 Christa Pfeifhofer-Obermair
Christa Pfeifhofer-Obermair Piotr Tymoszuk1†
Piotr Tymoszuk1† Verena Petzer
Verena Petzer Günter Weiss
Günter Weiss Manfred Nairz
Manfred Nairz- 1Department of Internal Medicine II, Infectious Diseases, Immunology, Rheumatology, Pneumology, Medical University of Innsbruck, Innsbruck, Austria
- 2Christian Doppler Laboratory for Iron Metabolism and Anemia Research, Medical University of Innsbruck, Innsbruck, Austria
Iron metabolism and tumor biology are intimately linked. Iron facilitates the production of oxygen radicals, which may either result in iron-induced cell death, ferroptosis, or contribute to mutagenicity and malignant transformation. Once transformed, malignant cells require high amounts of iron for proliferation. In addition, iron has multiple regulatory effects on the immune system, thus affecting tumor surveillance by immune cells. For these reasons, inconsiderate iron supplementation in cancer patients has the potential of worsening disease course and outcome. On the other hand, chronic immune activation in the setting of malignancy alters systemic iron homeostasis and directs iron fluxes into myeloid cells. While this response aims at withdrawing iron from tumor cells, it may impair the effector functions of tumor-associated macrophages and will result in iron-restricted erythropoiesis and the development of anemia, subsequently. This review summarizes our current knowledge of the interconnections of iron homeostasis with cancer biology, discusses current clinical controversies in the treatment of anemia of cancer and focuses on the potential roles of iron in the solid tumor microenvironment, also speculating on yet unknown molecular mechanisms.
Introduction
There are numerous interconnections between iron homeostasis and cancer biology. However, our knowledge of many of these links is largely descriptive and based on data obtained from in vitro models using immortalized cell lines or from in vivo animal models employing xenogeneic tumor cell transplantation. Many of the potential roles of iron in cancer, generally, and in the tumor microenvironment (TME), specifically, have therefore not been formally addressed in human tumor entities and patient cohorts yet.
One aspect of the interconnection between iron and cancer is based on the fact that excess labile iron is toxic and catalyzes the formation of reactive oxygen species (ROS) via Fenton-/Haber-Weiss chemistry (1). As a consequence, iron may drive the malignant transformation of cells by directly damaging DNA, eventually leading to mutagenic transformation, or through protein and lipid modifications within malignant cells, resulting in more aggressive tumor behavior (2). When iron-dependent lipid peroxidation exceeds the cell's glutathione-mediated anti-oxidative defense capacity, inactivation of glutathione peroxidase (GPX)-4 culminates in a unique form of iron-induced cell death known as ferroptosis (3). On the other hand, proliferation of neoplastic cells regularly occurs at an enhanced rate, requiring increased iron supply because DNA replication is an iron-dependent process (4, 5). DNA polymerases and helicases contain iron-sulfur groups, rendering DNA replication one of the numerous synthetic and metabolic pathways that rely on iron as essential co-factor (6). Therefore, the availability of iron to tumor cells may affect either cell survival or growth rate and the course of disease, consequently. In addition, cellular iron availability impacts on mitochondrial respiration, ATP (for adenosine triphosphate) and mitochondrial radical formation, but also controls cellular metabolism and aerobic glycolysis via its regulatory effects on citric acid cycle enzymes (7, 8). In addition, neovascularization is affected by iron because of its impact on hypoxia inducible factor (HIF) activation and vascular endothelial growth factor (VEGF) production and on the function of endothelial cells (EC) (9, 10). Also, tumor-associated macrophages (TAMs) and EC diversely interact in the TME, and some of these interactions are modulated by iron availability, impacting on tumor progression and metastasis formation (11–16).
Cancer biology and immune surveillance are inseparably interconnected (17). A central nexus of this linkage is the competition for iron between neoplastic cells and the immune system which takes place both at the systemic level and in the microenvironment (18). Presumably, immune-driven adaptations of iron homeostasis in the presence of inflammatory stimuli have evolved during evolution as mechanisms to fight off bacteria and other pathogens, most of which require iron as essential growth factor (19–21). However, similar regulations occur when cancer cells are detected by the immune system because pathogen-associated molecular patterns (PAMP) and danger-associated molecular patterns (DAMP) elicit identical responses. The adaptation of systemic iron homeostasis to these inflammatory stimuli is orchestrated by soluble mediators including cytokines, such as interleukin (IL)-6 and acute-phase reactants, such as hepcidin and α1-antitrypsin (22–27). In addition, ROS and reactive nitrogen species (RNS), generated to damage cancer cells, also affect the way immune cells handle iron at the systemic level and in the TME (28, 29). Increased iron uptake into myeloid cells along with reduced iron export result in iron storage and sequestration in the mononuclear phagocyte system (MPS). Iron accumulation in the MPS may affect innate immunity in either direction. Typically, T helper type-1 (TH1)-driven pathways are inhibited by macrophage iron overload (IO), whereas ROS-induced pro-inflammatory signaling events are stimulated by iron (30). Which of these pathways predominate in anti-tumor immunity remains to be determined, though, because many results have been obtained in non-neoplastic inflammatory models (31–34). As a side effect or iron sequestration in the MPS, this trace element is less available for hemoglobin (Hb) synthesis by erythroid progenitors (EPs) in the bone marrow. Taken together, multiple mechanisms contribute to the alterations of iron homeostasis observed in cancer patients, which progress to clinically evident anemia of cancer (AOC).
AOC is extremely common and occurs in ~40–70% of cancer patients (35, 36). Importantly, the anemia affects organ function, and a higher degree of AOC is associated with reduced quality of life and survival of cancer patients (37, 38). Therefore, treatment of AOC is warranted but the benefit-to-risk ratio has to be carefully considered on an individual basis because therapy-associated effects on the underlying malignancy have been observed, too. For example, treatment with iron, erythropoiesis-stimulating agents (ESAs) or packed red blood cells (RBCs), administered to treat or correct the AOC, all carry the potential to promote tumor cell proliferation or impair anti-tumor immunity and have been associated with a shortened survival or recurrence of cancer (39–41). Theoretically, this is also true of novel therapeutic options for anemia of chronic disease (ACD) or AOC, especially the ones that target the hepcidin-ferroportin (FPN)-1 axis or HIF activation. On the other hand, many cancer cells rely on GPX4 to evade ferroptosis, rendering it an attractive target for tumor therapy. To avoid unintended effects, we need prospective clinical outcome data from rigorously conducted prospective randomized controlled trials (RCTs) as well as a more profound understanding of the multiple interconnections between iron metabolism and tumor occurrence and progression. This review provides an overview of our current knowledge of some of these interconnections.
Ferroportin-1 Forms the “Iron Gate” to the Circulation
Iron homeostasis is tightly maintained because too little iron impairs cell metabolism and function whereas too much iron is potentially toxic (42, 43). In mammals, the major regulated step in systemic iron homeostasis is its transfer to the circulation at sites of iron absorption or recycling because no controlled excretory mechanism exists (44). The principal protein to mediate iron transfer from cells to the circulation is FPN1, the only ferrous iron exporter known. FPN1 is highly expressed in iron-recycling macrophage populations, such as red-pulp macrophages (RPMs) in the spleen and Kupffer cells (KCs) in the liver, at the basolateral surface of absorptive intestinal epithelial cells (IECs) in the duodenum and proximal jejunum and in the synzytiotrophoblast (45, 46). FPN1 mediates efflux of ferrous iron and cooperates with either of three multi-copper oxidases able to convert ferrous iron to its ferric form, i.e., hephaestin, ceruloplasmin, and zyklopen (47, 48). In the extracellular space, ferric iron is bound by apo-transferrin (TF). TF is the key iron transport protein in plasma, accepts one or two iron atoms per molecule and distributes them as holo-TF throughout the body for cellular uptake by transferrin receptor (TFR)-1 and utilization, for example by the erythron (Figure 1).
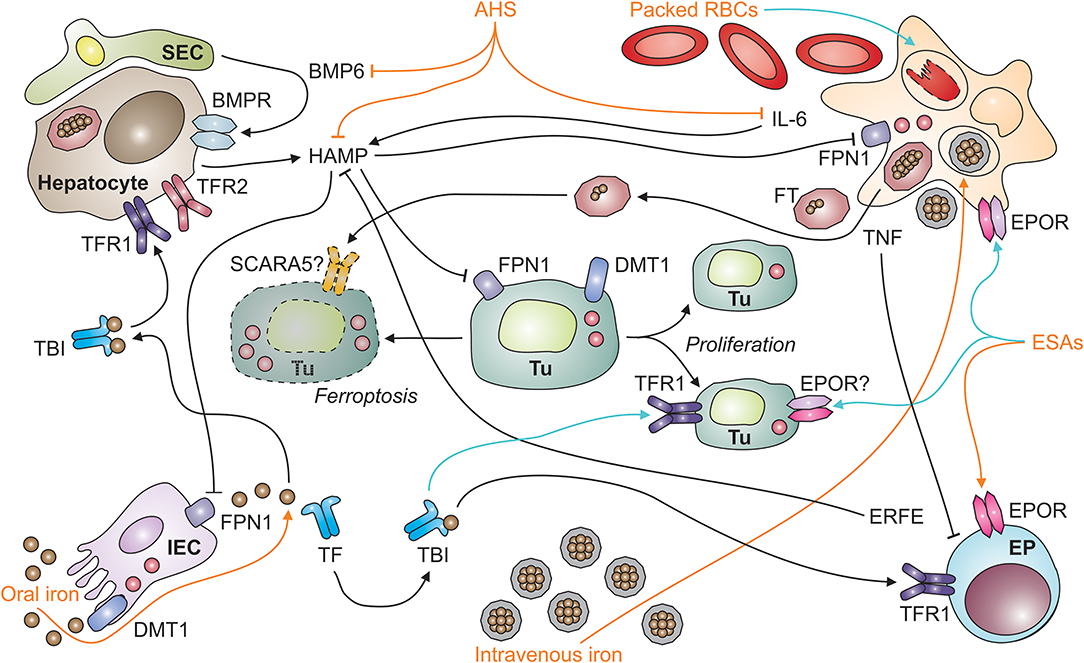
Figure 1. Systemic iron homeostasis in malignancy and potential effects of therapeutic intervention. After absorption by intestinal epithelial cells (IECs; depicted in the left lower corner) in the duodenum and upper jejunum, iron is loaded onto transferrin (TF) and distributed throughout the body as TBI (for transferrin-bound iron). TBI levels in the circulation are sensed by hepatocytes (depicted in the left upper corner) via transferrin receptors-1 and−2 (TFR1 and TFR2). An increase in iron levels in plasma results in the secretion of hepcidin antimicrobial peptide (HAMP). HAMP is also induced upon tissue iron loading, which results in the release of bone-morphogenic protein (BMP)-6 by liver sinusoidal endothelial cells (SECs; left upper corner). In addition, the cytokine interleukin-6 (IL-6) stimulates HAMP production by hepatocytes in inflammatory conditions, such as neoplasia. HAMP binds to ferroportin (FPN)-1 and blocks its iron export function, particularly in macrophages (MΦ; right upper corner), which results in iron sequestration and storage in ferritin (FT). FT can also be secreted by macrophages and taken up from plasma via specific receptors, such as SCARA5 (for scavenger receptor class A member-5). In the setting of malignancy, macrophages and other types of immune cells also secrete tumor necrosis factor (TNF). Among numerous functions, TNF inhibits the proliferation of erythroid progenitors (EP; right lower corner) in the bone marrow and their responsiveness to erythropoietin (EPO) produced in the kidneys (not depicted). Therapeutic options (depicted in orange) for cancer-related anemia include oral and intravenous iron preparations, anti-hepcidin strategies (AHS), erythropoiesis-stimulating agents (ESAs), and packed red blood cells (RBCs). All of these medications have potential side effects (depicted in turquois) on immune cells. For example, intravenous iron and packed RBCs can result in macrophage iron overload and impair their anti-tumor immune functions or facilitate the proliferation of tumor cells (Tu; depicted in the center, including dying [left-hand side] and proliferating [right-hand side] tumor cells). Cell types are indicated in bold; processes in italic. BMPR, BMP receptor; DMT1, divalent metal transporter-1; ERFE, erythroferrone; EPOR, EPO receptor.
Given its position as a gatekeeper to the circulation, FPN1 expression is controlled by several mechanisms ranging from transcriptional to post-translational regulation: (i) Iron-containing heme moieties stimulate FPN1 transcription via the stress-sensitive transcription factor Nrf2 (for nuclear factor (erythroid-derived 2)-like 2) (49), while increased iron levels in cells can also mitigate FPN1 transcription by a thus far not specified mechanism (50, 51). (ii) FPN1 mRNA translation is fine-tuned by two miRNAs, miR-485-3p and miR-20a (42, 52). The latter mechanism may be of special relevance for tumor cells as exemplified by the fact that in non-small cell lung cancer (NSCLC), increased miR-20a levels are found in biopsies, and miR-20a represses FPN1 expression and enhances iron availability for in vitro proliferation (53). (iii) An iron-responsive element (IRE) is present in the 5′ untranslated region of FPN1's mRNA. Iron deficiency (ID) in the cytoplasm is detected by iron-regulatory proteins (IRPs)-1 and−2, which enhances their activity to bind to the IRE and thereby inhibit FPN1 translation. By virtue of this mechanism, iron export is turned off when intracellular iron is already scarce. The IRP/IRE system appears to be relevant for tumor cell proliferation, too, in that activation of IRP2 along with an increase in iron content is documented for prostate cancer cells in vitro (54). Unexpectedly, overexpression of IRP1 impairs the growth of lung cancer cells transplanted into nude mice despite increased TFR1 levels (55). (iv) Most importantly, the transport activity of FPN1 is regulated post-translationally by its ligand hepcidin (Figure 1).
Hepcidin Levels Regulate Systemic Iron Homeostasis
Hepcidin is the body's iron-regulatory hormone and it acts in a negative feedback manner: Hepcidin binds to FPN1, closes its iron transport pore and induces its retraction from the cell surface with the subsequent induction of its degradation, thus blocking cellular iron export via FPN1 (45, 56). Therefore, high hepcidin levels, as observed in malignancy-driven inflammation, reduce iron transport to the circulation and cause iron retention in the MPS. Hepcidin is primarily formed and secreted by hepatocytes and its expression underlies multiple regulatory mechanisms which integrate the partly opposing measured variables that indicate both, systemic iron supply and demand. For example, an increase in body iron stores stimulates bone morphogenic protein (BMP)-6 production by sinusoidal EC in the liver (57) (Figure 1). BMP6 then acts on adjacent hepatocytes and induces hepcidin via the SMAD (homologs of Sma and Mad (mothers against decapentaplegic) proteins) pathway (58). In contrast, elevated serum iron levels act on hepatocytes themselves which carry a sensory protein complex of TFR1, TFR2, HFE and hemojuvelin (HJV) on their cell surface (59). Iron sensing by this complex also activates SMAD signaling, and hepcidin transcription, consequently (60, 61). Therefore, increasing serum iron levels are rapidly sensed by hepatocytes and balanced out by hepcidin secretion. Proinflammatory cytokines typically stimulate hepcidin expression, too. This effect is best described for IL-6. Specifically, IL-6, produced by macrophages and many other cell types, induces STAT3 (for signal transducer and activator of transcription-3) phosphorylation and thus hepcidin transcription, which contributes to the pathogenesis of ACD, as outlined below (62–64).
In contrast to inflammatory signals, absolute ID attributable to bleeding episodes or insufficient iron absorption, results in a decrease of hepcidin levels. This downregulation promotes iron absorption and re-distribution from the MPS to secure iron delivery for Hb synthesis and cellular functionality (65–67). In the setting of anemia, additional factors contribute to a reduction of hepcidin synthesis. A central repressor is erythroferrone (ERFE), a mainly erythropoietin (EPO)-inducible protein secreted by EPs (68–70). Hepcidin also needs to be repressed in hypoxia so that iron can be directed to erythropoietic progenitors for Hb synthesis (71). Central factors suppressing hepcidin production in anemia or hypoxic conditions include EPO and growth differentiation factor (GDF)-15 as well as platelet-derived growth factor (PDGF)-BB, the latter inhibiting hepcidin transcription via CREB-H (for cyclic AMP response element-binding protein H) (71–75). The transcription factor CREB-H is also essential to induce hepcidin in response to endoplasmatic reticulum (ER) stress, and this transcriptional induction is modified by CHOP (for CCAAT-enhancer-binding protein homologous protein), STAT3 and SMAD5 (76–78). Because ER stress is also exerted by chemotherapeutics, ER stress-sensitive transcription factors provide another potential link between iron metabolism and cancer (79, 80). In conclusion, FPN1 and hepcidin form a functional unit that constitutes the central regulator of systemic iron homeostasis both under physiological and pathological conditions including cancer.
Systemic Iron Homeostasis in Tumor Patients
The recognition of tumor cells and of neoantigens provides a strong stimulus for the immune system (81, 82). Both, innate immune mechanisms and T cell responses against malignant cells result in the production of a myriad of cytokines, such as interferon (IFN)-γ, tumor necrosis factor (TNF), IL-1ß, IL-6, IL-10, IL-13, and IL-22, all of which also impact on iron fluxes in the MPS contributing to the development of hypoferremia and hyperferritinemia as typical immune-driven alterations of iron metabolism (83–90). For example, IFN-γ, produced by TH1 cells, NKT (for natural killer T) cells and other cell types, increases DMT1 (for divalent metal transporter-1) expression in the MPS while decreasing FPN1 levels, thus contributing to iron sequestration (91). TNF, secreted by many cell types including TH1 cells, monocytes and macrophages, has similar effects, causing an imbalance between iron import and export in favor of the former, whereas IL-10 may mainly stimulate ferritin (FT) translation and thus iron storage (84, 91, 92). These alterations divert iron fluxes into the MPS and make it unavailable for erythropoiesis, a condition known as functional ID (93), a major hallmark of AOC.
Anemia of Cancer
Both IFN-γ and TNF have other pleiotropic effects and also inhibit EPO expression in the kidney and erythropoiesis in the bone marrow, aggravating the degree of AOC (94, 95). The latter effects are based on the fact that IFN-γ inhibits the proliferation of EPs and that TNF induces ROS, which damage them (96). Moreover, it is feasible to assume that these and other mediators also cause functional alterations in the bone marrow microenvironment, for instance by shifting hematopoiesis toward the myelopoietic direction or by impairing iron transfer from erythroid island macrophages to EPs. In addition, absolute ID may be present in cancer patients because of bleeding episodes, for example from malignant ulcers in the gastrointestinal or urogenital tract, extensive blood sampling, surgery or other interventions (97, 98). Furthermore, many other mechanisms may contribute to the development of AOC depending on the specific tumor entity. In multiple myeloma (MM), for example, IL-6 is produced by stromal cells and malignant cells themselves within the bone marrow and it appears to stimulate hepcidin production distantly in the liver (99). Another pathogenetic factor often contributing to the occurrence of AOC is the infiltration of the bone marrow by malignant cells which may affect erythropoiesis by nutrient and space deprivation, by damage to hematopoietic stem cells and by disruption of the integrity and function of erythroid islands and stem cell niches (40). These mechanisms may be most relevant in hematopoietic malignancies, such as acute leukemia in which a rapid expansion of malignant clones in the bone marrow takes place. Also, radiotherapy and chemotherapeutics have toxic effects on the bone marrow, thereby affecting all lines of hematopoiesis including RBC production and circulatory half life. Moreover, disease- or therapy-associated hemolysis or microangiopathy, malnutrition and comorbidities, such as chronic kidney disease (CKD) can contribute to AOC and affect its frequency and severity.
In summary, the AOC can be considered a subgroup of the ACD because of their similar pathophysiology. AOC is extremely common, occurs in various types of malignancy and is present in ~40–70% of cancer patients (35, 36). Its prevalence is affected by the type of cancer and by anti-tumor treatment and, if left untreated, it worsens as the disease progresses. Importantly, the AOC is associated with a shorter survival of cancer patients, as its occurrence and severity is associated with a more advanced disease (37, 38).
Treatments Options for Cancer-Related Anemia
Anemia may negatively impact on the quality of life and cardio-vascular performance of cancer patients, necessitating treatment of AOC. To date, we lack information on whether correction of the anemia exerts beneficial, neutral or detrimental effects toward the course of the underlying malignant disease. Of note, the pivotal therapy of AOC is the cure of the causative malignancy, which often results in resolution of anemia over time. As this may not always be feasible, other treatments have been used or are in development to correct Hb levels. Therapeutic options for the AOC include ESAs alone or combined with iron supplementation, blood transfusions and newly emerging treatment options with anti-hepcidin strategies (AHS) and PHD (for prolyl HIF dioxygenases) inhibitors (Figure 1).
In the setting of cancer and normal kidney function, ESAs help to overcome the resistance of the erythron to EPO. This growth factor is often suppressed by cytokines and, in the setting of AOC, circulates at concentrations too low for the degree of anemia. However, EPs are not the only cell types responsive to EPO. EPO and ESAs also may have extra-erythropoietic effects because the EPO receptor (EPOR) is expressed by many cell types other than EPs (100). EPOR has also been detected on many malignant cells, including NSCLC and breast cancer cells, although some controversy exists on the specificity of antibodies used to detect EPOR by means of immunohistochemistry (101–104). In breast cancer, there appears to be a negative correlation between EPOR expression and disease outcome, possibly because EPO counteracts p53-dependent apoptosis through induction of anti-apoptotic Bcl-XL (for B-cell lymphoma-extra large) (105, 106). In addition, EPO may stimulate the expression of TFR1, which increases TBI uptake into cells and can enhance the availability of iron for proliferation of malignant cells (107). EPOR is also present on immune cells including macrophages, B cells and T cells and may therefore exert diverse immune-modulatory effects (108, 109). In macrophages, EPOR ligation inhibits the activation of the NF-κB (for Nuclear factor kappa-light-chain-enhancer of activated B cells) subunit p65. As a consequence, pro-inflammatory effector pathways are impaired in EPO-stimulated macrophages (110). Similarly, EPO also reduces the autoreactivity of T cells. On the other hand, EPO beneficially affects the B and T cell responses against malignant cells in a mouse model of MM, suggesting differential effects of EPO and EPOR in various disease entities (111, 112).
PHD inhibitors form a novel class of drugs for the treatment of anemia. Their efficacy has been investigated in patients with CKD. However, their safety profile in cancer patients or in elderly individuals at increased risk of cancer remains to be assessed. A recent work dealing with the safety and efficacy of the PHD inhibitor roxadustat in a murine model of spontaneous mammary carcinoma demonstrates an improvement of cancer-related anemia together with increased VEGF production in the malignant tissue. In spite of that, no net boosting of tumor onset or progression is observed. The lack of reports concerning other tumor types and safety of iron and PHD application calls for further research and the long-term outcomes of currently ongoing phase III studies with different PHD inhibitors are awaited (66).
Transfusion-Related Immune Modulation
Blood transfusions may affect the outcome of cancer, too (113–115). This may be attributable to the fact that packed RBCs inevitably contain damaged cells and other immune-modulatory compounds. Stringent quality control, short-term storage and the depletion of leukocytes and platelets aim at reducing the risk of transfusion-related immune modulation (TRIM). Nevertheless, RBC damage may occur during and after transfusion, for instance due to mechanical stress or minor blood group incompatibilities, which releases free Hb, heme and iron. Free Hb impairs immunity and is a predictor of mortality in patients with sepsis and during extracorporal membrane oxygenation, partly because free heme elicits apoptosis (116–118). However, whether these observations are also relevant to other inflammatory conditions, such as neoplasia is unknown. Similarly, NTBI is implicated in TRIM in preterm infants, but this requires verification in adults and in the setting of malignancy (119). RBC transfusions may, however, directly deliver various forms of iron to tumor cells. Furthermore, RBCs themselves affect immune cell functions. For example, they inhibit T cell proliferation via direct cell-cell contact and affect the functions of dendritic cells (DCs) (120, 121). Importantly, monocytes and iron-recycling macrophages in the spleen and liver take up damaged RBCs via scavenger receptors, degrade Hb using lysosomal enzymes and heme oxygenase (HMOX)-1 and export iron via FPN1 (122, 123). FPN1+ iron-recycling monocytes may provide iron to tumor cells in their vicinity, especially in the liver, where these monocytes reside in large quantities, or after loading of ferric iron onto TF. In addition to RBCs and their compounds, anti-inflammatory cytokines and immune-modulatory extracellular vesicles may be present in blood products and affect anti-tumor immunity either systemically or in the TME. Therefore, both soluble and cellular factors may contribute to the immune-modulatory effects of RBC transfusions in cancer (Figure 1).
Iron Supplementation in Cancer Patients
Iron supplementation is a treatment option for anemic cancer patients with absolute ID. In the setting of immune activation secondary to cancer or other diseases, though, absolute ID is difficult to define as higher cut-offs for serum FT have to be employed. To date, no gold standard biomarker test is available which can differentiate between absolute and functional ID in the setting of inflammation (124–126). However, current clinical practice guidelines propose that a serum FT < 100 ng/ml indicates absolute ID and a TSAT < 20% together with a serum FT > 100 ng/ml characterizes functional ID in cancer patients (127). Oral iron formulations often cause gastrointestinal side effects and may be ineffective in AOC, especially when hepcidin levels are high, limiting oral bioavailability (128, 129). In addition, in vitro experiments suggest that at least two forms of oral iron, ferric citrate and ferric EDTA (for ethylenediaminetetraacetic acid), at supra-physiological concentrations can activate the EGF (for epidermal growth factor) receptor and MAP (for mitogen-activated protein) kinase signaling cascade in colon cancer cells, suggesting that these seemingly harmless compounds may have oncogenic potential when used at excess dosages (130). Intravenous iron preparations are an alternative to oral iron and constitute iron-carbohydrate nanoparticles which are engulfed and handled by myeloid cells (131) (Figure 1). Efficacy and long-term safety of intravenous iron formulations for the treatment of AOC are still under debate and should be evaluated in prospective RCTs. Most studies available suggest that intravenous iron supplementation in solid tumor patients is well-tolerated, replenishes iron stores and increases Hb levels, thus reducing the need for ESA or blood transfusion (132). However, data on effects of iron supplementation on tumor biology and long-term outcome of cancer patients are still scarce and a matter of serious concern. Until such data become available, a restrictive iron supplementation regimen is warranted for symptomatic patients with AOC but application thresholds for Hb and FT remain to be defined based on clinical data (133, 134).
Anti-Hepcidin Strategies (AHS)
AHS target hepcidin production, its circulatory concentrations or its effect on FPN1, thereby aiming to restore iron delivery from the MPS to the circulation. While these effects mean to provide iron for erythropoiesis, they also may supply iron to tumor cells or affect anti-tumor immunity. As for hepcidin production, BMPR/SMAD and IL-6R/STAT3 signaling constitute two attractive pathways to pharmacologically suppress hepcidin production.
In vitro, BMP6 has both stimulatory and inhibitory effects on tumor cells. BMP6 facilitates bone metastasis in an in vivo model (135). By contrast, treatment with BMP6 inhibits proliferation of breast cancer cells in vitro (136). Moreover, BMP6, secreted by prostate cancer cells, stimulates macrophages to produce IL-1α which in turn promotes tube formation by EC, a prerequisite for tumor neovascularization (137). Furthermore, BMP6 stimulates RNS production by macrophages but this has not been reported for TAMs yet. Given BMP6's opposing effects, neutralizing antibodies administered to treat AOC may affect the underlying malignancy in either direction. Similar to BMP6 neutralization, BMPR phosphorylation can be inhibited to decrease hepcidin production. The small molecule LDN-193189 is efficient in doing so and ameliorates ACD in a non-neoplastic rat model (27). ERFE analogs or other hypoxia- or erythropoiesis-driven inhibitors of hepcidin production, such as PDGF-BB, GDF-15 or PHD inhibitors may have similar effects and may become promising treatment options for ACD and AOC. Notably, endogenous heparins inhibit BMP/SMAD signaling, too. Based on this observation, heparin derivatives that lack anti-thrombotic but retain hepcidin-repressing effects have been developed. These compounds are effective in the treatment of ACD (138). Again, no prospective clinical data are available on the course of the underlying malignancy when hepcidin is suppressed to treat AOC.
Blockade of IL-6 signaling with tocilizumab, a humanized antibody directed against its cytokine receptor, is efficient in treating ACD in an animal model of arthritis and in Castelman's disease patients in an RCT (139, 140). In a retrospective analysis of patients with rheumatoid arthritis, long-term treatment with tocilizumab did not affect the expected rate of malignancies (141). Furthermore, the concerted blockade of IL-6 receptor and IL-8 receptor inhibits breast cancer metastasis in mouse xenograft models, suggesting that inhibition of hepcidin induction via the IL-6/STAT3 pathway may be a rather safe approach to treat AOC, at least in some tumor entities (142).
Direct neutralization of hepcidin can be achieved with antibodies, anticalins, or Spiegelmers, all of which are being evaluated for the treatment of ACD. Thus far, the hepcidin-neutralizing antibody LY2787106 is efficient in mobilizing iron in the setting of AOC (143). However, without further follow-up, the long-term safety of either of these treatment strategies for cancer patients is difficult to predict. Therefore, we need large RCTs with relevant clinical endpoints to assess the efficacy and safety of recently developed treatment options for AOC and to define optimal therapeutic start- and endpoints in a prospective fashion.
A Quadriga of Iron-Binding Proteins Is Present in Plasma
There are at least four iron-binding proteins in plasma which carry the potential to either deliver iron to tumor cells or deprive it from them: TF, lactoferrin (LF), FT, and lipocalin (LCN)-2 can bind iron in different forms and their content of and affinity for iron as well as the distribution of corresponding cell surface receptors between normal nucleated cells and tumor cells may determine whether the latter can benefit from these proteins as iron sources (Figure 2). In addition, non-protein bound labile iron, not detectable in healthy individuals, can occur in the extracellular space in malignancies, especially in patients with acute myeloid leukemia or myelodysplastic syndrome (MDS) during conditioning for allogenic hemopoietic stem cell transplantation (144, 145).
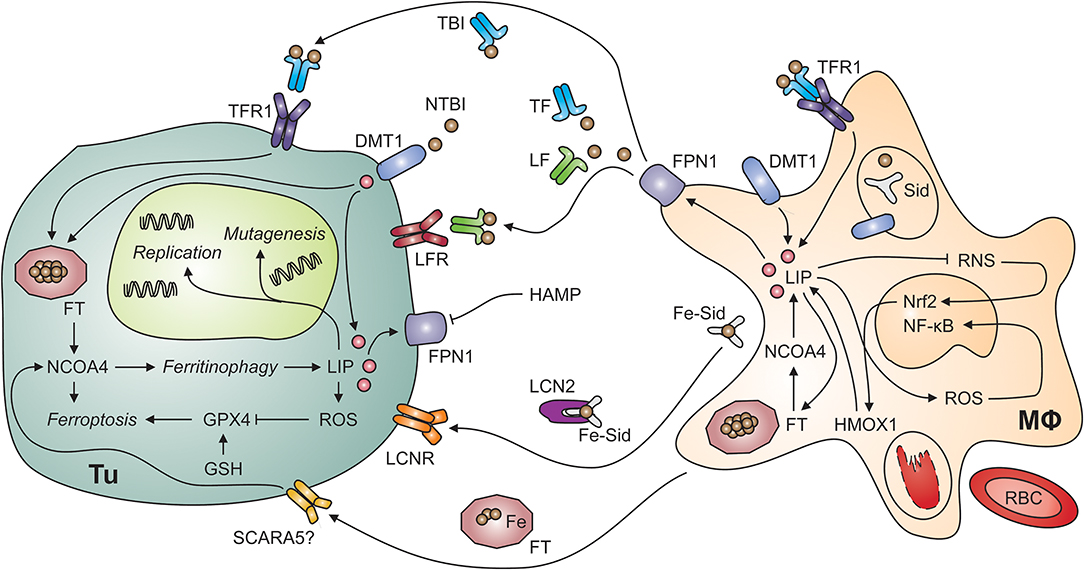
Figure 2. Putative interactions of tumor cells and tumor-associated macrophages. Myeloid cells, including tumor-associated macrophages (MΦ; right-hand side) possess multiple mechanisms to acquire iron including the uptake of non-transferrin-bound iron (NTBI) via divalent metal transporter (DMT)-1, endocytosis of transferrin-bound iron (TBI) via transferrin receptor (TFR)-1 and phagocytosis of aged or damaged red blood cells (RBCs). RBCs contain large amounts of iron incorporated in hemoglobin. The degradation of hemoglobin by proteases and heme oxygenase (HMOX)-1 releases free iron into the labile iron pool (LIP). An increase in the LIP then facilitates the production of reactive oxygen species (ROS), which activate nuclear factor-kappa B. In contrast, labile iron impairs the generation of reactive nitrogen species (RNS) via nitric oxide synthase-2. In the extracellular space, iron is present in at least four molecular forms, i.e., bound to transferrin (TF), bound to lactoferrin (LF), bound to siderophores (Sid) and lipocalin (Lcn)-2, and incorporated in ferritin (FT). All these forms may supply iron to tumor cells (Tu; left-hand side) because of their expression of specific receptors, possibly including the scavenger receptor A member (SCARA)-5, which binds FT. In addition, non-transferrin-bound iron (NTBI) may be present in the tumor microenvironment and acquired via DMT1. Once in the tumor cell's cytosol, labile iron can stimulate cell growth and DNA replication, induce mutagenesis or result in ferroptosis. Ferroptosis is a specific form of programmed cell death which is initiated by an increase in the LIP and in ROS production. ROS inactivate glutathione peroxidase (GPX)-4 after depletion of glutathione (GSH). Similarly, the degradation of FT by ferritinophagy, an autophagic process requiring the FT chaperone NCOA4 (for nuclear receptor coactivator-4), frees iron and can induce ferroptosis. On the other hand, iron can also be exported from the cytosol via ferroportin (FPN)-1 on tumor cells. However, this process is reduced when levels of hepcidin antimicrobial peptide (HAMP) in the circulation or in the tumor microenvironment are high. Cell types are indicated in bold; processes in italic. DNA, deoxyribonucleic acid; Nrf2, nuclear factor (erythroid-derived 2)-like-2.
Since TF is the key iron transport protein circulating in plasma, TFR1-mediated iron uptake is a simple and efficient iron acquisition strategy for malignant cells (Figure 2). Not surprisingly, overexpression of TFR1 has been found in many tumor entities including breast cancer, esophageal cancer, melanoma and glioblastoma cells (146–149). In addition, compounds targeting TFR1 are under consideration for molecular imaging and therapy of various tumor entities including B and T cell lymphomas (150, 151).
LF is a TF homolog with two binding site for ferric iron whose affinity exceed TF's by far (152). It is the predominant iron-binding protein in breast milk, saliva and tear fluid but also present in plasma. LF delivers iron to cells expressing specific receptors, one of which is GAPDH (for glyceraldehyde-3-phosphate dehydrogenase), a multifunctional protein present in the cell membrane of macrophages and other cell types (153, 154). GAPDH can also be secreted for subsequent autocrine uptake of LF or TF into cells (154, 155). Although malignant cells express GAPDH, uptake of LF-bound iron may be of minor importance for their iron supply because LF's predominant effect on breast, colorectal and other cancer cells in vitro is to impair signaling and cell cycle progression, to inhibit proliferation and to induce cell death (156–158). In line with these anti-neoplastic effects, mice lacking LF are more susceptible to dysplasia in the colon elicited by chronic inflammation (159). In addition, LF affects immune cell function and the composition of the intestinal microbiome but the impact of these observations on cancer biology are unknown (160, 161). However, it is interesting to note that a conjugate of LF and doxorubicin has beneficial effects in a prostate cancer model in vivo (162). Taken together, LF appears to have diverse anti-neoplastic effects and its therapeutic potential warrants further studies.
Macrophages secrete FT, which subsequently circulates in plasma in an iron-poor form (163). Serum FT can deliver iron to cells which carry corresponding receptors on their surface (Figure 2). TFR1 mediates binding and uptake of ferritin heavy chain (FTH) by some human cell types including erythroid cells, and TIM2 (for T cell immunoglobulin and mucin domain protein-2) fulfills a similar function in mouse cells (164, 165). Analogously, SCARA5 (for scavenger receptor class A member 5) is the receptor for FTL (166). Tumor cells may express FT receptors allowing them to utilize serum FT as iron source. Nevertheless, the expression of SCARA5 on tumor cells inhibits cell proliferation and migration because it impairs several signaling events including VEGF expression and ERK (for extracellular signal-regulated kinases) activation (167). High serum FT levels in cancer patients are associated with more aggressive clinical course and poor treatment response but it is unknown whether high FT actively contributes to disease progression or is a biomarker reflecting systemic immune activation (168), which is well-known to be associated with a poor prognosis of cancer (169).
LCN2 may contribute to both iron uptake and iron release by several cell types including cancer cells and TAMs (Figure 2). Among many functions, LCN2 acts as soluble scavenger for siderophores (170, 171). Siderophores are small iron-chelating molecules originally identified in bacteria but also produced by higher organisms including fungi and mammals (172–174). They bind ferric iron with extraordinarily high affinity and catecholate-type siderophores themselves are bound by LCN2. Thereafter, complexes of iron, a catecholate-siderophore and LCN2 are recognized by cell surface receptors including LCNR and megalin and thus mediate iron import into cells, which may contribute to the survival of thyroid carcinoma and other malignant cells (175, 176). However, LCNR has the intriguing property of providing bi-directional iron transport capacity because it can also accept intracellular LCN2 after binding of an iron-laden siderophore in the cytoplasm (177). Due to the fact that the oncogenic tyrosin kinase BCR-ABL (for breakpoint cluster region-Abelson) represses LCNR expression, iron-deprivation via the LCN2-LCNR pathway is inactive in BCR-ABL+ CML (chronic myeloid leukemia) cells, rendering them resistant to apoptosis (178). Expression of LCN2 is also associated with epithelial-mesenchymal transition (EMT), invasion, progression and metastatic spread of many types of cancer cells including those of breast, cholangiocellular, intestinal or prostate origin but model-specific differences exist (179–183). In addition, LCN2 forms a complex with matrix metalloproteinase (MMP)-9 (184). This stabilizes MMP9 activity and may contribute to metastatic spread (185). In mice with breast cancer, the oncogene HER (for human epidermal growth factor receptor) induces LCN2 expression and antibody-mediated blockade by trastuzumab antagonizes this effect (186). In prostate cancer cells, NF-κB as well as mutation or loss of p53 result in enhanced LCN2 expression (187). In addition, pro-inflammatory cytokines, such as TNF, IL-1ß, and IL-17 stimulate LCN2 expression via NF-κB (188, 189). The latter pathway opens the possibility that the immune response directed against tumor cells in fact induces a LCN2-dependent survival strategy but the biological or clinical significance of this putative paracrine mechanism has not been experimentally addressed yet. On the other hand, LCN2 has immune-modulatory effects itself which may help to limit tumor growth (190–195). Of note, LCN2 also stimulates VEGF expression via HIF and thus tumor angiogenesis (196). It is possible that LCN2-mediated iron depletion is an underlying mechanism but this has to be experimentally addressed (197). In conclusion, LCN2 has diverse functions in the cross-talk of different cell types in the TME and in cellular iron metabolism.
Cellular Iron Homeostasis
While circulating hepcidin levels have a major impact on the iron content of FPN1-expressing cells, additional mechanisms exist to maintain cellular iron homeostasis by balancing iron uptake, release and storage. The relative contribution of mechanisms for iron uptake is slightly different between cell types, though. For many of these, including EPs, enterocytes, B and T cells, TFR1 is assumed to be the quantitatively most important iron acquisition protein. TFR1 enables receptor-mediated endocytosis of TBI from the circulating iron pool (42). The endosome containing holo-TF and TFR1 is acidified, which results in dissociation of the complex into its components, TFR1, TF, and iron. Thereafter, ferric iron is converted to its divalent form by a reductase, such as STEAP3 (for six-transmembrane epithelial antigen of the prostate-3) before it can be shifted to the cytoplasm via DMT1 (198). DMT1 is also present in the cell surface membrane, where it mediates the uptake of ferrous iron in cooperation with the reductase duodenal cytochrome B (DcytB). After entry into the cytoplasm, ferrous iron is stabilized by a chaperone, PCBP2 (for Poly(RC) binding protein-2), until further utilization (199, 200). In the cytoplasm, labile iron can be incorporated into FT for storage. FT is a multimer that consists of 24 FTH and FTL subunits, assembled in cell-type specific proportions. In EPs and macrophages, FTH is the predominant subunit, possibly because these cells are exposed to the fastest iron fluxes and FTH is the only subunit carrying the ferroxidase activity essential for iron storage (201). In order to make FT-stored iron metabolically available again, FT needs to be degraded by an autophagosomal mechanism that relies on NCOA4 (for nuclear receptor coactivator-4) as cargo receptor (202, 203). After re-entry into the cytoplasmatic labile iron pool (LIP), iron can be used for metabolic or synthetic purposes, such as incorporation into iron-containing enzymes. Excess iron can also be exported out of the cytoplasm by the activity of FPN1. As mentioned above, the expression of many iron-handling proteins, such as TFR1, DMT1, FT, and FPN1 is fine-tuned at the post-transcriptional and translational level via IRP/IRE interaction (204, 205). The IRP system is of different importance for various cell types, though. For instance, it is essential for IECs whereas proper macrophage function in the absence of infectious agents does not require IRP (206, 207). However, putative functions of IRPs in TAMs have not been experimentally tested to date.
In many macrophage populations including TAMs, the uptake of aged or damaged RBCs by scavenger receptors contributes to iron acquisition to a substantial extent (208, 209). In addition, free Hb and heme are cleared from the circulation in complex with the plasma proteins haptoglobin and hemopexin, respectively (210, 211). This uptake is enabled by receptor-mediated endocytosis via CD163 and CD91, protects from oxidative damage by free Hb and heme and supplies macrophages with iron.
Ferroptosis, a Unique Form of Iron-Induced Cell Death
Ferroptosis is a form of regulated cell death sui generis that is elicited by iron-induced lipid peroxidation (3). The early events of ferroptosis are interconnected with ferritinophagy because NCOA4-facilitated FT degradation can provide free labile iron (212) (Figure 2). In the subsequent series of events culminating in iron-induced cell death, reduced glutathione peroxidase (GPX)-4 activity results in glutathione depletion and thus in increased susceptibility to oxidative stress. The tumor suppressor protein p53 induces ferroptosis via modulation of cysteine/glutamate metabolism with a direct impact on cellular oxidative stress, and p53 mutations, encountered in many types of cancer, result in the loss of p53-driven ferroptotic activity and tumor cell survival (213). It is thus not surprising that GPX4, ferroptosis and cancer are linked in several tumor entities including breast cancer, sarcomas and B cell lymphomas (214). In addition, the established anti-neoplastic drug sorafenib induces ferroptosis in hepatocellular carcinoma cells in vitro and novel compounds targeting GPX4 are effective in renal cell carcinoma and diffuse large B-cell lymphoma in vivo (215). Therefore, pharmacological modification of ferroptosis is a novel pathway for the treatment of both hematologic and solid malignancies.
Iron Regulates Cell Proliferation, Metabolism, and Signaling in Cancer
As stated above, iron is essential for cell division and basic metabolic processes and hence indispensable for living cells including malignant ones. From a canonical point of view, cancer cells frequently demonstrate excessive proliferation rates and high metabolic turnover, and are thus believed to require more iron than their non-malignant counterparts. This notion is supported by concomitant visualization of iron content in tumor tissue and the physiological storage organs spleen and liver. For example, iron accumulation in mammary tumors of mice go hand in hand with depletion of systemic stores, suggesting active iron mobilization to satisfy cancer cell needs (216). Overexpression of FT, TFR1, and DMT1, dysregulation of IRPs and of the FPN1-hepcidin axis in favor of an iron-loading phenotype of cancer cells is linked to accelerated tumor progression (217–226). In line with this concept, application of iron chelators, dietetic iron depletion, and interference with the hepcidin-FPN1 dyad to withdraw iron from malignant cells is successful in cancer therapy in vivo and in vitro (227–233). Of note, phlebotomy has been demonstrated to reduce the prevalence of and death from cancer, too (234, 235). Other reports, however, contradict this point of view and demonstrate that increasing cellular and systemic iron stores may, paradoxically, keep tumor progression at bay (236–238). Hence, it is likely that an equilibrium of iron levels that meets metabolic needs but still does not cause cellular damage, impair oncogenic signaling or induce ferroptosis has to be established in cancer cells to sustain disease progression.
There are several mechanisms by which iron influences tumor cell growth in a positive or negative manner: (i) as a catalyst in non-enzymatic reactions of ROS generation, (ii) as a cofactor of enzymes involved in cell division like ribonucleotide-diphosphate reductase, (iii) as regulator of cell cycle control proteins, (iv) as a participant in pro- and anti-oncogenic signaling, and, finally (v), as a key component of the hypoxic response and metabolic as well as epigenetic re-programming, mediated by 2-oxoglutarate dioxygenases (239, 240). In a pre-malignant setting and in tumor progression, ROS generation and DNA damage resulting from it, may increase mutation rate and lead to a (more) malignant phenotype. This phenomenon is, indeed, reported in the animal model of HFE-associated hereditary hemochromatosis (HH), Hfe−/− mice. There, high-iron diet increases the level of DNA damage in colon and mammary tissue and forms a possible mechanistic link between dietary iron and elevated colon carcinoma risk (241–244). On the other hand, IO and/or the occurrence of cellular labile iron in cancer cells may promote cell death via the same ROS-dependent mechanism, especially in combination with pro-apoptotic signaling and upon treatment with some chelating compounds (245, 246).
Stalling of the cell cycle and induction of apoptosis in iron-depleted cancer cells is a common observation made in multiple cancer cell lines (227, 231, 247, 248). The underlying mechanism is still not fully elucidated and can involve the distortion of cyclin expression and CDK (for cyclin-dependent kinase) activity pattern, as well as interference with the MDM2/p53 (for mouse double minute-2) pathway (231, 249–251). In addition, heme-iron directly interacts with p53 in normal hepatocytes and liver carcinoma cells and decreases its stability (252).
Iron has either attenuating or stimulatory effects on multiple signaling pathways. Iron affects NF-κB signaling by several mechanisms, such as increased peroxynitrite generation and subsequent tyrosine nitration in the inhibitory subunit of the NF-κB complex, IκBα, which may lead to its enhanced degradation (253). Iron on its own boosts IKK (for IκB kinase) activity as well (254). Of note, such enhancement of NF-κB, the master regulator of tumor-promoting inflammatory environment, may critically contribute to carcinogenesis (255). Iron plays a pivotal role in the stimulation of pro-inflammatory and oncogenic STAT3 signaling in colonic and hepatocellular carcinoma, too (256–258). And iron binding and activation of CDK1 with subsequent JAK2 (for Janus kinase-2) stimulation is a possible mechanistic explanation for this interplay. Iron is also shown to be involved in EMT, which is crucial for metastasis. This process, in turn, is under control of TGF-ß (for transforming growth factor-ß), WNT (for wingless-related integration site) and NOTCH signaling; all of which are stimulated by cellular iron loading and inhibited by its chelation (259–262).
The family of 2-oxoglutarate-dependent dioxygenases encompasses enzymes possessing ferrous ions in a non-heme and non-sulfur-cluster form in their active centers, requiring the citrate cycle intermediate 2-oxoglutarate as a cofactor for their activity and utilizing O2 as oxidizing substrate for catalyzed reactions. With those three components determining their activity, such proteins can be regarded as universal sensors of iron levels, energy status and oxygen levels in cells (263). Two most prominent subfamilies of 2-oxoglutarate-dependent dioxygenases are (i) Jumonji-type (JMJ) histone demethylases and (ii) HIF prolyl/aspartyl hydroxygenases. The link of the HIF hydroxygenases to iron metabolism of the normal and cancer cell will be discussed below in more detail.
JMJ histone demethylases are a group of enzymes sharing common structural motifs and a specificity for histone methyl-lysines (264, 265). As such, JMJ proteins are responsible for remodeling of the epigenetic landscape and tuning of the expression pattern in response to changes in energy status of the cell, oxygenation and iron levels (266–273). Importantly, a broad spectrum of the subfamily members displays overexpression and aberrant activity leading to oncogene expression, metabolic re-programming and cell cycle dysregulation in diverse malignancies (264, 274–279). In line, pre-clinical studies with murine models of prostate, mammary and lung cancer demonstrated high anti-neoplastic activity of JMJ-type demethylase inhibitors stressing the importance of JMJ-controlled epigenetic changes for malignancy (279–283). In addition, in mammary and ovarian cancer models, such blockers demonstrate interesting properties as they are particularly effective in targeting cancer stem cells, which often cannot be effectively reached by conventional chemo- and radiotherapy (282–284). Although ferrous iron constitutes a vital component of the JMJ-type enzymes, there are only few reports published to date, which systematically focus on the interplay between cellular iron status, JMJ enzyme activity and changes of epigenetic landscape of tumor cells. It is expected though that ID, e.g., due to chelation, should dampen activity of the JMJ proteins and, as a consequence, increase the general abundance of methyl-lysine repressing marks leading to downregulation of oncogene expression. There is indeed a major increase in histone methylation after iron chelation with deferroxamine (DFO), linking them to alterations in p21 and p53 expression (272). Interestingly, particular iron chelators specifically inhibit JMJ activity (285, 286); whole-genome epigenetic studies and reports on their anti-neoplastic properties are, however, still missing.
Iron and the Hypoxic Response Are Linked in Normal and Malignant Cells
Systemic and cellular adaptation to changeable concentrations of oxygen in the environment and within the tissue stays under control of evolutionary-conserved pathway of iron- and 2-oxoglutarate-dependent prolyl/aspartyl hydrogenases, PHDs, FIH (for factor inhibiting HIF), and HIFs (287–289). Under physiological O2 concentrations, both PHDs and FIH retain their full activity and hydroxylate proline residues in the oxygen-dependent domain (ODD) and asparagine residues in the transactivation domain (TAD) within HIF1α and HIF2α transcription factors. The PHD-mediated ODD hydroxylation causes ubiquitination by VHL (for von Hippel Lindau) Ub-ligase and targets the proteins for degradation. The TAD hydroxylation catalyzed by FIH, in turn, has no effect on protein levels but strongly inhibits its transcriptional activity. Under hypoxic conditions, activity of PHDs and FIH is diminished, enabling HIF1α and HIF2α accumulation, dimerization with the HIF1β coactivator, nuclear translocation and induction of the hypoxic response at the transcriptional level (289–292). Expression of HIF1α and HIF2α is tissue specific with substantial overlap in the repertoire of regulated genes. In tumor biology, HIF1α is believed to be of specific importance. The repertoire of HIF1α-induced genes encompasses angiogenic factors, such as VEGF and PDGF molecules, stroma and immune differentiation signals and a variety of amino acid and sugar transporters as well as enzymes of oxygen-independent energy metabolism (287, 289, 293–296). Activation of HIF1α at the early stages of carcinogenesis is crucial for the survival of the malignant cell since rapid cell division leads to shortages in oxygen and nutrient availability. HIF1α activation enables recruitment of blood vessels to the rudimentary neoplasm (so called angiogenic switch) and shifts the energy metabolism toward oxygen-independent glycolysis (so called glycolytic switch), with both these features being regarded as key properties of cancer (289, 295, 296).
Apart from oxygen concentration, iron availability is another factor determining activity of PHD and FIH hydroxylases, since redox-active iron ions are part of their catalytic centers. Along this line, modulation of cellular iron levels by chelation (e.g., with DFO) or iron supplementation can stimulate or inhibit the activity of these enzymes, respectively (263, 290, 297). Interestingly, the size of iron stores is inversely correlated with the degree of HIF-dependent respiratory response in humans as well (298). In DCs, FT has a pivotal role in the regulation of HIF1α as induction of FT by inflammatory stimuli decreases the LIP and hence iron available for PHD metalation and activity (299). As a result, scavenging of labile iron by FT causes HIF1α accumulation even at physiological O2 concentrations. Whether analogous mechanisms are active in other cell types, particularly malignant cells, remains to be investigated. Few reports, however, give some hints that the interplay of iron and HIF1α can bear significance for cancer biology. For impacting on cancer cell and tissue iron levels by means of TFR1 downregulation or dietary ID leads to increased HIF1α activity culminating in increased VEGF formation and cancer graft vascularization (10). In addition, dietary ID can accelerate mammary tumor growth and metastasis, and the activation of NOTCH signaling and HIF1α by ID is discussed as explanation of these paradoxical effects (237). In addition, low dietary iron intake affects tumor growth and susceptibility toward anti-VEGF therapy, and low iron diet results in a substantially better vascularization of the tumors (300). The net tumor growth is, however, decelerated following dietary ID as a result of slower tumor cell proliferation.
The question whether the functional iron/HIF1α interaction bears biological importance pertains to the management of AOC in the clinical setting as well. On one hand side, iron supplementation in any form (oral or intravenous) in cancer patients may bear a risk of faster progression due to stimulation of cancer cell proliferation as demonstrated in numerous pre-clinical reports (232, 259, 300). On the other, the same treatment may, in addition to improving quality of life of the patient, impair tumor metabolic adaptation and vascularization resulting in better outcome (10, 301). A novel class of drugs for the treatment of anemia, so called PHD inhibitors, which cause HIF1α stabilization, is discussed above (302, 303).
Iron Handling by Tumor-Associated Macrophages and Its Implication for Carcinogenesis
Tumors can be considered as organ-like structures with complex interactions between transformed and non-transformed cells. Stroma cells are needed to support the malignant potential of tumor cells, but tumors are additionally infiltrated by a wide range of immune cells, such as TAMs (304). TAMs can represent up to 50% of a tumor's mass and studies evaluated a significant link between TAM number and density with a poor prognosis of the underlying malignant disease (305–307).
Following the conventional M1/M2 classification, TAMs are typically M2-like cells and as such characterized by high expression of HMOX1, mannose receptor and scavenger receptor-A (308–313). M2-like TAMs preferentially home to hypoxic areas of the tumor, where their pro-tumoral activities are promoted (314). However, in cancer induced by chronic inflammation, TAMs can display an inflammatory M1-like phenotype or overlapping M1/M2 characteristics (315–317). In addition, specific microenvironmental signals may be important for different TAM activation states within the same tumor (294). In mammary tumors, two different microenvironments are infiltrated by different TAM subsets: (i) Sessile TAMs with high phagocytic capacity and expression of M2-like markers are present. (ii) Migratory TAMs are observed and produce EGF to attract cancer cells (318–321). The diversity and heterogeneity of TAMs in breast, lung, pancreas, brain and liver cancers is increasingly appreciated, too (322). It is thus likely that some TAM populations support tumor progression and development of metastasis, whereas others have anti-tumoral activities. Further insight in the microenvironmental stimuli, including iron, effector molecules and signaling pathways, that drive TAM heterogeneity within a tumor may be important to establish options to specifically target, inhibit or destroy pro-tumoral TAM populations in cancer patients.
As already stressed, macrophages residing in normal tissues can be regarded as “gate-keepers” of iron metabolism, i.e., cells which take up iron, store it in excess and export it to cover the needs of the surrounding cells. This role can also be supposed for TAMs in the TME and macrophage polarization dictates the way they are handling iron: whereas FThigh, FPN1low M1-like macrophages are predisposed to iron withdrawal, restriction and storage, the FTlow, FPN1high M2-like subtype promotes iron export and iron redistribution to the extracellular space. TAMs display a strongly M2-polarized phenotype in most types of malignancies and are ascribed such “iron-donating” features that may contribute to their tumor-promoting properties (323). In line with this model, human M2-skewed macrophages boost proliferation of human cancer cells in vitro in an iron- and FPN1-dependent manner (324). Data obtained with animal models and human tumor tissue further stress the “iron gate-keeper” and “iron donating” phenotype of TAMs. In murine primary lesions of the prostate or breast as well as in lung and brain metastasis, iron-storing macrophages can be visualized by magnetic resonance imaging (MRI) and histology (325, 326). In the human setting, iron deposition and expression of the iron turnover machinery (i.e., FPN1, hepcidin, TFR1, and FT) is significantly enriched in the macrophage and lymphocyte compartment (327, 328). Some indirect hints for the preferential active iron uptake by TAMs in the TME can be inferred from MRI, microscopy and cytometry studies using iron nanoparticles to specifically label this cell type (329–332). An interesting additional phenomenon is the fact that TAM-derived FT acts on malignant mammary epithelium as a growth factor (333). These growth-promoting effects of FT are, however, independent of its iron content.
The canonical view on iron homeostasis stresses the central role of the sole iron exporter FPN1 and its antagonist, hepcidin. In accord with it, FPN1, highly expressed on alternatively activated TAMs, should constitute the exclusive pipeline of the macrophage-tumor cell iron transfer. Its functionality may, however, not operate optimally under high systemic hepcidin levels in cancer patients and in the inflammatory, hepcidin-rich TME (219, 223, 327, 334). Importantly, increased hepcidin is unequivocally linked to accelerated tumor progression in experimental animals and in breast cancer individuals (219, 224, 225). This paradox suggests either a hepcidin-independent regulation of FPN1 in TAMs or the existence of alternative iron transport routes in the TME. The former possibility is supported by the data obtained in human breast carcinoma (327). There, elevated FPN1 levels in the macrophage infiltrate can be discerned in ductal carcinoma in situ (DCIS) and ductal carcinoma as compared to normal breast tissue. Concomitantly, the same stages of breast carcinogenesis demonstrate substantially increased hepcidin. The existence of FPN1-independent iron export can, in turn, be inferred from few reports identifying TAMs as the main source of LCN2 (335–337). Two reports dealing with the macrophage-epithelium iron transfer in mammary carcinoma describe a critical contribution of macrophage-secreted LCN2 to optimal proliferation and iron supply of cancer cells in vitro (338, 339) (Figure 2).
The great majority of literature on TAMs and iron refers to research on breast cancer. However, iron transfer in the TME may be subject to mechanisms specific to a given tumor entity. An interesting phenomenon, apparently contradicting the “iron-donating” phenotype of TAMs is observed in a murine lung carcinoma model. There, TAMs dwelling in hemorrhagic regions surprisingly demonstrate excessive iron loading and the classical M1 phenotype with a notable upregulation of NOS2 and toxicity against malignant cells (332). Mechanistically, such properties are provoked by ingestion of damaged RBCs reaching the tumor parenchyma via leaky tumor vasculature. Of great clinical interest, the M1 phenotype can also be induced by treatment of tumor-bearing mice with iron microparticles resulting in net tumor suppression (332).
Summarizing, TAMs, in parallel to “physiological” tissue-resident macrophages, can be regarded as the key nexus of iron homeostasis in the TME. These cells, however, can function both as tumor-promoting donors of the element and/or as iron-laden tumor cytotoxic leukocytes. For it remains open whether damaged RBCs, ingested apoptotic cells, which are widespread in the malignant milieu, TAM-derived siderophore-bound iron, FT or TBI from the systemic circulation pose such an iron source (332, 337).
Iron Controls T Cell Function
The microenvironment of solid tumors also contains tumor-infiltrating immune cells (TILs), including B cells and T cells, natural killer (NK) cells, neutrophils, myeloid-derived suppressor cells (MDSCs), and TAMs, and the function of all of these cells may be affected by iron (340–344).
For example, iron has immunosuppressive effects on T cell responses. According to early studies, the immune system and its circulating components are involved in the recognition and binding of metals as protection against metal toxicity, and the use of metals, such as iron, by bacteria or transformed cells (345). In line, in patients with thalassemia, iron influences the expansion of different T cell subsets (346). Furthermore, in patients with HFE-associated HH, a decrease in T cell numbers and activation defects can be observed, which may be causally linked to the toxic effects of free iron and oxidative stress (347). Additionally, abnormalities in the relative proportions of CD4+ and CD8+ subpopulations are described (348, 349). A similar impairment is observed in individuals receiving blood transfusions in the setting of TRIM and following intravenous iron infusions for the treatment of ID anemia or AOC as discussed above (350–353).
In general, there are different types of cancer in which iron has been implicated (354). However, very few reports address the influence of iron on cells of the adaptive immune system. The majority of studies deal with patients suffering from breast cancer. In these patients, a link between dysregulation of iron metabolism and progression of cancer exists (355). Experimental data indicate that a chronic failure in iron-dependent redox balance leads to the loss of tumor suppressors, oncogene expression and triggering of pro-oncogenic signaling, such as WNT and NF-κB pathways (259, 356–359). Several studies also point out that elevated iron stores are associated with increased risk for cancer development (360–362). However, many of these studies use serum FT as an indicator of iron loading which may be misleading because even in subclinical inflammation, FT levels may be increased by the action of cytokines, thus not accurately reflecting iron stores. Therefore, the association of high FT levels with the risk of cancer may in part reflect an inflammatory state as an expression of incipient cancer rather than a causative role of iron loading. Increased serum FT has been found to be associated with breast cancer risk, and FT levels in cancer-tissue are significantly increased in cancer specimens and correlate with higher degrees of tumor cell proliferation (363, 364). Functionally, FT secreted by TAMs is proposed to act as tumor growth factor and immunosuppressant (333, 365). However, the opposite functions of this protein on anti-tumor immunity are reported as well. The subcellular localization of FTH is important in triple negative breast cancer (366). This may be attributable to the fact that cytoplasmic FTH in tumor tissues regulates the MHC-I part of the antigen processing and presentation pathway and subsequently attracts CD8+ T cells to target tumor cells, whereas nuclear FTH supports the survival of cancer cells. Consequently, animal studies indicate that low iron nutrition and application of iron chelators moderate tumor growth and inhibit metastasis (367). Of note, FT can act as a pro-inflammatory mediator independent of iron availability, thereby affecting protein kinase C- and NF-κB-mediated signaling processes (368).
It is well-known that the proliferation of T cells requires iron, and intracellular iron stored in FT is thought to sustain proliferation of immune cells (369–371). In addition, a mutation in the gene encoding TFR1, TFRC, results in impaired T as well as B cell function (372). In line, ID reduces T cell numbers and impairs the activity of NK cells (373). Furthermore, iron chelation inhibits the production of IFN-γ, IL-2, and GM-CSF (for granulocyte-macrophage colony-stimulating factor) by T cells (374). On the other hand, patients suffering from IO secondary to ß-thalassemia have decreased CD4+ and increased CD8+ T cell numbers, while patients with HFE-associated HH show a trend to lower CD8+ T cells dependent on their HLA haplotype (347, 375– 377). Along with this, genetic deletion of FTH reduces the number of mature B cells and peripheral T cells in all lymphoid organs as a result of increased LIP and enhanced ROS formation (378). Therefore, a balanced iron metabolism is central for proper T cell function, but to which extent dysbalances affect the clinical course of cancer awaits further investigation.
Putative Roles of Iron in Cancer Biology Beyond Cancer and Immune Cells
Iron may influence cancer biology independent of its effects on cancer and immune cells, for example by altering the microbiome or the function of stromal cells in the TME. The role of the microbiome in cancer development is increasingly appreciated (379). This is especially true of the pathogenesis of colon cancer, which is thought to be strongly affected by the intraluminal microflora of the gastrointestinal tract (380). Of note, the composition and the iron content of the diet influences the diversity of the gastrointestinal microbiome, which may have secondary effects on IECs and the mucosal immune system. (381–385). These alterations may impair GI barrier function, undermine colonization resistance and increase the risk of colon cancer, but probiotic supplementation may prevent these adverse effects (386). In addition, heme has pro-oxidative properties and acts as DAMP which is recognized by TLR4 (for toll-like receptor-4) and cryopyrin (387, 388). As a consequence, pro-inflammatory signaling is initiated in IECs and ECs, whereas myeloid cell functions tend to be impaired by heme excess (389–392).
Of interest, the microbiome also increases the potential of heme to cause lipid peroxidation, whereas depletion of bacterial commensals with broad-spectrum antibiotics reverses this effect (393). In addition, antibiotic treatment suppresses bacteria that impair the intestinal mucus barrier thereby preventing heme's proliferation-inducing effect on IECs (394). Moroever, heme causes mutations in genes promoting colon cancer, such as APC (for adenomatous polyposis coli) and KRAS (395). These data suggest that an interaction between diet, the microbiome and the intestinal epithelium determines the susceptibility to colon cancer, indeed.
Various species of enterobacteriaceae produce siderophores in the GI lumen. Siderophores scavenge iron, which results in iron depletion of IECs and subsequent HIF1α activation (396). Apparently, activation of HIF1α promotes GI inflammation and alters WNT signaling but does not directly contribute to the pathogenesis of colon cancer (397–399). In contrast, HIF2α activation in IECs stimulates their proliferation rate and promotes neutrophil recruitment to the TME, thereby facilitating the occurrence of colon carcinoma (400, 401). This is also relevant for systemic iron homeostasis because HIF2α controls DMT1 and DcytB expression and induces FPN1 expression in response to ID (402–404). In addition, activation of HIFs results in VEGF production, which acts on ECs and is a prerequisite for tumor angiogenesis. Thus, the HIF-VEGF axis is another potential pathway linking intestinal dysbiosis to pro-oncogenic behavior in neoplastic and stromal cells.
In addition, LCN2 affects the balance between bacteria that do or do not utilize LCN2-susceptible siderophores, such as catecholate-type ones (405). In the absence of LCN2, Alistipes species outcompete other commensals because of their ability to secrete enterobactin, resulting in dysbiosis that promotes colorectal carcinogenesis (406).
In conclusion, dietary iron affects IECs, mucosal immune cells and the microbiome and modulates their interaction, thus promoting colorectal carcinogenesis.
Discussion
Our tools to manipulate systemic iron homeostasis have been evolving over the last couple of years. Medications for the treatment of ACD have direct or indirect effects on iron homeostasis and include AHS, calcium channel blockers, cytokine antagonists, PHD inhibitors, kinase inhibitors, ESAs and multiple iron preparations (302, 407–411). However, not all of these compounds are well-studied in cancer patients yet. Also, the local effects that these medications may have in the TME and therefore, in the medium-term, on the underlying malignant disease are largely unknown. We thus need to gain further insight into the effects of such treatments on the composition of the TME, the immune control of cancer, the metabolic re-programming of immune and cancer cells, their impact on cellular stress and proliferative/apoptotic/ferroptotic responses, along with off-target effects of such treatments linked to e.g., tumor vascularization and development of distant metastasis. We are also in a need to gather further knowledge on the effect of AOC correction by any treatment on the subsequent course of the malignant disease along with a personalized view depending on the tumor entities and specific factors of the individual patient.
It is only in recent years that we have begun to unscramble the complex, reciprocal and countless interconnections between iron metabolism and cancer as reviewed herein. Observations made in in vitro systems, co-culture models, organoids, and small animal models need to be carefully translated to the human setting. Novel technology, such as laser dissection microscopy, multi-laser flow cytometry and single-cell RNA sequencing on human cancer tissue may help in this translation process. Eventually, we face the challenge to close existing knowledge gaps and connect the dots to see the complete picture of the many roles of iron in cancer occurrence, progression and treatment for the sake of improved care for hemato-oncologic patients.
Author Contributions
CP-O wrote and edited the manuscript. PT wrote and edited the manuscript. VP discussed and edited the manuscript. GW provided the concept and wrote the manuscript. MN provided the concept, wrote the manuscript and drew the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
CP-O is employed by the Medical University of Innsbruck, which also supported this publication. CP-O and PT are supported by the ‘Österreichische Krebshilfe Tirol', projects 17006 and 15024. VP and GW are supported by the Austrian Research Funds, FWF-Doctoral Program-HOROS-W1253.
Abbreviations
ACD, anemia of chronic disease; AHS, anti-hepcidin strategy; AOC, anemia of cancer; APC, adenomatous polyposis coli; ATP, adenosine triphosphate; BMP6, bone morphogenic protein-6; CD, cluster of differentiation; CHOP, CCAAT-enhancer-binding protein homologous protein; CDK, cyclin-dependent kinase; CKD, chronic kidney disease; CML, chronic myeloid leukemia; CREB-H, cyclic AMP response element-binding protein H; DAMP, danger-associated molecular pattern; DC, dendritic cell; DCIS, ductal carcinoma in situ; DFO, deferroxamine; DMT1, divalent metal transporter-1 AKA SLC11A2; DNA, deoxyribonucleic acid; EC, endothelial cell; EDTA, ethylenediaminetetraacetic acid; EGF, epidermal growth factor; EMT, epithelial-mesenchymal transition; EP, erythroid progenitor; EPO, erythropoietin; ER, endoplasmatic reticulum; ERFE, erythroferrone; ERK, extracellular signal-regulated kinases; ESA, erythropoiesis-stimulating agent; FIH, factor inhibiting HIF; FPN1, ferroportin-1 AKA SLC40A1; FT, ferritin; FTL, ferritin light chain; FTH, ferritin heavy chain; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GDF15, growth differentiation factor-15; GM-CSF, granulocyte-macrophage colony-stimulating factor; GPX4, glutathione peroxidase-4; Hb, hemoglobin; HFE, hemochromatosis-associated gene/protein; HJV, hemojuvelin; HH, hereditary hemochromatosis; HMOX1, heme oxygenase-1; ID, iron deficiency; IEC, intestinal epithelial cell; IFN-γ, interferon gamma; IKK, IκB kinase; IL, interleukin; IO, iron overload; IRE, iron-responsive element; IRP, iron-regulatory protein; JAK, Janus kinase; JMJ, Jumonji-type protein; LCN2, lipocalin-2 AKA NGAL (for Neutrophil gelatinase-associated lipocalin); LCNR, lipocalin-2 receptor AKA SLC22A17; LF, lactoferrin; LIP, labile iron pool; LPS, lipopolysaccharide; MAP kinase, mitogen-activated protein kinase; MDM2, mouse double minute-2; MDS, myelodysplastic syndrome; MDSC, myeloid-derived suppressor cell; MM, multiple myeloma; MRI, magnetic resonance imaging; mRNA, messenger ribonucleic acid; miRNA, micro ribonucleic acid; NCOA4, nuclear receptor coactivator-4; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOS2, nitric oxide synthase-2 AKA inducible NOS; NSCLC, non-small cell lung cancer; NK, natural killer; NKT cell, natural killer T cell; NTBI, non-transferrin-bound iron; ODD, oxygen-dependent domain; PAMP, pathogen-associated molecular pattern; PCBP2, poly(RC) binding protein-2; PDGF, platelet-derived growth factor; PHD, prolyl HIF dioxygenases; RBC, red blood cell; RCT, randomized controlled trial; RNS, reactive nitrogen species; ROS, reactive oxygen species; SCARA5, scavenger receptor class A member-5; SLC, solute carrier; SMAD, homologs of Sma and Mad (mothers against decapentaplegic) proteins; STAT, signal transducer and activator of transcription; STEAP3, six-transmembrane early antigen of the prostate-3; TAD, transactivation domain; TAM, tumor-associated macrophage; TBI, transferrin-bound iron; TC cell, cytotoxic T cell; TFR, transferrin receptor; TGF-ß, transforming growth factor-ß; TH cell, T helper cell; TIL, tumor-infiltrating lymphocyte; TIM2, T cell immunoglobulin and mucin domain protein-2; TLR4, toll-like receptor-4; TREG cell, regulatory T cell; TME, tumor microenvironment; TNF, tumor necrosis factor; TRIM, transfusion-related immune modulation; VEGF, vascular endothelial growth factor; VHL, von Hippel-Lindau tumor suppressor; WNT, wingless-related integration site.
References
1. Henle ES, Linn S. Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J Biol Chem. (1997) 272:19095–8.
2. Thanan R, Oikawa S, Yongvanit P, Hiraku Y, Ma N, Pinlaor S, et al. Inflammation-induced protein carbonylation contributes to poor prognosis for cholangiocarcinoma. Free Radic Biol Med. (2012) 52:1465–72. doi: 10.1016/j.freeradbiomed.2012.01.018
3. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
4. Zhang C. Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control. Protein Cell (2014) 5:750–60. doi: 10.1007/s13238-014-0083-7
5. Puig S, Ramos-Alonso L, Romero AM, Martinez-Pastor MT. The elemental role of iron in DNA synthesis and repair. Metallomics (2017) 9:1483–500. doi: 10.1039/c7mt00116a
6. Netz DJ, Stith CM, Stumpfig M, Kopf G, Vogel D, Genau HM, et al. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat Chem Biol. (2011) 8:125–32. doi: 10.1038/nchembio.721
7. Oexle H, Gnaiger E, Weiss G. Iron-dependent changes in cellular energy metabolism: influence on citric acid cycle and oxidative phosphorylation. Biochim Biophys Acta (1999) 1413:99–107.
8. Volani C, Doerrier C, Demetz E, Haschka D, Paglia G, Lavdas AA, et al. Dietary iron loading negatively affects liver mitochondrial function. Metallomics (2017) 9:1634–44. doi: 10.1039/c7mt00177k
9. Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology (2005) 69 Suppl. 3:4–10. doi: 10.1159/000088478
10. Eckard J, Dai J, Wu J, Jian J, Yang Q, Chen H, et al. Effects of cellular iron deficiency on the formation of vascular endothelial growth factor and angiogenesis. Iron deficiency and angiogenesis. Cancer Cell Int. (2010) 10:28. doi: 10.1186/1475-2867-10-28
11. Syed SN, Jung M, Weigert A, Brune B. S1P Provokes tumor lymphangiogenesis via macrophage-derived mediators such as IL-1beta or lipocalin-2. Mediators Inflamm. (2017) 2017:7510496. doi: 10.1155/2017/7510496
12. Jung M, Oren B, Mora J, Mertens C, Dziumbla S, Popp R, et al. Lipocalin 2 from macrophages stimulated by tumor cell-derived sphingosine 1-phosphate promotes lymphangiogenesis and tumor metastasis. Sci Signal. (2016) 9:ra64. doi: 10.1126/scisignal.aaf3241
13. Yamagata Y, Tomioka H, Sakamoto K, Sato K, Harada H, Ikeda T, et al. CD163-positive macrophages within the tumor stroma are associated with lymphangiogenesis and lymph node metastasis in oral squamous cell carcinoma. J Oral Maxillofac Surg. (2017) 75:2144–53. doi: 10.1016/j.joms.2017.03.009
14. Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol. (2014) 5:75. doi: 10.3389/fphys.2014.00075
15. Wang Q, He Z, Huang M, Liu T, Wang Y, Xu H, et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2alpha. Nat Commun. (2018) 9:559. doi: 10.1038/s41467-018-03050-0
16. He H, Xu J, Warren CM, Duan D, Li X, Wu L, et al. Endothelial cells provide an instructive niche for the differentiation and functional polarization of M2-like macrophages. Blood (2012) 120:3152–62. doi: 10.1182/blood-2012-04-422758
17. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer (2016) 16:447–62. doi: 10.1038/nrc.2016.54
18. Thevenod F. Iron and its role in cancer defense: a double-edged sword. Met Ions Life Sci. (2018) 18. doi: 10.1515/9783110470734-021
19. Schaible UE, Kaufmann SH. Iron and microbial infection. Nat Rev Microbiol. (2004) 2:946–53. doi: 10.1038/nrmicro1046
20. Nairz M, Haschka D, Demetz E, Weiss G. Iron at the interface of immunity and infection. Front Pharmacol. (2014) 5:152. doi: 10.3389/fphar.2014.00152
21. Weiss G, Carver PL. Role of divalent metals in infectious disease susceptibility and outcome. Clin Microbiol Infect. (2018) 24:16–23. doi: 10.1016/j.cmi.2017.01.018
22. Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. (2004) 113:1271–6. doi: 10.1172/JCI20945
23. Peyssonnaux C, Zinkernagel AS, Datta V, Lauth X, Johnson RS, Nizet V. TLR-4 dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood (2006) 107:3727–32. doi: 10.1182/blood-2005-06-2259
24. Graziadei I, Weiss G, Bohm A, Werner-Felmayer G, Vogel W. Unidirectional upregulation of the synthesis of the major iron proteins, transferrin-receptor and ferritin, in HepG2 cells by the acute-phase protein alpha1-antitrypsin. J Hepatol. (1997) 27:716–25.
25. Schaefer B, Haschka D, Finkenstedt A, Petersen BS, Theurl I, Henninger B, et al. Impaired hepcidin expression in alpha-1-antitrypsin deficiency associated with iron overload and progressive liver disease. Hum Mol Genet. (2015) 24:6254–63. doi: 10.1093/hmg/ddv348
26. Theurl I, Aigner E, Theurl M, Nairz M, Seifert M, Schroll A, et al. Regulation of iron homeostasis in anemia of chronic disease and iron deficiency anemia: diagnostic and therapeutic implications. Blood (2009) 113:5277–86. doi: 10.1182/blood-2008-12-195651
27. Theurl I, Schroll A, Sonnweber T, Nairz M, Theurl M, Willenbacher W, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood (2011) 118:4977–84. doi: 10.1182/blood-2011-03-345066
28. Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol. (2005) 202:199–211. doi: 10.1016/j.taap.2004.06.021
29. Millonig G, Ganzleben I, Peccerella T, Casanovas G, Brodziak-Jarosz L, Breitkopf-Heinlein K, et al. Sustained submicromolar H2O2 levels induce hepcidin via signal transducer and activator of transcription 3 (STAT3). J Biol Chem. (2012) 287:37472–82. doi: 10.1074/jbc.M112.358911
30. Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. (2015) 264:182–203. doi: 10.1111/imr.12266
31. Nairz M, Schroll A, Sonnweber T, Weiss G. The struggle for iron–a metal at the host-pathogen interface. Cell Microbiol. (2010) 12:1691–702. doi: 10.1111/j.1462-5822.2010.01529.x
32. Nairz M, Fritsche G, Brunner P, Talasz H, Hantke K, Weiss G. Interferon-gamma limits the availability of iron for intramacrophage Salmonella typhimurium. Eur J Immunol. (2008) 38:1923–36. doi: 10.1002/eji.200738056
33. Skaar EP, Raffatellu M. Metals in infectious diseases and nutritional immunity. Metallomics (2015) 7:926–8. doi: 10.1039/c5mt90021b
34. Zhou Y, Que KT, Zhang Z, Yi ZJ, Zhao PX, You Y, et al. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med. (2018) 7:4012–22. doi: 10.1002/cam4.1670
35. Ludwig H, Evstatiev R, Kornek G, Aapro M, Bauernhofer T, Buxhofer-Ausch V, et al. Iron metabolism and iron supplementation in cancer patients. Wien Klin Wochenschr. (2015) 127:907–19. doi: 10.1007/s00508-015-0842-3
36. Steurer M, Wagner H, Gastl G. Prevalence and management of anaemia in haematologic cancer patients receiving cyclic nonplatinum chemotherapy: results of a prospective national chart survey. Wien Klin Wochenschr. (2004) 116:367–72. doi: 10.1007/BF03040915
37. Ludwig H, Muldur E, Endler G, Hubl W. Prevalence of iron deficiency across different tumors and its association with poor performance status, disease status and anemia. Ann Oncol. (2013) 24:1886–92. doi: 10.1093/annonc/mdt118
38. Caro JJ, Salas M, Ward A, Goss G. Anemia as an independent prognostic factor for survival in patients with cancer: a systemic, quantitative review. Cancer (2001) 91:2214–21. doi: 10.1002/1097-0142(20010615)91:12 < 2214::AID-CNCR1251>3.0.CO;2-P
39. Bohlius J, Schmidlin K, Brillant C, Schwarzer G, Trelle S, Seidenfeld J, et al. Recombinant human erythropoiesis-stimulating agents and mortality in patients with cancer: a meta-analysis of randomised trials. Lancet (2009) 373:1532–42. doi: 10.1016/S0140-6736(09)60502-X
40. Mori K, Lee HT, Rapoport D, Drexler IR, Foster K, Yang J, et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J Clin Invest. (2005) 115:610–21. doi: 10.1172/JCI23056
41. Goodnough LT, Levy JH, Murphy MF. Concepts of blood transfusion in adults. Lancet (2013) 381:1845–54. doi: 10.1016/S0140-6736(13)60650-9
42. Muckenthaler MU, Rivella S, Hentze MW, Galy B. A red carpet for iron metabolism. Cell (2016) 168:344–61. doi: 10.1016/j.cell.2016.12.034
43. Nairz M, Weiss G. Molecular and clinical aspects of iron homeostasis: from anemia to hemochromatosis. Wien Klin Wochenschr. (2006) 118:442–62. doi: 10.1007/s00508-006-0653-7
44. Evstatiev R, Gasche C. Iron sensing and signalling. Gut (2010) 61:933–52. doi: 10.1136/gut.2010.214312
45. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science (2004) 306:2090–3. doi: 10.1126/science.1104742
46. Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. (1999) 21:195–9. doi: 10.1038/5979
47. Chen H, Attieh ZK, Syed BA, Kuo YM, Stevens V, Fuqua BK, et al. Identification of zyklopen, a new member of the vertebrate multicopper ferroxidase family, and characterization in rodents and human cells. J Nutr. (2010) 140:1728–35. doi: 10.3945/jn.109.117531
48. Wessling-Resnick M. Iron imports. III. Transfer of iron from the mucosa into circulation. Am J Physiol Gastrointest Liver Physiol. (2006) 290:G1–6. doi: 10.1152/ajpgi.00415.2005
49. Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, et al. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position−7007 of the FPN1 promoter. Haematologica (2009) 95:1261–8. doi: 10.3324/haematol.2009.020123
50. Beaumont C. Multiple regulatory mechanisms act in concert to control ferroportin expression and heme iron recycling by macrophages. Haematologica (2010) 95:1233–6. doi: 10.3324/haematol.2010.025585
51. Zoller H, Theurl I, Koch R, Kaser A, Weiss G. Mechanisms of iron mediated regulation of the duodenal iron transporters divalent metal transporter 1 and ferroportin 1. Blood Cells Mol Dis. (2002) 29:488–97. doi: 10.1006/bcmd.2002.0587
52. Sangokoya C, Doss JF, Chi JT. Iron-responsive miR-485-3p regulates cellular iron homeostasis by targeting ferroportin. PLoS Genet. (2013) 9:e1003408. doi: 10.1371/journal.pgen.1003408PGENETICS-D-12-00445
53. Babu KR, Muckenthaler MU. miR-20a regulates expression of the iron exporter ferroportin in lung cancer. J Mol Med (Berl). (2016) 94:347–59. doi: 10.1007/s00109-015-1362-3
54. Deng Z, Manz DH, Torti SV, Torti FM. Iron-responsive element-binding protein 2 plays an essential role in regulating prostate cancer cell growth. Oncotarget (2017) 8:82231–43. doi: 10.18632/oncotarget.19288
55. Chen G, Fillebeen C, Wang J, Pantopoulos K. Overexpression of iron regulatory protein 1 suppresses growth of tumor xenografts. Carcinogenesis (2007) 28:785–91. doi: 10.1093/carcin/bgl210
56. Aschemeyer S, Qiao B, Stefanova D, Valore EV, Sek AC, Ruwe TA, et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood (2018) 131:899–910. doi: 10.1182/blood-2017-05-786590
57. Canali S, Zumbrennen-Bullough KB, Core AB, Wang CY, Nairz M, Bouley R, et al. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood (2016) 129:405–14. doi: 10.1182/blood-2016-06-721571
58. Core AB, Canali S, Babitt JL. Hemojuvelin and bone morphogenetic protein (BMP) signaling in iron homeostasis. Front Pharmacol. (2014) 5:104. doi: 10.3389/fphar.2014.00104
59. Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. (2008) 7:205–14. doi: 10.1016/j.cmet.2007.11.016
60. Casanovas G, Mleczko-Sanecka K, Altamura S, Hentze MW, Muckenthaler MU. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J Mol Med (Berl). (2009) 87:471–80. doi: 10.1007/s00109-009-0447-2
61. Nili M, Shinde U, Rotwein P. Soluble repulsive guidance molecule c/hemojuvelin is a broad spectrum bone morphogenetic protein (BMP) antagonist and inhibits both BMP2- and BMP6-mediated signaling and gene expression. J Biol Chem. (2010) 285:24783–92. doi: 10.1074/jbc.M110.130286
62. Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood (2006) 108:3204–9. doi: 10.1182/blood-2006-06-027631
63. Pietrangelo A, Dierssen U, Valli L, Garuti C, Rump A, Corradini E, et al. STAT3 is required for IL-6-gp130-dependent activation of hepcidin in vivo. Gastroenterology (2007) 132:294–300. doi: 10.1053/j.gastro.2006.10.018
64. Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood (2007) 109:353–8. doi: 10.1182/blood-2006-07-033969
65. Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. (2002) 110:1037–44. doi: 10.1172/JCI15686
66. Nairz M, Theurl I, Wolf D, Weiss G. Iron deficiency or anemia of inflammation?: Differential diagnosis and mechanisms of anemia of inflammation. Wien Med Wochenschr. (2016) 166:411–23. doi: 10.1007/s10354-016-0505-7
67. Camaschella C. Iron-deficiency anemia. N Engl J Med. (2015) 372:1832–43. doi: 10.1056/NEJMra1401038
68. Nai A, Rubio A, Campanella A, Gourbeyre O, Artuso I, Bordini J, et al. Limiting hepatic Bmp-Smad signaling by matriptase-2 is required for erythropoietin-mediated hepcidin suppression in mice. Blood (2015) 127:2327–36. doi: 10.1182/blood-2015-11-681494
69. Kautz L, Jung G, Nemeth E, Ganz T. Erythroferrone contributes to recovery from anemia of inflammation. Blood (2014) 124:2569–74. doi: 10.1182/blood-2014-06-584607
70. Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. (2014) 46:678–84. doi: 10.1038/ng.2996
71. Sonnweber T, Nachbaur D, Schroll A, Nairz M, Seifert M, Demetz E, et al. Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor BB. Gut (2013) 63:1951–9. doi: 10.1136/gutjnl-2013-305317
72. Goetze O, Schmitt J, Spliethoff K, Theurl I, Weiss G, Swinkels DW, et al. Adaptation of iron transport and metabolism to acute high-altitude hypoxia in mountaineers. Hepatology (2013) 58:2153–62. doi: 10.1002/hep.26581
73. Robach P, Recalcati S, Girelli D, Gelfi C, Aachmann-Andersen NJ, Thomsen JJ, et al. Alterations of systemic and muscle iron metabolism in human subjects treated with low-dose recombinant erythropoietin. Blood (2009) 113:6707–15. doi: 10.1182/blood-2008-09-178095
74. Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. (2007) 13:1096–101. doi: 10.1038/nm1629
75. Theurl I, Finkenstedt A, Schroll A, Nairz M, Sonnweber T, Bellmann-Weiler R, et al. Growth differentiation factor 15 in anaemia of chronic disease, iron deficiency anaemia and mixed type anaemia. Br J Haematol. (2009) 148:449–55. doi: 10.1111/j.1365-2141.2009.07961.x
76. Vecchi C, Montosi G, Zhang K, Lamberti I, Duncan SA, Kaufman RJ, et al. ER stress controls iron metabolism through induction of hepcidin. Science (2009) 325:877–80. doi: 10.1126/science.1176639
77. Oliveira SJ, Pinto JP, Picarote G, Costa VM, Carvalho F, Rangel M, et al. ER stress-inducible factor CHOP affects the expression of hepcidin by modulating C/EBPalpha activity. PLoS ONE (2009) 4:e6618. doi: 10.1371/journal.pone.0006618
78. Canali S, Vecchi C, Garuti C, Montosi G, Babitt JL, Pietrangelo A. The SMAD pathway is required for hepcidin response during endoplasmic reticulum stress. Endocrinology (2016) 157:3935–45. doi: 10.1210/en.2016-1258
79. Kim JL, Lee DH, Na YJ, Kim BR, Jeong YA, Lee SI, et al. Iron chelator-induced apoptosis via the ER stress pathway in gastric cancer cells. Tumour Biol. (2016) 37:9709–19. doi: 10.1007/s13277-016-4878-4
80. Merlot AM, Shafie NH, Yu Y, Richardson V, Jansson PJ, Sahni S, et al. Mechanism of the induction of endoplasmic reticulum stress by the anti-cancer agent, di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT): activation of PERK/eIF2alpha, IRE1alpha, ATF6 and calmodulin kinase. Biochem Pharmacol. (2016) 109:27–47. doi: 10.1016/j.bcp.2016.04.001
81. van der Woude LL, Gorris MAJ, Halilovic A, Figdor CG, de Vries IJM. Migrating into the tumor: a roadmap for T cells. Trends Cancer (2017) 3:797–808. doi: 10.1016/j.trecan.2017.09.006
82. Haabeth OA, Tveita AA, Fauskanger M, Schjesvold F, Lorvik KB, Hofgaard PO, et al. How do CD4(+) T cells detect and eliminate tumor cells that either lack or express MHC class II molecules? Front Immunol. (2014) 5:174. doi: 10.3389/fimmu.2014.00174
83. Lee P, Peng H, Gelbart T, Wang L, Beutler E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc Natl Acad Sci USA. (2005) 102:1906–10. doi: 10.1073/pnas.0409808102
84. Tilg H, Ulmer H, Kaser A, Weiss G. Role of IL-10 for induction of anemia during inflammation. J Immunol. (2002) 169:2204–9. doi: 10.4049/jimmunol.169.4.2204
85. Weiss G, Bogdan C, Hentze MW. Pathways for the regulation of macrophage iron metabolism by the anti-inflammatory cytokines IL-4 and IL-13. J Immunol. (1997) 158:420–5.
86. Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol. (2014) 26:38–47. doi: 10.1016/j.smim.2014.01.008
87. Lim C, Savan R. The role of the IL-22/IL-22R1 axis in cancer. Cytokine Growth Factor Rev. (2014) 25:257–71. doi: 10.1016/j.cytogfr.2014.04.005
88. Weiss G, Fuchs D, Hausen A, Reibnegger G, Werner ER, Werner-Felmayer G, et al. Iron modulates interferon-gamma effects in the human myelomonocytic cell line THP-1. Exp Hematol. (1992) 20:605–10.
89. Libregts SF, Gutierrez L, de Bruin AM, Wensveen FM, Papadopoulos P, van Ijcken W, et al. Chronic IFN-gamma production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF-1/PU.1 axis. Blood (2011) 118:2578–88. doi: 10.1182/blood-2010-10-315218
90. Wang CQ, Udupa KB, Lipschitz DA. Interferon-gamma exerts its negative regulatory effect primarily on the earliest stages of murine erythroid progenitor cell development. J Cell Physiol. (1995) 162:134–8.
91. Ludwiczek S, Aigner E, Theurl I, Weiss G. Cytokine-mediated regulation of iron transport in human monocytic cells. Blood (2003) 101:4148–54. doi: 10.1182/blood-2002-08-2459
92. Shanmugam NK, Ellenbogen S, Trebicka E, Wang L, Mukhopadhyay S, Lacy-Hulbert A, et al. Tumor necrosis factor alpha inhibits expression of the iron regulating hormone hepcidin in murine models of innate colitis. PLoS ONE (2012) 7:e38136. doi: 10.1371/journal.pone.0038136
93. Weiss G. Modification of iron regulation by the inflammatory response. Best Pract Res Clin Haematol. (2005) 18:183–201. doi: 10.1016/j.beha.2004.09.001
94. Jelkmann W. Proinflammatory cytokines lowering erythropoietin production. J Interferon Cytokine Res. (1998) 18:555–9.
96. Means RT Jr., Krantz SB. Inhibition of human erythroid colony-forming units by gamma interferon can be corrected by recombinant human erythropoietin. Blood (1991) 78:2564–7.
97. de Castro J, Gascon P, Casas A, Munoz-Langa J, Alberola V, Cucala M, et al. Iron deficiency in patients with solid tumours: prevalence and management in clinical practice. Clin Transl Oncol. (2014) 16:823–8. doi: 10.1007/s12094-013-1155-5
98. Aapro M, Osterborg A, Gascon P, Ludwig H, Beguin Y. Prevalence and management of cancer-related anaemia, iron deficiency and the specific role of i.v. iron. Ann Oncol. (2018) 23:1954–62. doi: 10.1093/annonc/mds112
99. Sharma S, Nemeth E, Chen YH, Goodnough J, Huston A, Roodman GD, et al. Involvement of hepcidin in the anemia of multiple myeloma. Clin Cancer Res. (2008) 14:3262–7. doi: 10.1158/1078-0432.CCR-07-4153
100. Brines M, Cerami A. Erythropoietin-mediated tissue protection: reducing collateral damage from the primary injury response. J Intern Med. (2008) 264:405–32. doi: 10.1111/j.1365-2796.2008.02024.x
101. Dagnon K, Pacary E, Commo F, Antoine M, Bernaudin M, Bernaudin JF, et al. Expression of erythropoietin and erythropoietin receptor in non-small cell lung carcinomas. Clin Cancer Res. (2005) 11:993–9.
102. Volgger B, Kurz K, Zoschg K, Theurl I, Ciresa-Konig A, Marth C, et al. Importance of erythropoetin receptor expression in tumour tissue for the clinical course of breast cancer. Anticancer Res. (2010) 30:3721–6.
103. Elliott S, Swift S, Busse L, Scully S, Van G, Rossi J, et al. Epo receptors are not detectable in primary human tumor tissue samples. PLoS ONE (2013) 8:e68083. doi: 10.1371/journal.pone.0068083
104. Swift S, Ellison AR, Kassner P, McCaffery I, Rossi J, Sinclair AM, et al. Absence of functional EpoR expression in human tumor cell lines. Blood (2009) 115:4254–63. doi: 10.1182/blood-2009-10-248674
105. Dolznig H, Habermann B, Stangl K, Deiner EM, Moriggl R, Beug H, et al. Apoptosis protection by the Epo target Bcl-X(L) allows factor-independent differentiation of primary erythroblasts. Curr Biol. (2002) 12:1076–85. doi: 10.1016/S0960-9822(02)00930-2
106. Baltaziak M, Koda M, Wincewicz A, Sulkowska M, Kanczuga-Koda L, Sulkowski S. Relationships of P53 and Bak with EPO and EPOR in human colorectal cancer. Anticancer Res. (2009) 29:4151–6.
107. Weiss G, Houston T, Kastner S, Johrer K, Grunewald K, Brock JH. Regulation of cellular iron metabolism by erythropoietin: activation of iron-regulatory protein and upregulation of transferrin receptor expression in erythroid cells. Blood (1997) 89:680–7.
108. Nairz M, Sonnweber T, Schroll A, Theurl I, Weiss G. The pleiotropic effects of erythropoietin in infection and inflammation. Microbes Infect. (2011) 14:238–46. doi: 10.1016/j.micinf.2011.10.005
109. Kimata H, Yoshida A, Ishioka C, Masuda S, Sasaki R, Mikawa H. Human recombinant erythropoietin directly stimulates B cell immunoglobulin production and proliferation in serum-free medium. Clin Exp Immunol. (1991) 85:151–6.
110. Nairz M, Schroll A, Moschen AR, Sonnweber T, Theurl M, Theurl I, et al. Erythropoietin contrastingly affects bacterial infection and experimental colitis by inhibiting nuclear factor-kappaB-inducible immune pathways. Immunity (2011) 34:61–74. doi: 10.1016/j.immuni.2011.01.002
111. Katz O, Gil L, Lifshitz L, Prutchi-Sagiv S, Gassmann M, Mittelman M, et al. Erythropoietin enhances immune responses in mice. Eur J Immunol. (2007) 37:1584–93. doi: 10.1002/eji.200637025
112. Mittelman M, Neumann D, Peled A, Kanter P, Haran-Ghera N. Erythropoietin induces tumor regression and antitumor immune responses in murine myeloma models. Proc Natl Acad Sci USA. (2001) 98:5181–6. doi: 10.1073/pnas.081275298
113. Tzounakas VL, Seghatchian J, Grouzi E, Kokoris S, Antonelou MH. Red blood cell transfusion in surgical cancer patients: Targets, risks, mechanistic understanding and further therapeutic opportunities. Transfus Apher Sci. (2017) 56:291–304. doi: 10.1016/j.transci.2017.05.015
114. Goubran H, Sheridan D, Radosevic J, Burnouf T, Seghatchian J. Transfusion-related immunomodulation and cancer. Transfus Apher Sci. (2017) 56:336–40. doi: 10.1016/j.transci.2017.05.019
115. Edgren G, Hjalgrim H, Reilly M, Tran TN, Rostgaard K, Shanwell A, et al. Risk of cancer after blood transfusion from donors with subclinical cancer: a retrospective cohort study. Lancet (2007) 369:1724–30. doi: 10.1016/S0140-6736(07)60779-X
116. Adamzik M, Hamburger T, Petrat F, Peters J, de Groot H, Hartmann M. Free hemoglobin concentration in severe sepsis: methods of measurement and prediction of outcome. Crit Care (2012) 16:R125. doi: 10.1186/cc11425
117. Omar HR, Mirsaeidi M, Socias S, Sprenker C, Caldeira C, Camporesi EM, et al. Plasma free hemoglobin is an independent predictor of mortality among patients on extracorporeal membrane oxygenation support. PLoS ONE (2015) 10:e0124034. doi: 10.1371/journal.pone.0124034
118. Larsen R, Gouveia Z, Soares MP, Gozzelino R. Heme cytotoxicity and the pathogenesis of immune-mediated inflammatory diseases. Front Pharmacol. (2012) 3:77. doi: 10.3389/fphar.2012.00077
119. Stark MJ, Keir AK, Andersen CC. Does non-transferrin bound iron contribute to transfusion related immune-modulation in preterms? Arch Dis Child Fetal Neonatal Ed. (2012) 98:F424–9. doi: 10.1136/archdischild-2012-303353
120. Bernard A, Meier C, Ward M, Browning T, Montgomery A, Kasten M, et al. Packed red blood cells suppress T-cell proliferation through a process involving cell-cell contact. J Trauma (2010) 69:320–9. doi: 10.1097/TA.0b013e3181e401f0
121. Yi T, Li J, Chen H, Wu J, An J, Xu Y, et al. Splenic dendritic cells survey red blood cells for missing self-CD47 to trigger adaptive immune responses. Immunity (2015) 43:764–75. doi: 10.1016/j.immuni.2015.08.021
122. Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med. (2016) 22:945–51. doi: 10.1038/nm.4146
123. Knutson MD, Oukka M, Koss LM, Aydemir F, Wessling-Resnick M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci USA. (2005) 102:1324–8. doi: 10.1073/pnas.0409409102
124. Weiss G. Anemia of chronic disorders: new diagnostic tools and new treatment strategies. Semin Hematol. (2015) 52:313–20. doi: 10.1053/j.seminhematol.2015.07.004
125. Goodnough LT, Nemeth E, Ganz T. Detection, evaluation, and management of iron-restricted erythropoiesis. Blood (2010) 116:4754–61. doi: 10.1182/blood-2010-05-286260
126. van Santen S, de Mast Q, Oosting JD, van Ede A, Swinkels DW, van der Ven AJ. Hematologic parameters predicting a response to oral iron therapy in chronic inflammation. Haematologica (2014) 99:e171–3. doi: 10.3324/haematol.2014.106799
127. Aapro M, Beguin Y, Bokemeyer C, Dicato M, Gascon P, Glaspy J, et al. Management of anaemia and iron deficiency in patients with cancer: ESMO clinical practice guidelines. Ann Oncol. (2018) 29(Supplement_4):iv96–110. doi: 10.1093/annonc/mdx758
128. Steinmetz HT. The role of intravenous iron in the treatment of anemia in cancer patients. Ther Adv Hematol. (2012) 3:177–91. doi: 10.1177/2040620712440071
129. Theurl M, Nairz M, Schroll A, Sonnweber T, Asshoff M, Haschka D, et al. Hepcidin as a predictive factor and therapeutic target in erythropoiesis-stimulating agent treatment for anemia of chronic disease in rats. Haematologica (2013) 99:1516–24. doi: 10.3324/haematol.2013.099481
130. Scheers NM, Pereira DIA, Faria N, Powell JJ. Ferric citrate and ferric EDTA but not ferrous sulfate drive amphiregulin-mediated activation of the MAP kinase ERK in gut epithelial cancer cells. Oncotarget (2018) 9:17066–77. doi: 10.18632/oncotarget.24899
131. Koskenkorva-Frank TS, Weiss G, Koppenol WH, Burckhardt S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic Biol Med. (2013) 65:1174–94. doi: 10.1016/j.freeradbiomed.2013.09.001
132. Lebrun F, Klastersky J, Levacq D, Wissam Y, Paesmans M. Intravenous iron therapy for anemic cancer patients: a review of recently published clinical studies. Support Care Cancer (2017) 25:2313–9. doi: 10.1007/s00520-017-3672-1
133. Gemici C, Yetmen O, Yaprak G, Ozden S, Tepetam H, Ozyurt H, et al. Is there any role of intravenous iron for the treatment of anemia in cancer? BMC Cancer (2016) 16:661. doi: 10.1186/s12885-016-2686-2
134. Park S, Jung CW, Kim K, Kim SJ, Kim WS, Jang JH. Iron deficient erythropoiesis might play key role in development of anemia in cancer patients. Oncotarget (2015) 6:42803–12. doi: 10.18632/oncotarget.5658
135. Dai J, Keller J, Zhang J, Lu Y, Yao Z, Keller ET. Bone morphogenetic protein-6 promotes osteoblastic prostate cancer bone metastases through a dual mechanism. Cancer Res. (2005) 65:8274–85. doi: 10.1158/0008-5472.CAN-05-1891
136. Hu F, Meng X, Tong Q, Liang L, Xiang R, Zhu T, et al. BMP-6 inhibits cell proliferation by targeting microRNA-192 in breast cancer. Biochim Biophys Acta (2013) 1832:2379–90. doi: 10.1016/j.bbadis.2013.08.011
137. Kwon SJ, Lee GT, Lee JH, Iwakura Y, Kim WJ, Kim IY. Mechanism of pro-tumorigenic effect of BMP-6: neovascularization involving tumor-associated macrophages and IL-1a. Prostate (2014) 74:121–33. doi: 10.1002/pros.22734
138. Poli M, Girelli D, Campostrini N, Maccarinelli F, Finazzi D, Luscieti S, et al. Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood (2010) 117:997–1004. doi: 10.1182/blood-2010-06-289082
139. Song SN, Tomosugi N, Kawabata H, Ishikawa T, Nishikawa T, Yoshizaki K. Down-regulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multicentric Castleman disease. Blood (2010) 116:3627–34. doi: 10.1182/blood-2010-03-271791
140. Hashizume M, Uchiyama Y, Horai N, Tomosugi N, Mihara M. Tocilizumab, a humanized anti-interleukin-6 receptor antibody, improved anemia in monkey arthritis by suppressing IL-6-induced hepcidin production. Rheumatol Int. (2010) 30:917–23. doi: 10.1007/s00296-009-1075-4
141. Rubbert-Roth A, Sebba A, Brockwell L, Kelman A, Porter-Brown B, Pulley J, et al. Malignancy rates in patients with rheumatoid arthritis treated with tocilizumab. RMD Open (2015) 2:e000213. doi: 10.1136/rmdopen-2015-000213
142. Jayatilaka H, Tyle P, Chen JJ, Kwak M, Ju J, Kim HJ, Lee JSH, et al. Synergistic IL-6 and IL-8 paracrine signalling pathway infers a strategy to inhibit tumour cell migration. Nat Commun. (2017) 8:15584. doi: 10.1038/ncomms15584
143. Vadhan-Raj S, Abonour R, Goldman JW, Smith DA, Slapak CA, Ilaria RL Jr., et al. A first-in-human phase 1 study of a hepcidin monoclonal antibody, LY2787106, in cancer-associated anemia. J Hematol Oncol. (2017) 10:73. doi: 10.1186/s13045-017-0427-x
144. Wermke M, Eckoldt J, Gotze KS, Klein SA, Bug G, de Wreede LC, et al. Enhanced labile plasma iron and outcome in acute myeloid leukaemia and myelodysplastic syndrome after allogeneic haemopoietic cell transplantation (ALLIVE): a prospective, multicentre, observational trial. Lancet Haematol. (2018) 5:e201–10. doi: 10.1016/S2352-3026(18)30036-X
145. Esposito BP, Breuer W, Sirankapracha P, Pootrakul P, Hershko C, Cabantchik ZI. Labile plasma iron in iron overload: redox activity and susceptibility to chelation. Blood (2003) 102:2670–7. doi: 10.1182/blood-2003-03-0807
146. Jian J, Yang Q, Huang X. Src regulates Tyr(20) phosphorylation of transferrin receptor-1 and potentiates breast cancer cell survival. J Biol Chem. (2011) 286:35708–15. doi: 10.1074/jbc.M111.271585
147. Chan KT, Choi MY, Lai KK, Tan W, Tung LN, Lam HY, et al. Overexpression of transferrin receptor CD71 and its tumorigenic properties in esophageal squamous cell carcinoma. Oncol Rep. (2014) 31:1296–304. doi: 10.3892/or.2014.2981
148. Cabral Filho PE, Cardoso AL, Pereira MI, Ramos AP, Hallwass F, Castro MM, et al. CdTe quantum dots as fluorescent probes to study transferrin receptors in glioblastoma cells. Biochim Biophys Acta (2016) 1860(1 Pt A):28–35. doi: 10.1016/j.bbagen.2015.09.021
149. Tao J, Liu YQ, Li Y, Peng JL, Li L, Liu J, et al. Hypoxia: dual effect on the expression of transferrin receptor in human melanoma A375 cell line. Exp Dermatol. (2007) 16:899–904. doi: 10.1111/j.1600-0625.2007.00601.x
150. Shimosaki S, Nakahata S, Ichikawa T, Kitanaka A, Kameda T, Hidaka T, et al. Development of a complete human IgG monoclonal antibody to transferrin receptor 1 targeted for adult T-cell leukemia/lymphoma. Biochem Biophys Res Commun. (2017) 485:144–51. doi: 10.1016/j.bbrc.2017.02.039
151. Rodriguez JA, Luria-Perez R, Lopez-Valdes HE, Casero D, Daniels TR, Patel S, et al. Lethal iron deprivation induced by non-neutralizing antibodies targeting transferrin receptor 1 in malignant B cells. Leuk Lymphoma (2011) 52:2169–78. doi: 10.3109/10428194.2011.596964
152. Mazurier J, Spik G. Comparative study of the iron-binding properties of human transferrins. I. Complete and sequential iron saturation and desaturation of the lactotransferrin. Biochim Biophys Acta (1980) 629:399–408.
153. Raje CI, Kumar S, Harle A, Nanda JS, Raje M. The macrophage cell surface glyceraldehyde-3-phosphate dehydrogenase is a novel transferrin receptor. J Biol Chem. (2007) 282:3252–61. doi: 10.1074/jbc.M608328200
154. Rawat P, Kumar S, Sheokand N, Raje CI, Raje M. The multifunctional glycolytic protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a novel macrophage lactoferrin receptor. Biochem Cell Biol. (2012) 90:329–38. doi: 10.1139/o11-058
155. Chauhan AS, Rawat P, Malhotra H, Sheokand N, Kumar M, Patidar A, et al. Secreted multifunctional Glyceraldehyde-3-phosphate dehydrogenase sequesters lactoferrin and iron into cells via a non-canonical pathway. Sci Rep. (2015) 5:18465. doi: 10.1038/srep18465
156. Duarte DC, Nicolau A, Teixeira JA, Rodrigues LR. The effect of bovine milk lactoferrin on human breast cancer cell lines. J Dairy Sci. (2011) 94:66–76. doi: 10.3168/jds.2010-3629
157. Xu XX, Jiang HR, Li HB, Zhang TN, Zhou Q, Liu N. Apoptosis of stomach cancer cell SGC-7901 and regulation of Akt signaling way induced by bovine lactoferrin. J Dairy Sci. (2010) 93:2344–50. doi: 10.3168/jds.2009-2926
158. Gibbons JA, Kanwar RK, Kanwar JR. Lactoferrin and cancer in different cancer models. Front Biosci (Schol Ed). (2011) 3:1080–8.
159. Ye Q, Zheng Y, Fan S, Qin Z, Li N, Tang A, et al. Lactoferrin deficiency promotes colitis-associated colorectal dysplasia in mice. PLoS ONE (2014) 9:e103298. doi: 10.1371/journal.pone.0103298
160. Kruzel ML, Zimecki M, Actor JK. Lactoferrin in a context of inflammation-induced pathology. Front Immunol. (2017) 8:1438. doi: 10.3389/fimmu.2017.01438
161. Sherman MP, Sherman J, Arcinue R, Niklas V. Randomized control trial of human recombinant lactoferrin: a substudy reveals effects on the fecal microbiome of very low birth weight infants. J Pediatr. (2016) 173 Suppl.:S37–42. doi: 10.1016/j.jpeds.2016.02.074
162. Shankaranarayanan JS, Kanwar JR, Al-Juhaishi AJ, Kanwar RK. Doxorubicin conjugated to immunomodulatory anticancer lactoferrin displays improved cytotoxicity overcoming prostate cancer chemo resistance and inhibits tumour development in TRAMP mice. Sci Rep. (2016) 6:32062. doi: 10.1038/srep32062
163. Cohen LA, Gutierrez L, Weiss A, Leichtmann-Bardoogo Y, Zhang DL, Crooks DR, et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood (2009) 116:1574–84. doi: 10.1182/blood-2009-11-253815
164. Chen TT, Li L, Chung DH, Allen CD, Torti SV, Torti FM, et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J Exp Med. (2005) 202:955–65. doi: 10.1084/jem.20042433
165. Han J, Seaman WE, Di X, Wang W, Willingham M, Torti FM, et al. Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells. PLoS ONE (2011) 6:e23800. doi: 10.1371/journal.pone.0023800
166. Li JY, Paragas N, Ned RM, Qiu A, Viltard M, Leete T, et al. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev Cell (2009) 16:35–46. doi: 10.1016/j.devcel.2008.12.002
167. You K, Su F, Liu L, Lv X, Zhang J, Zhang Y, et al. SCARA5 plays a critical role in the progression and metastasis of breast cancer by inactivating the ERK1/2, STAT3, and AKT signaling pathways. Mol Cell Biochem. (2017) 435:47–58. doi: 10.1007/s11010-017-3055-4
168. Alkhateeb AA, Leitzel K, Ali SM, Campbell-Baird C, Evans M, Fuchs EM, et al. Elevation in inflammatory serum biomarkers predicts response to trastuzumab-containing therapy. PLoS ONE (2012) 7:e51379. doi: 10.1371/journal.pone.0051379
169. Sucher R, Schroecksnadel K, Weiss G, Margreiter R, Fuchs D, Brandacher G. Neopterin, a prognostic marker in human malignancies. Cancer Lett. (2010) 287:13–22. doi: 10.1016/j.canlet.2009.05.008
170. Behnsen J, Raffatellu M. Siderophores: more than stealing iron. MBio (2016) 7:e01906–16. doi: 10.1128/mBio.01906-16
171. Smith KD. Iron metabolism at the host pathogen interface: lipocalin 2 and the pathogen-associated iroA gene cluster. Int J Biochem Cell Biol. (2007) 39:1776–80. doi: 10.1016/j.biocel.2007.07.003
172. Schrettl M, Bignell E, Kragl C, Sabiha Y, Loss O, Eisendle M, et al. Distinct roles for intra- and extracellular siderophores during Aspergillus fumigatus infection. PLoS Pathog. (2007) 3:1195–207. doi: 10.1371/journal.ppat.0030128
173. Bao G, Clifton M, Hoette TM, Mori K, Deng SX, Qiu A, et al. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat Chem Biol. (2010) 6:602–9. doi: 10.1038/nchembio.402
174. Devireddy LR, Hart DO, Goetz DH, Green MR. A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell (2010) 141:1006–17. doi: 10.1016/j.cell.2010.04.040
175. Iannetti A, Pacifico F, Acquaviva R, Lavorgna A, Crescenzi E, Vascotto C, et al. The neutrophil gelatinase-associated lipocalin (NGAL), a NF-kappaB-regulated gene, is a survival factor for thyroid neoplastic cells. Proc Natl Acad Sci USA. (2008) 105:14058–63. doi: 10.1073/pnas.0710846105
176. Hvidberg V, Jacobsen C, Strong RK, Cowland JB, Moestrup SK, Borregaard N. The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase-associated lipocalin with high affinity and mediates its cellular uptake. FEBS Lett. (2005) 579:773–7. doi: 10.1016/j.febslet.2004.12.031
177. Nairz M, Theurl I, Schroll A, Theurl M, Fritsche G, Lindner E, et al. Absence of functional Hfe protects mice from invasive Salmonella enterica serovar Typhimurium infection via induction of lipocalin-2. Blood (2009) 114:3642–51. doi: 10.1182/blood-2009-05-223354
178. Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell (2005) 123:1293–305. doi: 10.1016/j.cell.2005.10.027
179. Rodvold JJ, Mahadevan NR, Zanetti M. Lipocalin 2 in cancer: when good immunity goes bad. Cancer Lett. (2011) 316:132–8. doi: 10.1016/j.canlet.2011.11.002
180. Yang J, Bielenberg DR, Rodig SJ, Doiron R, Clifton MC, Kung AL, et al. Lipocalin 2 promotes breast cancer progression. Proc Natl Acad Sci USA. (2009) 106:3913–8. doi: 10.1073/pnas.0810617106
181. Reilly PT, Teo WL, Low MJ, Amoyo-Brion AA, Dominguez-Brauer C, Elia AJ, et al. Lipocalin 2 performs contrasting, location-dependent roles in APCmin tumor initiation and progression. Oncogene (2012) 32:1233–9. doi: 10.1038/onc.2012.159
182. Cramer EP, Glenthoj A, Hager M, Juncker-Jensen A, Engelholm LH, Santoni-Rugiu E, et al. No effect of NGAL/lipocalin-2 on aggressiveness of cancer in the MMTV-PyMT/FVB/N mouse model for breast cancer. PLoS ONE (2012) 7:e39646. doi: 10.1371/journal.pone.0039646
183. Kim SL, Lee ST, Min IS, Park YR, Lee JH, Kim DG, et al. Lipocalin 2 negatively regulates cell proliferation and epithelial to mesenchymal transition through changing metabolic gene expression in colorectal cancer. Cancer Sci. (2017) 108:2176–86. doi: 10.1111/cas.13389
184. Chakraborty S, Kaur S, Guha S, Batra SK. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim Biophys Acta (2012) 1826:129–69. doi: 10.1016/j.bbcan.2012.03.008
185. Du ZP, Wu BL, Xie YM, Zhang YL, Liao LD, Zhou F, et al. Lipocalin 2 promotes the migration and invasion of esophageal squamous cell carcinoma cells through a novel positive feedback loop. Biochim Biophys Acta (2015) 1853(10 Pt A):2240–50. doi: 10.1016/j.bbamcr.2015.07.007
186. Kumandan S, Mahadevan NR, Chiu K, DeLaney A, Zanetti M. Activation of the unfolded protein response bypasses trastuzumab-mediated inhibition of the PI-3K pathway. Cancer Lett. (2012) 329:236–42. doi: 10.1016/j.canlet.2012.11.014
187. Chappell WH, Candido S, Abrams SL, Russo S, Ove R, Martelli AM, et al. Roles of p53, NF-kappaB and the androgen receptor in controlling NGAL expression in prostate cancer cell lines. Adv Biol Regul. (2018) 69:43–62. doi: 10.1016/j.jbior.2018.05.002
188. Karlsen JR, Borregaard N, Cowland JB. Induction of neutrophil gelatinase-associated lipocalin expression by co-stimulation with interleukin-17 and tumor necrosis factor-alpha is controlled by IkappaB-zeta but neither by C/EBP-beta nor C/EBP-delta. J Biol Chem. (2010) 285:14088–100. doi: 10.1074/jbc.M109.017129
189. Cowland JB, Muta T, Borregaard N. IL-1beta-specific up-regulation of neutrophil gelatinase-associated lipocalin is controlled by IkappaB-zeta. J Immunol. (2006) 176:5559–66. doi: 10.4049/jimmunol.176.9.5559
190. Singh V, Yeoh BS, Xiao X, Kumar M, Bachman M, Borregaard N, et al. Interplay between enterobactin, myeloperoxidase and lipocalin 2 regulates E. coli survival in the inflamed gut. Nat Commun. (2015) 6:7113. doi: 10.1038/ncomms8113
191. Schroll A, Eller K, Feistritzer C, Nairz M, Sonnweber T, Moser PA, et al. Lipocalin-2 ameliorates granulocyte functionality. Eur J Immunol. (2011) 42:3346–57. doi: 10.1002/eji.201142351
192. Wieser V, Tymoszuk P, Adolph TE, Grander C, Grabherr F, Enrich B, et al. Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J Hepatol. (2015) 64:872–80. doi: 10.1016/j.jhep.2015.11.037
193. Sickinger S, Maier H, Konig S, Vallant N, Kofler M, Schumpp P, et al. Lipocalin-2 as mediator of chemokine expression and granulocyte infiltration during ischemia and reperfusion. Transpl Int. (2013) 26:761–9. doi: 10.1111/tri.12116
194. Liu Z, Petersen R, Devireddy L. Impaired neutrophil function in 24p3 null mice contributes to enhanced susceptibility to bacterial infections. J Immunol. (2013) 190:4692–706. doi: 10.4049/jimmunol.1202411
195. Li H, Feng D, Cai Y, Liu Y, Xu M, Xiang X, et al. Hepatocytes and neutrophils cooperatively suppress bacterial infection by differentially regulating lipocalin-2 and neutrophil extracellular traps. Hepatology (2018) 68:1604–20. doi: 10.1002/hep.29919
196. Yang J, McNeish B, Butterfield C, Moses MA. Lipocalin 2 is a novel regulator of angiogenesis in human breast cancer. FASEB J. (2013) 27:45–50. doi: 10.1096/fj.12-211730
197. Ikeda Y, Tajima S, Yoshida S, Yamano N, Kihira Y, Ishizawa K, et al. Deferoxamine promotes angiogenesis via the activation of vascular endothelial cell function. Atherosclerosis (2011) 215:339–47. doi: 10.1016/j.atherosclerosis.2011.01.009
198. Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, et al. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet. (2005) 37:1264–9. doi: 10.1038/ng1658
199. Lane DJ, Richardson DR. Chaperone turns gatekeeper: PCBP2 and DMT1 form an iron-transport pipeline. Biochem J. (2014) 462:e1–3. doi: 10.1042/BJ20140720
200. Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science (2008) 320:1207–10. doi: 10.1126/science.1157643
201. Wang W, Grier DD, Woo J, Ward M, Sui G, Torti SV, et al. Ferritin H is a novel marker of early erythroid precursors and macrophages. Histopathology (2013) 62:931–40. doi: 10.1111/his.12101
202. Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. (2014) 16:1069–79. doi: 10.1038/ncb3053
203. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature (2014) 509:105–9. doi: 10.1038/nature13148
204. Muckenthaler MU, Galy B, Hentze MW. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr. (2008) 28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521
205. Meyron-Holtz EG, Ghosh MC, Rouault TA. Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science (2004) 306:2087–90. doi: 10.1126/science.1103786
206. Galy B, Ferring-Appel D, Kaden S, Grone HJ, Hentze MW. Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell Metab. (2008) 7:79–85. doi: 10.1016/j.cmet.2007.10.006
207. Nairz M, Ferring-Appel D, Casarrubea D, Sonnweber T, Viatte L, Schroll A, et al. Iron regulatory proteins mediate host resistance to salmonella infection. Cell Host Microbe (2015) 18:254–61. doi: 10.1016/j.chom.2015.06.017
208. Nairz M, Theurl I, Swirski FK, Weiss G. “Pumping iron”–how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflugers Arch. (2017) 469:397–418. doi: 10.1007/s00424-017-1944-8
209. Knutson M, Wessling-Resnick M. Iron metabolism in the reticuloendothelial system. Crit Rev Biochem Mol Biol. (2003) 38:61–88. doi: 10.1080/713609210
210. Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood (2005) 106:2572–9. doi: 10.1182/blood-2005-03-1185
211. Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature (2001) 409:198–201. doi: 10.1038/35051594
212. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. (2016) 26:1021–32. doi: 10.1038/cr.2016.95
213. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature (2015) 520:57–62. doi: 10.1038/nature14344
214. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature (2017) 547:453–7. doi: 10.1038/nature23007
215. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
216. Freitas I, Boncompagni E, Vaccarone R, Fenoglio C, Barni S, Baronzio GF. Iron accumulation in mammary tumor suggests a tug of war between tumor and host for the microelement. Anticancer Res. (2007) 27:3059–65.
217. Nimeus E, Malmstrom J, Johnsson A, Marko-Varga G, Ferno M. Proteomic analysis identifies candidate proteins associated with distant recurrences in breast cancer after adjuvant chemotherapy. J Pharm Biomed Anal. (2007) 43:1086–93. doi: 10.1016/j.jpba.2006.09.019
218. Jiang XP, Elliott RL, Head JF. Manipulation of iron transporter genes results in the suppression of human and mouse mammary adenocarcinomas. Anticancer Res. (2010) 30:759–65.
219. Zhang S, Chen Y, Guo W, Yuan L, Zhang D, Xu Y, et al. Disordered hepcidin-ferroportin signaling promotes breast cancer growth. Cell Signal. (2014) 26:2539–50. doi: 10.1016/j.cellsig.2014.07.029
220. Boult J, Roberts K, Brookes MJ, Hughes S, Bury JP, Cross SS, et al. Overexpression of cellular iron import proteins is associated with malignant progression of esophageal adenocarcinoma. Clin Cancer Res. (2008) 14:379–87. doi: 10.1158/1078-0432.CCR-07-1054
221. Brookes MJ, Hughes S, Turner FE, Reynolds G, Sharma N, Ismail T, et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut (2006) 55:1449–60. doi: 10.1136/gut.2006.094060
222. Wang W, Deng Z, Hatcher H, Miller LD, Di X, Tesfay L, et al. IRP2 regulates breast tumor growth. Cancer Res. (2014) 74:497–507. doi: 10.1158/0008-5472.CAN-13-1224
223. Ward DG, Roberts K, Brookes MJ, Joy H, Martin A, Ismail T, et al. Increased hepcidin expression in colorectal carcinogenesis. World J Gastroenterol. (2008) 14:1339–45. doi: 10.3748/wjg.14.1339
224. Pinnix ZK, Miller LD, Wang W, D'Agostino R Jr., Kute T, Willingham MC, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. (2010) 2:43ra56. doi: 10.1126/scisignal.3001127
225. Miller LD, Coffman LG, Chou JW, Black MA, Bergh J, D'Agostino R Jr., et al. An iron regulatory gene signature predicts outcome in breast cancer. Cancer Res. (2011) 71:6728–37. doi: 10.1158/0008-5472.CAN-11-1870
226. Gu Z, Wang H, Xia J, Yang Y, Jin Z, Xu H, et al. Decreased ferroportin promotes myeloma cell growth and osteoclast differentiation. Cancer Res. (2015) 75:2211–21. doi: 10.1158/0008-5472.CAN-14-3804
227. Richardson DR, Kalinowski DS, Lau S, Jansson PJ, Lovejoy DB. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim Biophys Acta (2009) 1790:702–17. doi: 10.1016/j.bbagen.2008.04.003
228. Jiang XP, Wang F, Yang DC, Elliott RL, Head JF. Induction of apoptosis by iron depletion in the human breast cancer MCF-7 cell line and the 13762NF rat mammary adenocarcinoma in vivo. Anticancer Res. (2002) 22:2685–92.
229. Abeysinghe RD, Greene BT, Haynes R, Willingham MC, Turner J, Planalp RP, et al. p53-independent apoptosis mediated by tachpyridine, an anti-cancer iron chelator. Carcinogenesis (2001) 22:1607–14. doi: 10.1093/carcin/22.10.1607
230. Wang F, Elliott RL, Head JF. Inhibitory effect of deferoxamine mesylate and low iron diet on the 13762NF rat mammary adenocarcinoma. Anticancer Res. (1999) 19:445–50.
231. Kulp KS, Green SL, Vulliet PR. Iron deprivation inhibits cyclin-dependent kinase activity and decreases cyclin D/CDK4 protein levels in asynchronous MDA-MB-453 human breast cancer cells. Exp Cell Res. (1996) 229:60–8. doi: 10.1006/excr.1996.0343
232. Hrabinski D, Hertz JL, Tantillo C, Berger V, Sherman AR. Iron repletion attenuates the protective effects of iron deficiency in DMBA-induced mammary tumors in rats. Nutr Cancer (1995) 24:133–42. doi: 10.1080/01635589509514401
233. Hann HW, Stahlhut MW, Blumberg BS. Iron nutrition and tumor growth: decreased tumor growth in iron-deficient mice. Cancer Res. (1988) 48:4168–70.
234. Zacharski LR, Chow BK, Howes PS, Shamayeva G, Baron JA, Dalman RL, et al. Decreased cancer risk after iron reduction in patients with peripheral arterial disease: results from a randomized trial. J Natl Cancer Inst. (2008) 100:996–1002. doi: 10.1093/jnci/djn209
235. Zacharski LR. Anticoagulation, ferrotoxicity and the future of translational lung cancer research. Transl Lung Cancer Res. (2016) 5:280–7. doi: 10.21037/tlcr.2016.05.06
236. Bauckman K, Haller E, Taran N, Rockfield S, Ruiz-Rivera A, Nanjundan M. Iron alters cell survival in a mitochondria-dependent pathway in ovarian cancer cells. Biochem J. (2015) 466:401–13. doi: 10.1042/BJ20140878
237. Jian J, Yang Q, Shao Y, Axelrod D, Smith J, Singh B, et al. A link between premenopausal iron deficiency and breast cancer malignancy. BMC Cancer (2013) 13:307. doi: 10.1186/1471-2407-13-307
238. Fryknas M, Zhang X, Bremberg U, Senkowski W, Olofsson MH, Brandt P, et al. Iron chelators target both proliferating and quiescent cancer cells. Sci Rep. (2016) 6:38343. doi: 10.1038/srep38343
239. Abbas K, Breton J, Drapier JC. The interplay between nitric oxide and peroxiredoxins. Immunobiology (2008) 213:815–22. doi: 10.1016/j.imbio.2008.07.029
240. Cabantchik ZI, Kakhlon O, Epsztejn S, Zanninelli G, Breuer W. Intracellular and extracellular labile iron pools. Adv Exp Med Biol. (2002) 509:55–75. doi: 10.1007/978-1-4615-0593-8_4
241. Stevens RG, Morris JE, Cordis GA, Anderson LE, Rosenberg DW, Sasser LB. Oxidative damage in colon and mammary tissue of the HFE-knockout mouse. Free Radic Biol Med. (2003) 34:1212–6. doi: 10.1016/S0891-5849(03)00072-8
242. Ribeiro ML, Priolli DG, Miranda DDC, Arçari DP, Pedrazzoli JJr., Martinez CA. Analysis of oxidative DNA damage in patients with colorectal cancer. Clin Colorectal Cancer (2008) 7:267–72. doi: 10.3816/CCC.2008.n.034
243. Ishikawa S, Tamaki S, Ohata M, Arihara K, Itoh M. Heme induces DNA damage and hyperproliferation of colonic epithelial cells via hydrogen peroxide produced by heme oxygenase: a possible mechanism of heme-induced colon cancer. Mol Nutr Food Res. (2010) 54:1182–91. doi: 10.1002/mnfr.200900348
244. Chua AC, Klopcic B, Lawrance IC, Olynyk JK, Trinder D. Iron: an emerging factor in colorectal carcinogenesis. World J Gastroenterol. (2010) 16:663–72. doi: 10.3748/wjg.v16.i6.663
245. Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell (2004) 119:529–42. doi: 10.1016/j.cell.2004.10.017
246. Rao VA, Klein SR, Agama KK, Toyoda E, Adachi N, Pommier Y, et al. The iron chelator Dp44mT causes DNA damage and selective inhibition of topoisomerase IIalpha in breast cancer cells. Cancer Res. (2009) 69:948–57. doi: 10.1158/0008-5472.CAN-08-1437
247. Le NT, Richardson DR. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim Biophys Acta (2002) 1603:31–46. doi: 10.1016/S0304-419X(02)00068-9
248. Dongiovanni P, Fracanzani AL, Cairo G, Megazzini CP, Gatti S, Rametta R, et al. Iron-dependent regulation of MDM2 influences p53 activity and hepatic carcinogenesis. Am J Pathol. (2010) 176:1006–17. doi: 10.2353/ajpath.2010.090249
249. Fu D, Richardson DR. Iron chelation and regulation of the cell cycle: 2 mechanisms of posttranscriptional regulation of the universal cyclin-dependent kinase inhibitor p21CIP1/WAF1 by iron depletion. Blood (2007) 110:752–61. doi: 10.1182/blood-2007-03-076737
250. Gao J, Richardson DR. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents, IV: the mechanisms involved in inhibiting cell-cycle progression. Blood (2001) 98:842–50. doi: 10.1182/blood.V98.3.842
251. Liang SX, Richardson DR. The effect of potent iron chelators on the regulation of p53: examination of the expression, localization and DNA-binding activity of p53 and the transactivation of WAF1. Carcinogenesis (2003) 24:1601–14. doi: 10.1093/carcin/bgg116
252. Deng R, Wang SM, Yin T, Ye TH, Shen GB, Li L, et al. Inhibition of tumor growth and alteration of associated macrophage cell type by an HO-1 inhibitor in breast carcinoma-bearing mice. Oncol Res. (2013) 20:473–82.
253. Xiong S, She H, Takeuchi H, Han B, Engelhardt JF, Barton CH, et al. Signaling role of intracellular iron in NF-kappaB activation. J Biol Chem. (2003) 278:17646–54. doi: 10.1074/jbc.M210905200
254. She H, Xiong S, Lin M, Zandi E, Giulivi C, Tsukamoto H. Iron activates NF-kappaB in Kupffer cells. Am J Physiol Gastrointest Liver Physiol. (2002) 283:G719–26. doi: 10.1152/ajpgi.00108.2002
255. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. (2011) 12:715–23. doi: 10.1038/ni.2060
256. Chua AC, Klopcic BR, Ho DS, Fu SK, Forrest CH, Croft KD, et al. Dietary iron enhances colonic inflammation and IL-6/IL-11-Stat3 signaling promoting colonic tumor development in mice. PLoS ONE (2013) 8:e78850. doi: 10.1371/journal.pone.0078850
257. Xue X, Ramakrishnan SK, Weisz K, Triner D, Xie L, Attili D, et al. Iron uptake via DMT1 integrates cell cycle with JAK-STAT3 signaling to promote colorectal tumorigenesis. Cell Metab. (2016) 24:447–61. doi: 10.1016/j.cmet.2016.07.015
258. Wang J, Yin D, Xie C, Zheng T, Liang Y, Hong X, et al. The iron chelator Dp44mT inhibits hepatocellular carcinoma metastasis via N-Myc downstream-regulated gene 2 (NDRG2)/gp130/STAT3 pathway. Oncotarget (2014) 5:8478–91. doi: 10.18632/oncotarget.2328
259. Coombs GS, Schmitt AA, Canning CA, Alok A, Low IC, Banerjee N, et al. Modulation of Wnt/beta-catenin signaling and proliferation by a ferrous iron chelator with therapeutic efficacy in genetically engineered mouse models of cancer. Oncogene (2011) 31:213–25. doi: 10.1038/onc.2011.228
260. Brookes MJ, Boult J, Roberts K, Cooper BT, Hotchin NA, Matthews G, et al. A role for iron in Wnt signalling. Oncogene (2008) 27:966–75. doi: 10.1038/sj.onc.1210711
261. Chen Z, Zhang D, Yue F, Zheng M, Kovacevic Z, Richardson DR. The iron chelators Dp44mT and DFO inhibit TGF-beta-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1). J Biol Chem. (2012) 287:17016–28. doi: 10.1074/jbc.M112.350470
262. Zhang KH, Tian HY, Gao X, Lei WW, Hu Y, Wang DM, et al. Ferritin heavy chain-mediated iron homeostasis and subsequent increased reactive oxygen species production are essential for epithelial-mesenchymal transition. Cancer Res. (2009) 69:5340–8. doi: 10.1158/0008-5472.CAN-09-0112
263. Salminen A, Kauppinen A, Kaarniranta K. 2-Oxoglutarate-dependent dioxygenases are sensors of energy metabolism, oxygen availability, and iron homeostasis: potential role in the regulation of aging process. Cell Mol Life Sci. (2015) 72:3897–914. doi: 10.1007/s00018-015-1978-z
264. Johansson C, Tumber A, Che K, Cain P, Nowak R, Gileadi C, et al. The roles of Jumonji-type oxygenases in human disease. Epigenomics (2014) 6:89–120. doi: 10.2217/epi.13.79
265. Franci G, Ciotta A, Altucci L. The Jumonji family: past, present and future of histone demethylases in cancer. Biomol Concepts (2014) 5:209–24. doi: 10.1515/bmc-2014-0010
266. Seok S, Kim YC, Byun S, Choi S, Xiao Z, Iwamori N, et al. Fasting-induced JMJD3 histone demethylase epigenetically activates mitochondrial fatty acid beta-oxidation. J Clin Invest. (2018) 128:3144–59. doi: 10.1172/JCI97736
267. Su Y, Yu QH, Wang XY, Yu LP, Wang ZF, Cao YC, et al. JMJD2A promotes the Warburg effect and nasopharyngeal carcinoma progression by transactivating LDHA expression. BMC Cancer (2017) 17:477. doi: 10.1186/s12885-017-3473-4
268. Wang HJ, Hsieh YJ, Cheng WC, Lin CP, Lin YS, Yang SF, et al. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1alpha-mediated glucose metabolism. Proc Natl Acad Sci USA. (2014) 111:279–84. doi: 10.1073/pnas.1311249111
269. Sanchez-Fernandez EM, Tarhonskaya H, Al-Qahtani K, Hopkinson RJ, McCullagh JS, Schofield CJ, et al. Investigations on the oxygen dependence of a 2-oxoglutarate histone demethylase. Biochem J. (2013) 449:491–6. doi: 10.1042/BJ20121155
270. Cascella B, Mirica LM. Kinetic analysis of iron-dependent histone demethylases: alpha-ketoglutarate substrate inhibition and potential relevance to the regulation of histone demethylation in cancer cells. Biochemistry (2012) 51:8699–701. doi: 10.1021/bi3012466
271. Wellmann S, Bettkober M, Zelmer A, Seeger K, Faigle M, Eltzschig HK, et al. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem Biophys Res Commun. (2008) 372:892–7. doi: 10.1016/j.bbrc.2008.05.150
272. Pogribny IP, Tryndyak VP, Pogribna M, Shpyleva S, Surratt G, Gamboa da Costa G, et al. Modulation of intracellular iron metabolism by iron chelation affects chromatin remodeling proteins and corresponding epigenetic modifications in breast cancer cells and increases their sensitivity to chemotherapeutic agents. Int J Oncol. (2013) 42:1822–32. doi: 10.3892/ijo.2013.1855
273. Hickok JR, Vasudevan D, Antholine WE, Thomas DD. Nitric oxide modifies global histone methylation by inhibiting Jumonji C domain-containing demethylases. J Biol Chem. (2013) 288:16004–15. doi: 10.1074/jbc.M112.432294
274. Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci USA. (2012) 109:E3367–76. doi: 10.1073/pnas.1217394109
275. Poulard C, Rambaud J, Lavergne E, Jacquemetton J, Renoir JM, Tredan O, et al. Role of JMJD6 in breast tumourigenesis. PLoS ONE (2015) 10:e0126181. doi: 10.1371/journal.pone.0126181
276. Lee YF, Miller LD, Chan XB, Black MA, Pang B, Ong CW, et al. JMJD6 is a driver of cellular proliferation and motility and a marker of poor prognosis in breast cancer. Breast Cancer Res. (2012) 14:R85. doi: 10.1186/bcr3200
277. Li LL, Xue AM, Li BX, Shen YW, Li YH, Luo CL, et al. JMJD2A contributes to breast cancer progression through transcriptional repression of the tumor suppressor ARHI. Breast Cancer Res. (2014) 16:R56. doi: 10.1186/bcr3667
278. Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. (2013) 73:2936–42. doi: 10.1158/0008-5472.CAN-12-4300
279. Duan L, Rai G, Roggero C, Zhang QJ, Wei Q, Ma SH, et al. KDM4/JMJD2 histone demethylase inhibitors block prostate tumor growth by suppressing the expression of AR and BMYB-regulated genes. Chem Biol. (2015) 22:1185–96. doi: 10.1016/j.chembiol.2015.08.007
280. Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. (2013) 4:2035. doi: 10.1038/ncomms3035
281. Yan N, Xu L, Wu X, Zhang L, Fei X, Cao Y, et al. GSKJ4, an H3K27me3 demethylase inhibitor, effectively suppresses the breast cancer stem cells. Exp Cell Res. (2017) 359:405–14. doi: 10.1016/j.yexcr.2017.08.024
282. Xun J, Wang D, Shen L, Gong J, Gao R, Du L, et al. JMJD3 suppresses stem cell-like characteristics in breast cancer cells by downregulation of Oct4 independently of its demethylase activity. Oncotarget (2017) 8:21918–29. doi: 10.18632/oncotarget.15747
283. Metzger E, Stepputtis SS, Strietz J, Preca BT, Urban S, Willmann D, et al. KDM4 inhibition targets breast cancer stem-like cells. Cancer Res. (2017) 77:5900–12. doi: 10.1158/0008-5472.CAN-17-1754
284. Sakaki H, Okada M, Kuramoto K, Takeda H, Watarai H, Suzuki S, et al. GSKJ4, A selective Jumonji H3K27 demethylase inhibitor, effectively targets ovarian cancer stem cells. Anticancer Res. (2015) 35:6607–14.
285. Ruger N, Roatsch M, Emmrich T, Franz H, Schule R, Jung M, et al. Tetrazolylhydrazides as selective fragment-like inhibitors of the JumonjiC-domain-containing histone demethylase KDM4A. ChemMedChem (2015) 10:1875–83. doi: 10.1002/cmdc.201500335
286. Chang KH, King ONF, Tumber A, Woon ECY, Heightman TD, McDonough MA, et al. Inhibition of histone demethylases by 4-carboxy-2,2′-bipyridyl compounds. ChemMedChem (2011) 6:759–64. doi: 10.1002/cmdc.201100026
287. Choudhry H, Harris AL. Advances in hypoxia-inducible factor biology. Cell Metab. (2017) 27:281–98. doi: 10.1016/j.cmet.2017.10.005
288. Chan MC, Ilott NE, Schodel J, Sims D, Tumber A, Lippl K, et al. Tuning the transcriptional response to hypoxia by inhibiting hypoxia-inducible factor (HIF) prolyl and asparaginyl hydroxylases. J Biol Chem. (2016) 291:20661–73. doi: 10.1074/jbc.M116.749291
289. Kimbro KS, Simons JW. Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr Relat Cancer (2006) 13:739–49. doi: 10.1677/erc.1.00728
290. Safran M, Kim WY, O'Connell F, Flippin L, Gunzler V, Horner JW, et al. Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: assessment of an oral agent that stimulates erythropoietin production. Proc Natl Acad Sci USA. (2006) 103:105–10. doi: 10.1073/pnas.0509459103
291. Tarhonskaya H, Hardy AP, Howe EA, Loik ND, Kramer HB, McCullagh JS, et al. Kinetic investigations of the role of factor inhibiting hypoxia-inducible factor (FIH) as an oxygen sensor. J Biol Chem. (2015) 290:19726–42. doi: 10.1074/jbc.M115.653014
292. Haase VH. Hypoxic regulation of erythropoiesis and iron metabolism. Am J Physiol Renal Physiol. (2010) 299:F1–13. doi: 10.1152/ajprenal.00174.2010
293. Pavlakis K, Messini I, Vrekoussis T, Yiannou P, Keramopoullos D, Louvrou N, et al. The assessment of angiogenesis and fibroblastic stromagenesis in hyperplastic and pre-invasive breast lesions. BMC Cancer (2008) 8:88. doi: 10.1186/1471-2407-8-88
294. Van Overmeire E, Laoui D, Keirsse J, Van Ginderachter JA, Sarukhan A. Mechanisms driving macrophage diversity and specialization in distinct tumor microenvironments and parallelisms with other tissues. Front Immunol. (2014) 5:127. doi: 10.3389/fimmu.2014.00127
295. Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer (2008) 8:705–13. doi: 10.1038/nrc2468
296. Zwaans BM, Lombard DB. Interplay between sirtuins, MYC and hypoxia-inducible factor in cancer-associated metabolic reprogramming. Dis Model Mech. (2014) 7:1023–32. doi: 10.1242/dmm.016287
297. Callapina M, Zhou J, Schnitzer S, Metzen E, Lohr C, Deitmer JW, et al. Nitric oxide reverses desferrioxamine- and hypoxia-evoked HIF-1alpha accumulation–implications for prolyl hydroxylase activity and iron. Exp Cell Res. (2005) 306:274–84. doi: 10.1016/j.yexcr.2005.02.018
298. Frise MC, Cheng HY, Nickol AH, Curtis MK, Pollard KA, Roberts DJ, et al. Clinical iron deficiency disturbs normal human responses to hypoxia. J Clin Invest. (2016) 126:2139–50. doi: 10.1172/JCI85715
299. Siegert I, Schodel J, Nairz M, Schatz V, Dettmer K, Dick C, et al. Ferritin-mediated iron sequestration stabilizes hypoxia-inducible factor-1alpha upon LPS activation in the presence of ample oxygen. Cell Rep. (2015) 13:2048–55. doi: 10.1016/j.celrep.2015.11.005
300. Ohara T, Noma K, Urano S, Watanabe S, Nishitani S, Tomono Y, et al. A novel synergistic effect of iron depletion on antiangiogenic cancer therapy. Int J Cancer (2013) 132:2705–13. doi: 10.1002/ijc.27943
301. Jian J, Yang Q, Dai J, Eckard J, Axelrod D, Smith J, et al. Effects of iron deficiency and iron overload on angiogenesis and oxidative stress-a potential dual role for iron in breast cancer. Free Radic Biol Med. (2010) 50:841–7. doi: 10.1016/j.freeradbiomed.2010.12.028
302. Gupta N, Wish JB. Hypoxia-inducible factor prolyl hydroxylase inhibitors: a potential new treatment for anemia in patients with CKD. Am J Kidney Dis. (2016) 69:815–26. doi: 10.1053/j.ajkd.2016.12.011
303. Seeley TW, Sternlicht MD, Klaus SJ, Neff TB, Liu DY. Induction of erythropoiesis by hypoxia-inducible factor prolyl hydroxylase inhibitors without promotion of tumor initiation, progression, or metastasis in a VEGF-sensitive model of spontaneous breast cancer. Hypoxia (Auckl). (2017) 5:1–9. doi: 10.2147/HP.S130526
304. Laoui D, Van Overmeire E, Movahedi K, Van den Bossche J, Schouppe E, Mommer C, et al. Mononuclear phagocyte heterogeneity in cancer: different subsets and activation states reaching out at the tumor site. Immunobiology (2011) 216:1192–02. doi: 10.1016/j.imbio.2011.06.007
305. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. (2002) 196:254–65. doi: 10.1002/path.1027
306. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE (2012) 7:e50946. doi: 10.1371/journal.pone.0050946
307. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. (2006) 66:605–12. doi: 10.1158/0008-5472.CAN-05-4005
308. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
309. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. (2009) 27:451–83. doi: 10.1146/annurev.immunol.021908.132532
310. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
311. Ghassabeh GH, De Baetselier P, Brys L, Noel W, Van Ginderachter JA, Meerschaut S, et al. Identification of a common gene signature for type II cytokine-associated myeloid cells elicited in vivo in different pathologic conditions. Blood (2006) 108:575–83. doi: 10.1182/blood-2005-04-1485
312. Koning N, van Eijk M, Pouwels W, Brouwer MS, Voehringer D, Huitinga I, et al. Expression of the inhibitory CD200 receptor is associated with alternative macrophage activation. J Innate Immun. (2010) 2:195–200. doi: 10.1159/000252803
313. Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. (2010) 70:5728–39. doi: 10.1158/0008-5472.CAN-09-4672
314. Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell (2013) 24:695–709. doi: 10.1016/j.ccr.2013.11.007
315. Torroella-Kouri M, Silvera R, Rodriguez D, Caso R, Shatry A, Opiela S, et al. Identification of a subpopulation of macrophages in mammary tumor-bearing mice that are neither M1 nor M2 and are less differentiated. Cancer Res. (2009) 69:4800–9. doi: 10.1158/0008-5472.CAN-08-3427
316. Biswas SK, Allavena P, Mantovani A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Semin Immunopathol. (2013) 35:585–600. doi: 10.1007/s00281-013-0367-7
317. Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol. (2008) 83:1136–44. doi: 10.1189/jlb.0907611
318. Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. (2007) 67:2649–56. doi: 10.1158/0008-5472.CAN-06-1823
319. Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. (2005) 65:5278–83. doi: 10.1158/0008-5472.CAN-04-1853
320. Kedrin D, Gligorijevic B, Wyckoff J, Verkhusha VV, Condeelis J, Segall JE, et al. Intravital imaging of metastatic behavior through a mammary imaging window. Nat Methods (2008) 5:1019–21. doi: 10.1038/nmeth.1269
321. Egeblad M, Ewald AJ, Askautrud HA, Truitt ML, Welm BE, Bainbridge E, et al. Visualizing stromal cell dynamics in different tumor microenvironments by spinning disk confocal microscopy. Dis Model Mech. (2008) 1:155–67. doi: 10.1242/dmm.000596
322. Lahmar Q, Keirsse J, Laoui D, Movahedi K, Van Overmeire E, Van Ginderachter JA. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. Biochim Biophys Acta (2016) 1865:23–34. doi: 10.1016/j.bbcan.2015.06.009
323. Cairo G, Recalcati S, Mantovani A, Locati M. Iron trafficking and metabolism in macrophages: contribution to the polarized phenotype. Trends Immunol. (2011) 32:241–7. doi: 10.1016/j.it.2011.03.007
324. Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol. (2010) 40:824–35. doi: 10.1002/eji.200939889
325. Leftin A, Ben-Chetrit N, Klemm F, Joyce JA, Koutcher JA. Iron imaging reveals tumor and metastasis macrophage hemosiderin deposits in breast cancer. PLoS ONE (2017) 12:e0184765. doi: 10.1371/journal.pone.0184765
326. Leftin A, Zhao H, Turkekul M, de Stanchina E, Manova K, Koutcher JA. Iron deposition is associated with differential macrophage infiltration and therapeutic response to iron chelation in prostate cancer. Sci Rep. (2017) 7:11632. doi: 10.1038/s41598-017-11899-2
327. Marques O, Porto G, Rema A, Faria F, Cruz Paula A, Gomez-Lazaro M, et al. Local iron homeostasis in the breast ductal carcinoma microenvironment. BMC Cancer (2016) 16:187. doi: 10.1186/s12885-016-2228-y
328. Jezequel P, Campion L, Spyratos F, Loussouarn D, Campone M, Guerin-Charbonnel C, et al. Validation of tumor-associated macrophage ferritin light chain as a prognostic biomarker in node-negative breast cancer tumors: a multicentric 2004 national PHRC study. Int J Cancer (2012) 131:426–37. doi: 10.1002/ijc.26397
329. Daldrup-Link HE, Golovko D, Ruffell B, Denardo DG, Castaneda R, Ansari C, et al. MRI of tumor-associated macrophages with clinically applicable iron oxide nanoparticles. Clin Cancer Res. (2011) 17:5695–704. doi: 10.1158/1078-0432.CCR-10-3420
330. Shih YY, Hsu YH, Duong TQ, Lin SS, Chow KP, Chang C. Longitudinal study of tumor-associated macrophages during tumor expansion using MRI. NMR Biomed. (2011) 24:1353–60. doi: 10.1002/nbm.1698
331. Serkova NJ. Nanoparticle-based magnetic resonance imaging on tumor-associated macrophages and inflammation. Front Immunol. (2017) 8:590. doi: 10.3389/fimmu.2017.00590
332. Costa da Silva M, Breckwoldt MO, Vinchi F, Correia MP, Stojanovic A, Thielmann CM, et al. Iron induces anti-tumor activity in tumor-associated macrophages. Front Immunol. (2017) 8:1479. doi: 10.3389/fimmu.2017.01479
333. Alkhateeb AA, Han B, Connor JR. Ferritin stimulates breast cancer cells through an iron-independent mechanism and is localized within tumor-associated macrophages. Breast Cancer Res Treat. (2013) 137:733–44. doi: 10.1007/s10549-012-2405-x
334. Tesfay L, Clausen KA, Kim JW, Hegde P, Wang X, Miller LD, et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res. (2015) 75:2254–63. doi: 10.1158/0008-5472.CAN-14-2465
335. Jung M, Weigert A, Mertens C, Rehwald C, Brune B. Iron handling in tumor-associated macrophages-is there a new role for lipocalin-2? Front Immunol. (2017) 8:1171. doi: 10.3389/fimmu.2017.01171
336. Oren B, Urosevic J, Mertens C, Mora J, Guiu M, Gomis RR, et al. Tumour stroma-derived lipocalin-2 promotes breast cancer metastasis. J Pathol. 239:274–85. doi: 10.1002/path.4724
337. Jung M, Mertens C, Bauer R, Rehwald C, Brune B. Lipocalin-2 and iron trafficking in the tumor microenvironment. Pharmacol Res. (2017) 120:146–56. doi: 10.1016/j.phrs.2017.03.018
338. Mertens C, Mora J, Oren B, Grein S, Winslow S, Scholich K, et al. Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Oncoimmunology (2017) 7:e1408751. doi: 10.1080/2162402X.2017.1408751
339. Duan X, He K, Li J, Cheng M, Song H, Liu J, et al. Tumor associated macrophages deliver iron to tumor cells via Lcn2. Int J Physiol Pathophysiol Pharmacol. (2018) 10:105–14.
340. Standish LJ, Sweet ES, Novack J, Wenner CA, Bridge C, Nelson A, et al. Breast cancer and the immune system. J Soc Integr Oncol. (2008) 6:158–68.
341. DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. (2007) 9:212. doi: 10.1186/bcr1746
342. Goto S, Sato M, Kaneko R, Itoh M, Sato S, Takeuchi S. Analysis of Th1 and Th2 cytokine production by peripheral blood mononuclear cells as a parameter of immunological dysfunction in advanced cancer patients. Cancer Immunol Immunother. (1999) 48:435–42.
343. Fan Z, Yu P, Wang Y, Wang Y, Fu ML, Liu W, et al. NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood (2006) 107:1342–51. doi: 10.1182/blood-2005-08-3485
344. Curigliano G. Immunity and autoimmunity: revising the concepts of response to breast cancer. Breast (2011) 20 Suppl. 3:S71–4. doi: 10.1016/S0960-9776(11)70298-3
345. De Sousa M. Lymphoid cell positioning: a new proposal for the mechanism of control of lymphoid cell migration. Symp Soc Exp Biol. (1978) 32:393–410.
346. Grady RW, Akbar AN, Giardina PJ, Hilgartner MW, de Sousa M. Disproportionate lymphoid cell subsets in thalassaemia major: the relative contributions of transfusion and splenectomy. Br J Haematol. (1985) 59:713–24.
347. Macedo MF, Porto G, Costa M, Vieira CP, Rocha B, Cruz E. Low numbers of CD8+ T lymphocytes in hereditary haemochromatosis are explained by a decrease of the most mature CD8+ effector memory T cells. Clin Exp Immunol. (2010) 159:363–71. doi: 10.1111/j.1365-2249.2009.04066.x
348. Reimao R, Porto G, de Sousa M. Stability of CD4/CD8 ratios in man: new correlation between CD4/CD8 profiles and iron overload in idiopathic haemochromatosis patients. C R Acad Sci III (1991) 313:481–7.
349. Porto G, Vicente C, Teixeira MA, Martins O, Cabeda JM, Lacerda R, et al. Relative impact of HLA phenotype and CD4-CD8 ratios on the clinical expression of hemochromatosis. Hepatology (1997) 25:397–402. doi: 10.1002/hep.510250223
350. Cunningham-Rundles S, Giardina PJ, Grady RW, Califano C, McKenzie P, De Sousa M. Effect of transfusional iron overload on immune response. J Infect Dis. (2000) 182 Suppl. 1:S115–21. doi: 10.1086/315919
351. Porto G, De Sousa M. Iron overload and immunity. World J Gastroenterol. (2007) 13:4707–15. doi: 10.3748/wjg.v13.i35.4707
352. Mencacci A, Cenci E, Boelaert JR, Bucci P, Mosci P, Fe d'Ostiani C, et al. Iron overload alters innate and T helper cell responses to Candida albicans in mice. J Infect Dis. (1997) 175:1467–76.
354. Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer (2013) 13:342–55. doi: 10.1038/nrc3495
355. Hery C, Ferlay J, Boniol M, Autier P. Changes in breast cancer incidence and mortality in middle-aged and elderly women in 28 countries with Caucasian majority populations. Ann Oncol. (2008) 19:1009–18. doi: 10.1093/annonc/mdm593
356. Messa E, Carturan S, Maffe C, Pautasso M, Bracco E, Roetto A, et al. Deferasirox is a powerful NF-kappaB inhibitor in myelodysplastic cells and in leukemia cell lines acting independently from cell iron deprivation by chelation and reactive oxygen species scavenging. Haematologica (2010) 95:1308–16. doi: 10.3324/haematol.2009.016824
357. Benhar M, Engelberg D, Levitzki A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. (2002) 3:420–5. doi: 10.1093/embo-reports/kvf094
358. Kowdley KV. Liver transplantation: an “in vivo” model for the pathophysiology of hemochromatosis? Hepatology (2004) 39:1495–8. doi: 10.1002/hep.20269
359. Galaris D, Skiada V, Barbouti A. Redox signaling and cancer: the role of “labile” iron. Cancer Lett. (2008) 266:21–9. doi: 10.1016/j.canlet.2008.02.038
360. Stevens RG, Jones DY, Micozzi MS, Taylor PR. Body iron stores and the risk of cancer. N Engl J Med. (1988) 319:1047–52.
361. Knekt P, Reunanen A, Takkunen H, Aromaa A, Heliovaara M, Hakulinen T. Body iron stores and risk of cancer. Int J Cancer (1994) 56:379–82.
362. Stevens RG, Graubard BI, Micozzi MS, Neriishi K, Blumberg BS. Moderate elevation of body iron level and increased risk of cancer occurrence and death. Int J Cancer (1994) 56:364–9.
363. Guner G, Kirkali G, Yenisey C, Tore IR. Cytosol and serum ferritin in breast carcinoma. Cancer Lett. (1992) 67:103–12.
364. Elliott RL, Elliott MC, Wang F, Head JF. Breast carcinoma and the role of iron metabolism. A cytochemical, tissue culture, and ultrastructural study. Ann N Y Acad Sci. (1993) 698:159–66.
365. Alkhateeb AA, Connor JR. The significance of ferritin in cancer: anti-oxidation, inflammation and tumorigenesis. Biochim Biophys Acta (2013) 1836:245–54. doi: 10.1016/j.bbcan.2013.07.002
366. Liu NQ, De Marchi T, Timmermans AM, Beekhof R, Trapman-Jansen AM, Foekens R, et al. Ferritin heavy chain in triple negative breast cancer: a favorable prognostic marker that relates to a cluster of differentiation 8 positive (CD8+) effector T-cell response. Mol Cell Proteomics (2014) 13:1814–27. doi: 10.1074/mcp.M113.037176
367. Giurgiovich AJ, Diwan BA, Olivero OA, Anderson LM, Rice JM, Poirier MC. Elevated mitochondrial cisplatin-DNA adduct levels in rat tissues after transplacental cisplatin exposure. Carcinogenesis (1997) 18:93–6.
368. Ruddell RG, Hoang-Le D, Barwood JM, Rutherford PS, Piva TJ, Watters DJ, et al. Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB-regulated signaling in rat hepatic stellate cells. Hepatology (2009) 49:887–900. doi: 10.1002/hep.22716
369. Dorner MH, Silverstone A, Nishiya K, de Sostoa A, Munn G, de Sousa M. Ferritin synthesis by human T lymphocytes. Science (1980) 209:1019–21.
370. Golding S, Young SP. Iron requirements of human lymphocytes: relative contributions of intra- and extra-cellular iron. Scand J Immunol. (1995) 41:229–36.
371. Iscove NN, Melchers F. Complete replacement of serum by albumin, transferrin, and soybean lipid in cultures of lipopolysaccharide-reactive B lymphocytes. J Exp Med. (1978) 147:923–33.
372. Jabara HH, Boyden SE, Chou J, Ramesh N, Massaad MJ, Benson H, et al. A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat Genet. (2016) 48:74–8. doi: 10.1038/ng.3465
373. Oppenheimer SJ. Iron and its relation to immunity and infectious disease. J Nutr. (2001) 131:616S−33S. doi: 10.1093/jn/131.2.616S
374. Wang Z, Yin W, Zhu L, Li J, Yao Y, Chen F, et al. Iron drives T helper cell pathogenicity by promoting RNA-binding protein PCBP1-mediated proinflammatory cytokine production. Immunity (2018) 49:80–92.e7. doi: 10.1016/j.immuni.2018.05.008
376. Arosa FA, Oliveira L, Porto G, da Silva BM, Kruijer W, Veltman J, et al. Anomalies of the CD8+ T cell pool in haemochromatosis: HLA-A3-linked expansions of CD8+CD28− T cells. Clin Exp Immunol. (1997) 107:548–54.
377. Cruz E, Melo G, Lacerda R, Almeida S, Porto G. The CD8+ T-lymphocyte profile as a modifier of iron overload in HFE hemochromatosis: an update of clinical and immunological data from 70 C282Y homozygous subjects. Blood Cells Mol Dis. (2006) 37:33–9. doi: 10.1016/j.bcmd.2006.04.004
378. Vanoaica L, Richman L, Jaworski M, Darshan D, Luther SA, Kuhn LC. Conditional deletion of ferritin h in mice reduces B and T lymphocyte populations. PLoS ONE (2014) 9:e89270. doi: 10.1371/journal.pone.0089270
379. Tilg H, Adolph TE, Gerner RR, Moschen AR. The intestinal microbiota in colorectal cancer. Cancer Cell (2018) 33:954–64. doi: 10.1016/j.ccell.2018.03.004
380. Niederreiter L, Adolph TE, Tilg H. Food, microbiome and colorectal cancer. Dig Liver Dis. (2018) 50:647–52. doi: 10.1016/j.dld.2018.03.030
381. Ijssennagger N, Derrien M, van Doorn GM, Rijnierse A, van den Bogert B, Muller M, et al. Dietary heme alters microbiota and mucosa of mouse colon without functional changes in host-microbe cross-talk. PLoS ONE (2012) 7:e49868. doi: 10.1371/journal.pone.0049868
382. Qiao L, Feng Y. Intakes of heme iron and zinc and colorectal cancer incidence: a meta-analysis of prospective studies. Cancer Causes Control (2013) 24:1175–83. doi: 10.1007/s10552-013-0197-x
383. Zimmermann MB, Chassard C, Rohner F, N'Goran EK, Nindjin C, Dostal A, et al. The effects of iron fortification on the gut microbiota in African children: a randomized controlled trial in Cote d'Ivoire. Am J Clin Nutr. (2010) 92:1406–15. doi: 10.3945/ajcn.110.004564
384. Paganini D, Zimmermann MB. The effects of iron fortification and supplementation on the gut microbiome and diarrhea in infants and children: a review. Am J Clin Nutr. (2017) 106(Suppl. 6):1688S−93S. doi: 10.3945/ajcn.117.156067
385. Jaeggi T, Kortman GA, Moretti D, Chassard C, Holding P, Dostal A, et al. Iron fortification adversely affects the gut microbiome, increases pathogen abundance and induces intestinal inflammation in Kenyan infants. Gut (2015) 64:731–42. doi: 10.1136/gutjnl-2014-307720
386. Mendes MCS, Paulino DS, Brambilla SR, Camargo JA, Persinoti GF, Carvalheira JBC. Microbiota modification by probiotic supplementation reduces colitis associated colon cancer in mice. World J Gastroenterol. (2018) 24:1995–2008. doi: 10.3748/wjg.v24.i18.1995
387. Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. (2007) 282:20221–9. doi: 10.1074/jbc.M610737200
388. Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, et al. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci USA. (2014) 111:E4110–8. doi: 10.1073/pnas.1405023111
389. Erdei J, Toth A, Balogh E, Nyakundi BB, Banyai E, Ryffel B, et al. Induction of NLRP3 inflammasome activation by heme in human endothelial cells. Oxid Med Cell Longev. (2018) 2018:4310816. doi: 10.1155/2018/4310816
390. Martins R, Maier J, Gorki AD, Huber KV, Sharif O, Starkl P, et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat Immunol. (2016) 17:1361–72. doi: 10.1038/ni.3590
391. Wagener BM, Hu PJ, Oh JY, Evans CA, Richter JR, Honavar J, et al. Role of heme in lung bacterial infection after trauma hemorrhage and stored red blood cell transfusion: a preclinical experimental study. PLoS Med. (2018) 15:e1002522. doi: 10.1371/journal.pmed.1002522
392. Onyiah JC, Sheikh SZ, Maharshak N, Otterbein LE, Plevy SE. Heme oxygenase-1 and carbon monoxide regulate intestinal homeostasis and mucosal immune responses to the enteric microbiota. Gut Microbes (2014) 5:220–4. doi: 10.4161/gmic.27290
393. Martin OC, Lin C, Naud N, Tache S, Raymond-Letron I, Corpet DE, et al. Antibiotic suppression of intestinal microbiota reduces heme-induced lipoperoxidation associated with colon carcinogenesis in rats. Nutr Cancer (2015) 67:119–25. doi: 10.1080/01635581.2015.976317
394. Ijssennagger N, Belzer C, Hooiveld GJ, Dekker J, van Mil SW, Muller M, et al. Gut microbiota facilitates dietary heme-induced epithelial hyperproliferation by opening the mucus barrier in colon. Proc Natl Acad Sci USA. (2015) 112:10038–43. doi: 10.1073/pnas.1507645112
395. Gamage SMK, Dissabandara L, Lam AK, Gopalan V. The role of heme iron molecules derived from red and processed meat in the pathogenesis of colorectal carcinoma. Crit Rev Oncol Hematol. (2018) 126:121–8. doi: 10.1016/j.critrevonc.2018.03.025
396. Hartmann H, Eltzschig HK, Wurz H, Hantke K, Rakin A, Yazdi AS, et al. Hypoxia-independent activation of HIF-1 by enterobacteriaceae and their siderophores. Gastroenterology (2008) 134:756–67. doi: 10.1053/j.gastro.2007.12.008
397. Shah YM, Ito S, Morimura K, Chen C, Yim SH, Haase VH, et al. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology (2008) 134:2036–48, 2048 e2031–3. doi: 10.1053/j.gastro.2008.03.009
398. Santoyo-Ramos P, Likhatcheva M, Garcia-Zepeda EA, Castaneda-Patlan MC, Robles-Flores M. Hypoxia-inducible factors modulate the stemness and malignancy of colon cancer cells by playing opposite roles in canonical Wnt signaling. PLoS ONE (2014) 9:e112580. doi: 10.1371/journal.pone.0112580
399. Xue X, Ramakrishnan SK, Shah YM. Activation of HIF-1alpha does not increase intestinal tumorigenesis. Am J Physiol Gastrointest Liver Physiol. (2014) 307:G187–95. doi: 10.1152/ajpgi.00112.2014
400. Xue X, Taylor M, Anderson E, Hao C, Qu A, Greenson JK, et al. Hypoxia-inducible factor-2alpha activation promotes colorectal cancer progression by dysregulating iron homeostasis. Cancer Res. (2012) 72:2285–93. doi: 10.1158/0008-5472.CAN-11-3836
401. Triner D, Xue X, Schwartz AJ, Jung I, Colacino JA, Shah YM. Epithelial hypoxia-inducible factor 2alpha facilitates the progression of colon tumors through recruiting neutrophils. Mol Cell Biol. (2017) 37:e00481–16. doi: 10.1128/MCB.00481-16
402. Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. (2009) 9:152–64. doi: 10.1016/j.cmet.2008.12.012
403. Shah YM, Xie L. Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterology (2013) 146:630–42. doi: 10.1053/j.gastro.2013.12.031
404. Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest. (2007) 117:1926–32. doi: 10.1172/JCI31370
405. Deriu E, Liu JZ, Pezeshki M, Edwards RA, Ochoa RJ, Contreras H, et al. Probiotic bacteria reduce salmonella typhimurium intestinal colonization by competing for iron. Cell Host Microbe (2013) 14:26–37. doi: 10.1016/j.chom.2013.06.007
406. Moschen AR, Gerner RR, Wang J, Klepsch V, Adolph TE, Reider SJ, et al. Lipocalin 2 protects from inflammation and tumorigenesis associated with gut microbiota alterations. Cell Host Microbe (2016) 19:455–69. doi: 10.1016/j.chom.2016.03.007
407. Ludwiczek S, Theurl I, Muckenthaler MU, Jakab M, Mair SM, Theurl M, et al. Ca2+ channel blockers reverse iron overload by a new mechanism via divalent metal transporter-1. Nat Med. (2007) 13:448–54. doi: 10.1038/nm1542
408. Stauder R, Valent P, Theurl I. Anemia at older age: etiologies, clinical implications and management. Blood (2018) 131:505–14. doi: 10.1182/blood-2017-07-746446
409. Mair SM, Nairz M, Bellmann-Weiler R, Muehlbacher T, Schroll A, Theurl I, et al. Nifedipine affects the course of Salmonella enterica serovar Typhimurium infection by modulating macrophage iron homeostasis. J Infect Dis. (2011) 204:685–94. doi: 10.1093/infdis/jir395
410. Gasche C, Lomer MC, Cavill I, Weiss G. Iron, anaemia, and inflammatory bowel diseases. Gut (2004) 53:1190–7. doi: 10.1136/gut.2003.035758
Keywords: iron, anemia of cancer, ACD, hepcidin, ferroptosis, TAM
Citation: Pfeifhofer-Obermair C, Tymoszuk P, Petzer V, Weiss G and Nairz M (2018) Iron in the Tumor Microenvironment—Connecting the Dots. Front. Oncol. 8:549. doi: 10.3389/fonc.2018.00549
Received: 12 September 2018; Accepted: 06 November 2018;
Published: 26 November 2018.
Edited by:
Lionel Apetoh, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Amorette Barber, Longwood University, United StatesAnna Karolina Kozlowska, City of Hope National Medical Center, United States
Copyright © 2018 Pfeifhofer-Obermair, Tymoszuk, Petzer, Weiss and Nairz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manfred Nairz, manfred.nairz@i-med.ac.at
†These authors have contributed equally to this work