF-kappaB pathway in hepatocellular carcinoma cells. In Vivo 2012, 26, 671–681. [Google Scholar] [PubMed]
- Chou, T.C. The mass-action law based algorithm for cost-effective approach for cancer drug discovery and development. Am. J. Cancer Res. 2011, 1, 925–954. [Google Scholar]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Bień, K.; Sokołowska, J.; Bąska, P.; Nowak, Z.; Stankiewicz, W.; Krzyzowska, M. Fas/FasL pathway participates in regulation of antiviral and inflammatory response during mousepox infection of lungs. Mediat. Inflamm. 2015, 2015, 281613. [Google Scholar] [CrossRef]
- Wu, J.Y.; Lin, S.S.; Hsu, F.T.; Chung, J.G. Fluoxetine Inhibits DNA Repair and NF-kB-modulated Metastatic Potential in Non-Small Cell Lung Cancer. Anticancer Res. 2018, 38, 5201–5210. [Google Scholar] [CrossRef]
- Yen, T.H.; Hsieh, C.L.; Liu, T.T.; Huang, C.S.; Chen, Y.C.; Chuang, Y.C.; Lin, S.S.; Hsu, F.T. Amentoflavone Induces Apoptosis and Inhibits NF-kB-modulated Anti-Apoptotic Signaling in Glioblastoma Cells. In Vivo 2018, 32, 279–285. [Google Scholar]
- Lee, C.F.; Chiang, N.N.; Lu, Y.H.; Huang, Y.S.; Yang, J.S.; Tsai, S.H.; Lu, C.C.; Chen, F.A. Benzyl isothiocyanate (BITC) triggers mitochondria-mediated apoptotic machinery in human cisplatin-resistant oral cancer CAR cells. Biomedicine 2018, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Casao, A.; Mata-Campuzano, M.; Ordas, L.; Cebrian-Perez, J.A.; Muino-Blanco, T.; Martinez-Pastor, F. Cleaved PARP-1, an Apoptotic Marker, can be Detected in Ram Spermatozoa. Reprod. Domest Anim. 2015, 50, 688–691. [Google Scholar] [CrossRef]
- Hsu, F.T.; Sun, C.C.; Wu, C.H.; Lee, Y.J.; Chiang, C.H.; Wang, W.S. Regorafenib Induces Apoptosis and Inhibits Metastatic Potential of Human Bladder Carcinoma Cells. Anticancer Res. 2017, 37, 4919–4926. [Google Scholar]
- Weng, M.C.; Li, M.H.; Chung, J.G.; Liu, Y.C.; Wu, J.Y.; Hsu, F.T.; Wang, H.E. Apoptosis induction and AKT/NF-κB inactivation are associated with regorafenib-inhibited tumor progression in non-small cell lung cancer in vitro and in vivo. Biomed. Pharmacother. 2019, 116, 109032. [Google Scholar] [CrossRef]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.-H.; Chiang, I.-T.; Hsu, F.-T. Protein Kinase B Inactivation Is Associated with Magnolol-Enhanced Therapeutic Efficacy of Sorafenib in Hepatocellular Carcinoma In Vitro and In Vivo. Cancers 2020, 12, 87. https://doi.org/10.3390/cancers12010087
Chen J-H, Chiang I-T, Hsu F-T. Protein Kinase B Inactivation Is Associated with Magnolol-Enhanced Therapeutic Efficacy of Sorafenib in Hepatocellular Carcinoma In Vitro and In Vivo. Cancers. 2020; 12(1):87. https://doi.org/10.3390/cancers12010087
Chicago/Turabian StyleChen, Jiann-Hwa, I-Tsang Chiang, and Fei-Ting Hsu. 2020. "Protein Kinase B Inactivation Is Associated with Magnolol-Enhanced Therapeutic Efficacy of Sorafenib in Hepatocellular Carcinoma In Vitro and In Vivo" Cancers 12, no. 1: 87. https://doi.org/10.3390/cancers12010087