 Teresa Y. Lee1
Teresa Y. Lee1 Ubaldo E. Martinez-Outschoorn1
Ubaldo E. Martinez-Outschoorn1 Russell J. Schilder1Christine H. Kim2Scott D. Richard2Norman G. Rosenblum2
Russell J. Schilder1Christine H. Kim2Scott D. Richard2Norman G. Rosenblum2 Jennifer M. Johnson1*
Jennifer M. Johnson1*- 1Department of Medical Oncology, Thomas Jefferson University, Philadelphia, PA, United States
- 2Department of Obstetrics and Gynecology, Thomas Jefferson University, Philadelphia, PA, United States
Endometrial cancer is the most common gynecologic malignancy in developed countries. Its increasing incidence is thought to be related in part to the rise of metabolic syndrome, which has been shown to be a risk factor for the development of hyperestrogenic and hyperinsulinemic states. This has consequently lead to an increase in other hormone-responsive cancers as well e.g., breast and ovarian cancer. The correlation between obesity, hyperglycemia, and endometrial cancer has highlighted the important role of metabolism in cancer establishment and persistence. Tumor-mediated reprogramming of the microenvironment and macroenvironment can range from induction of cytokines and growth factors to stimulation of surrounding stromal cells to produce energy-rich catabolites, fueling the growth, and survival of cancer cells. Such mechanisms raise the prospect of the metabolic microenvironment itself as a viable target for treatment of malignancies. Metformin is a biguanide drug that is a first-line treatment for type 2 diabetes that has beneficial effects on various markers of the metabolic syndrome. Many studies suggest that metformin shows potential as an adjuvant treatment for uterine and other cancers. Here, we review the evidence for metformin as a treatment for cancers of the endometrium. We discuss the available clinical data and the molecular mechanisms by which it may exert its effects, with a focus on how it may alter the tumor microenvironment. The pleiotropic effects of metformin on cellular energy production and usage as well as intercellular and hormone-based interactions make it a promising candidate for reprogramming of the cancer ecosystem. This, along with other treatments aimed at targeting tumor metabolic pathways, may lead to novel treatment strategies for endometrial cancer.
Endometrial Cancer
Cancer of the endometrium is the fifth most common malignancy in women worldwide, with 455,000 new cases diagnosed worldwide in 2015 (1). The incidence is rising, and is noted to be much higher in developed than developing countries (1, 2). The American Cancer Society estimates that 63,230 new cases will be diagnosed in the United States in 2018 (representing 7% of cancer diagnoses in women), with 11,350 predicted deaths (3). The majority of cases arise in the post-menopausal period, but up to 14% of cases occur in women age 40 or younger (4). The principal risk factor for development of endometrial cancer is exposure to endogenous and exogenous estrogens, which is influenced by factors such as age at menarche and menopause, parity, use of unopposed estrogen therapy or other hormonal therapies (e.g., tamoxifen), and a host of metabolic factors including obesity. 5–25% of cases are also associated with high risk germline mutations, particularly those affecting DNA mismatch repair pathways, leading to early onset of disease (5).
The most common classification system divides endometrial cancers into two subtypes (6). Type I cancers are low-grade, diploid, endometrioid, and hormone-receptor positive, carrying a better prognosis. They frequently display mutations in phosphate and tensin homolog (PTEN). Type II cancers (which include the serous, clear cell, mixed cell, undifferentiated, and carcinosarcoma histologies) are high grade, non-endometrioid, aneuploid, and hormone-negative, with higher rates of metastasis and worse prognosis. They tend to occur in older patients and are more likely to have mutations in the tumor suppressor p53. Type II cancers make up only 10% of endometrial cancers but account for almost 50% of relapses and deaths (7), suggesting a fundamental biologic difference between the two subsets.
Most cases are diagnosed at an early stage due to the early detection sign of abnormal bleeding. Standard treatment for apparent stage I endometrial cancer consists of surgical resection (primary hysterectomy with bilateral salpingo-oophorectomy with possible lymph node mapping). Disease that is confirmed to be uterine-confined with low risk features, can be treated with surgery only and has a >90% relapse-free survival rate at 5 years (8). Radiation decreases local relapse rates but does not affect relapse at distant sites or increase overall survival (9, 10). Trials of adjuvant chemotherapy alone with cyclophosphamide, doxorubicin, and cisplatin demonstrated no significant improvement in progression-free survival, overall survival, or relapse (11). Combined adjuvant chemoradiation has shown a slight increase in progression-free survival but not overall survival (11).
For metastatic or recurrent disease, management may include surgery or radiation (if localized to a single site); those with unresectable disease may sometimes receive primary chemotherapy followed by cytoreductive surgery. For disease not amenable to local therapy, a carboplatin-paclitaxel combination is increasingly used as a first-line alternative to the traditional cisplatin, paclitaxel, and doxorubicin (7). With respect to hormonal therapy in advanced disease, a 33% response rate was noted after alternating tamoxifen and medroxyprogesterone (11–13). In recurrent or metastatic disease, progestogens, tamoxifen alternated with megestrol, gonadotropin-releasing hormone analogues, selective estrogen receptor modulators, and aromatase inhibitors have been used with response rates ranging from 11 to 56% (13, 14). Ultimately, the response rates for recurrent advanced disease are low, and there are no standard second line therapies (13, 15).
This lack of effective treatment for advanced stage endometrial cancer has led to exploration of alternative therapeutic modalities. In particular, numerous studies have examined the effectiveness of targeted therapies acting on the phosphoinositide 3-Kinase (PI3K)/Protein kinase B (Akt)/mammalian target of rapamycin (mTOR) pathway, epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), and vascular endothelial growth factor (VEGF), reviewed elsewhere (11). The results of single-agent mTOR inhibitor treatment, or EGFR and HER2 inhibitors have been disappointing, with response rates of 0–12%. Anti-angiogenic drugs such as bevacizumab, sunitinib, brivanib, and lenvatinib have resulted in slightly higher objective response rates of 14–19%. Studies of additional targets, including fibroblast growth factor receptor (FGFR), luteinizing hormone releasing hormone (LHRH), poly ADP-ribose polymerase (PARP), and Programmed Death-1/Programmed Death Ligand-1 (PD-1/PD-L1) are underway. At this point, no targeted therapies have been approved. Therefore, further interest has been focused on other factors that could contribute to development and progression of endometrial cancer.
Associations With Metabolic Syndrome, Obesity, and Metabolism
Among the risk factors associated with endometrial cancer, metabolic syndrome (a constellation of obesity, hyperglycemia, hypertension, and hyperlipidemia) has attracted a large amount of interest in recent years. Multiple associative studies have suggested that the metabolic syndrome is a risk factor for development of many different types of cancers (16–18), including endometrial cancer (19–24). A meta-analysis of 6 studies from North America, Europe, and China estimated a relative risk (RR) for endometrial cancer of 1.89 in patients with metabolic syndrome (95% confidence interval [CI] 1.34–2.67, p = 0.001) (25). Another meta-analysis of 7 European cohorts reported a 56% increase in endometrial cancer risk per increase of one standard deviation in a composite metabolic risk score derived from sex- and cohort-specific means in body mass index (BMI), blood pressure, plasma cholesterol, triglycerides, and glucose (18). Apart from incidence, Ni and colleagues reported increased endometrial cancer stage, grade, vascular invasion, tumor size, and lymphatic metastasis in patients with metabolic syndrome, as well as decreased overall survival (26).
The individual components of the metabolic syndrome have also been studied in relation to endometrial cancer risk, but it is unknown if their contribution is additive or synergistic. In particular, obesity has been noted to be strongly associated with risk of endometrial cancer in several case-control studies and meta-analyses (21–25, 27, 28). Multiple measures of adiposity, including BMI, waist circumference, waist-to-hip-ratio, and hip circumference, have been found to be directly associated with endometrial cancer incidence. Increased waist circumference and BMI have also been shown to be significantly associated with increased risk of overall mortality from endometrial cancer (29, 30). Other studies have demonstrated positive albeit less robust association between endometrial cancer and the other components of the metabolic syndrome: hypertension (21–24), hyperlipidemia (21–24), and hyperglycemia or diabetes mellitus (19, 21–25, 31, 32). The association between diabetes and endometrial cancer appears to be partially confounded by co-existing overweight/obesity (33, 34). However, elevated risk of endometrial cancer in patients with diabetes has been reported even after adjustment for BMI, with one meta-analysis including 29 cohort studies reporting a summary relative risk of 1.89 [95% CI, 1.46–2.45, p < 0.001] (32). This study also noted a small increased risk of disease-specific mortality in diabetic patients with endometrial cancer (RR 1.32, 95% CI, 1.10–1.60; p = 0.003).
The major driver of increased risk of endometrial and other hormone-responsive cancers in obesity is thought to be the generation of a hyper-estrogenic state caused by the presence of the aromatase enzyme in adipose tissue (35). This enzyme catalyzes conversion of androgens to estrogens, making adipose tissue a key source of estrogens in post-menopausal women. In addition, adiposity has been associated with other factors that may drive tumorigenesis in general, including increased inflammation, depressed immune function, and chronic insulin resistance and hyperinsulinemia. Endometrial cancer patients have been shown to have increased markers of insulin resistance, including higher fasting insulin levels and elevated non-fasting and fasting C-peptide levels (36, 37). Supporting this link between abnormal glucose metabolism and cancer risk is the observation that better diabetic control is associated with decreased endometrial cancer risk (21). Ultimately, these data suggest that abnormal metabolism, including insulin resistance and hyperglycemia, may play a role in the development of endometrial cancer and thus represent a possible therapeutic target.
Metformin Repurposing and Epidemiologic Data From Endometrial Cancer
In recent years there has been growing interest in drug repurposing or repositioning, a process which seeks to identify new pharmacologic properties (e.g., anti-tumorigenic) of existing medications for use as primary or adjuvant treatments for other conditions (38, 39). These drugs are already well-studied in terms of tolerability and side effects, often inexpensive, and amenable to retrospective and associative studies as many patients are already taking them for other indications. The association between obesity, diabetes, hyperinsulinemia, and endometrial cancer has led to the hypothesis that medications which target glucose metabolism such as metformin may be effective in preventing or treating such malignancies. One drug that has received a significant amount of attention in this arena has been metformin [1,1-dimethylbiguanide] which is a first line oral antihyperglycemic agent used in the treatment of type 2 diabetes (40). Broadly, its effects include lowering of blood glucose concentrations, increasing insulin sensitization, and reducing plasma fasting insulin levels. Furthermore, unlike with some oral hypoglycemic medications and insulin, metformin users show a tendency toward sustained weight loss (41). The low toxicity of metformin makes it especially interesting as a potential adjunctive therapy, or even as monotherapy for patients with contraindications to chemotherapy or considerations such as the desire to preserve fertility.
Many investigators have sought to examine the effect of metformin exposure on the development of endometrial cancer (Table 1). Multiple epidemiologic studies have reported lower overall cancer incidence in metformin users, reviewed by several groups (47–53). Studies evaluating the relationship between metformin use and endometrial cancer incidence specifically have yielded more conflicting results. Three cohort studies and two case-control studies found no decrease in the risk of endometrial cancer in metformin users compared to nonusers (34, 42, 43, 45, 46). However, these studies show considerable heterogeneity in factors such as study size, indication for metformin use, and duration and method of measurement of metformin exposure (e.g., prescriptions vs. self-report). Notably, a large study of 478,921 Taiwanese women with diabetes showed a significantly decreased incidence of endometrial cancer (hazard ratio [HR] 0.675, 95% CI 0.614–0.742) in metformin users compared to never users (44). When stratified by duration of use or cumulative doses, the decrease in incidence demonstrated a dose-response effect. Additionally, a meta-analysis by Tang and colleagues found that metformin use was associated with a decreased risk of endometrial cancer incidence (RR 0.87, 95% CI 0.80–0.95) (54).
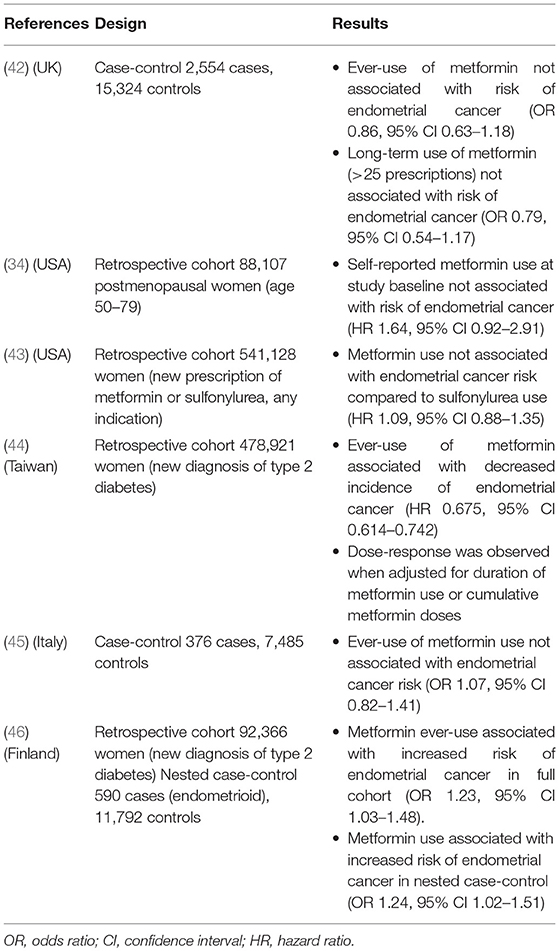
Table 1. Studies of metformin use and incidence of endometrial cancer.
Other associative studies have focused instead on the relationship between metformin exposure and endometrial cancer outcomes (Table 2). Metformin use in diabetic patients with endometrial cancer was associated with improved overall survival compared to those not taking metformin in two separate studies, including one involving patients with stage III–IV or recurrent endometrial cancer receiving chemotherapy (56, 59). The study by Ko also found improved recurrence-free survival in patients taking metformin. In contrast, some did not find any effect of metformin exposure on survival parameters (57, 58, 61). Still others have reported effects only on certain subgroups of patients. For example, Nevadunsky found increased survival for metformin users only among patients with non-endometrioid but not endometrioid forms of endometrial cancer (55), while Hall reported a significantly lower recurrence rate of only endometrioid endometrial cancers among metformin users (60). As with the incidence research, these studies are limited by heterogeneity and sample size. However, a 2017 meta-analysis including 6 of the above studies supports a higher overall survival rate in metformin-users with endometrial cancer compared to non-metformin users and non-diabetic patients (HR 0.82, 95% CI 0.70–0.95, I2 = 40%) (62). Finally, a meta-analysis of 28 studies reported that metformin use was associated with decreased all-cause mortality in patients with concurrent diabetes for several cancer types, including endometrial (RR 0.49, 95% CI 0.32, 0.73, p < 0.001) (63).
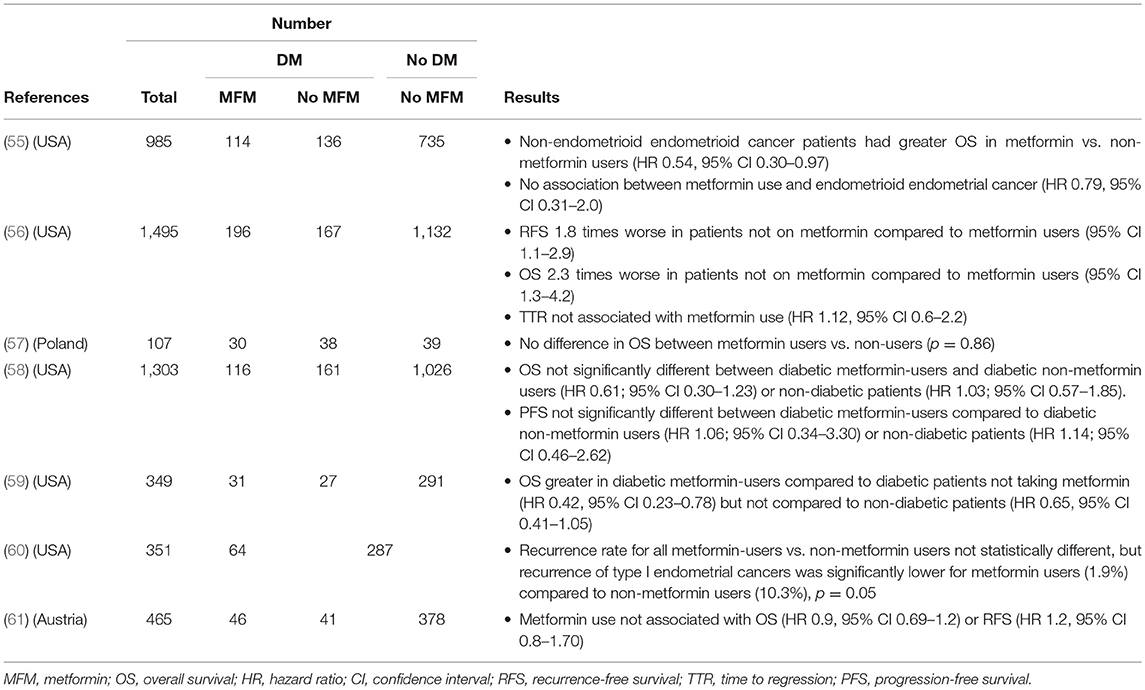
Table 2. Observational studies of metformin exposure in endometrial cancer.
Cellular and Molecular Mechanisms of Metformin Inhibition of Endometrial Cancer
Metabolic alterations in endometrial cancer have been described not only on a systemic but also on a cellular and molecular level. For example, Byrne and colleagues examined microarray data from women with women with type I endometrial cancer and demonstrated that tumor-derived endometrium showed enrichment of genes related to glycolysis and lipogenesis compared to normal endometrium (64). They also reported that multiple human endometrial cancer cell lines showed strong upregulation of the glucose transporter GLUT6 as well as activation of AKT compared to nonmalignant cells. In vitro metabolic profiling demonstrated that these changes were associated with upregulation of glycolysis, decreased glucose oxidation, and increased de novo lipogenesis. Finally, the authors demonstrated that endometrial cancer cell cultures experience cytotoxicity when exposed to a variety of inhibitors targeting metabolic pathways, including the glycolysis inhibitors 2-deoxy-D-glucose [2-DG] and 3-bromopyruvate (BrPA), the lipogenesis inhibitor 5-(tetradecyloxy)-2-furoic acid (TOFA), the fatty acid oxidation inhibitor etomixir, and the pleiotropic metabolic inhibitor metformin. Further support for the importance of glucose metabolism on endometrial cancer cell growth comes from Han and colleagues, who studied the growth of two endometrial cancer cell lines (ECC-1 and Ishikawa cells) under low, normal, or high glucose conditions (65). High glucose conditions (corresponding to physiologic hyperglycemia) led to increased cell proliferation, in vitro colony formation, and increased expression of the GLUT1 glucose transporter along with increased glucose uptake. High glucose also increased phosphorylation of lactate dehydrogenase A (LDHA) and decreased levels of pyruvate dehydrogenase (PDH), suggesting an increase in glycolytic activity. Conversely, low glucose conditions led to increased cell apoptosis, cell cycle arrest, decreased adhesion, and invasion. All of these data support the idea that the metabolic vulnerabilities of endometrial cancer may make it susceptible to therapies such as metformin.
Multiple studies have demonstrated the ability of metformin to inhibit proliferation of both type I and type II human endometrial cancer cell lines in culture (66–73). Metformin treatment of endometrial cancer cell lines upregulates markers of cell cycle arrest (66, 68, 69, 74), apoptosis (66, 67, 69, 72, 73, 75, 76), and autophagy (69, 77), while decreasing markers associated with senescence (66, 74) and inhibiting cell migration (68, 71, 76). The anticancer effects of metformin treatment may not be limited to direct effects on endometrial cancer cells, but may also result from changes to the systemic milieu. Polycystic ovary syndrome (PCOS) is a condition which is associated with endometrial hyperplasia and predisposition to endometrial cancer (78). Endometrial cancer cell lines incubated with sera from PCOS patients showed increased migration and markers of invasiveness such as activity of matrix metalloproteinases (MMP)−2 and −9 compared to cells incubated with sera from healthy controls. In contrast, sera from PCOS patients treated with metformin for 6 months showed attenuation of this effect, with decreased migration and MMP-2/9 activity compared to cells treated with sera from PCOS patients not on metformin (79).
The molecular mechanisms of metformin's effects in endometrial cancer cells are diverse and continue to be an active area of investigation (Figure 1). Its general mechanisms are complex and multifactorial and are reviewed in detail elsewhere (80). Multiple groups have demonstrated that metformin's ability to inhibit oxidative phosphorylation (OXPHOS) at the mitochondrial level is an important mediator of its biologic activity (81, 82). The end result is a decrease in proton gradient across the inner mitochondrial membrane, ultimately leading to reduction in proton-driven synthesis of adenosine triphosphate (ATP) and an increase in the ratio of cellular adenosine monophosphate (AMP) to ATP, caused by imbalance in the rate of ATP production vs. consumption. The decrease in ATP is theorized to be responsible for a key effect of metformin treatment, namely, phosphorylation and activation of the serine/threonine AMP-activated protein kinase (AMPK), a regulatory protein which plays a role in sensing and energy status of the cell and regulating cellular function under conditions of energy restriction (83, 84). This leads to AMP binding to AMPK and a conformational change that allows for phosphorylation/activation of AMPK by liver kinase B1 (LKB1) (85). Activation of AMPK switches cells to a catabolic state via AMPK-mediated phosphorylation and inhibition of key enzymes and transcription factors involved in ATP-consuming synthetic pathways (e.g., glucose, lipid and protein).
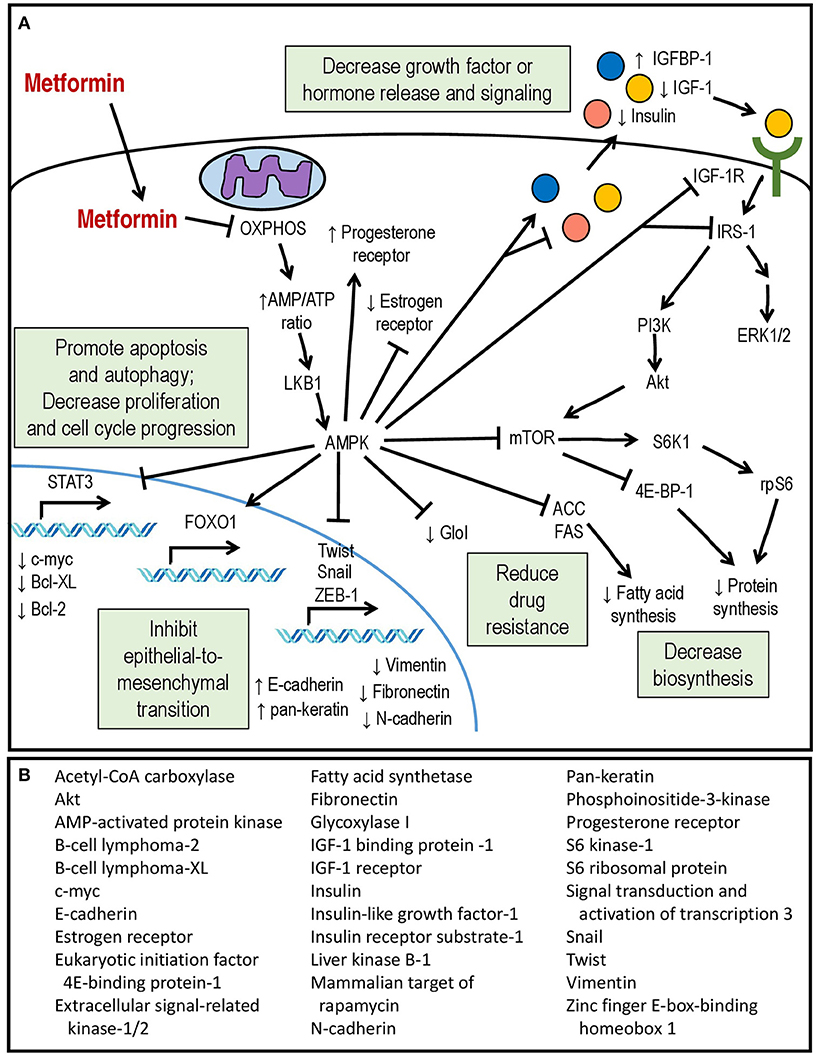
Figure 1. (A) Mechanisms of action of metformin within the endometrial cancer cell. (B) Downstream molecular targets of metformin showing differential expression or activity in endometrial cancer.
Among the known downstream effects of AMPK activation is decreased protein synthesis due to inhibition of the mTOR pathway, resulting in the inhibition of translation by the eukaryotic initiation factor 4E-binding protein-1 [4E-BP1] complex and decreased activity of the S6 kinase 1 (S6K1) responsible for phosphorylation of the ribosomal S6 protein (rpS6) (86, 87). Metformin inhibition of mTOR signaling in endometrial cancer cells has been confirmed by multiple groups (66, 74, 88–90). Other well-described effects of AMPK activation include phosphorylation and inactivation of Acetyl-CoA carboxylase (ACC) leading to downregulation of fatty acid synthesis (83, 91) as well as inhibition of signaling via the insulin-like growth factor-1 receptor (IGF-1R). Metformin inhibition of ACC has not been reported in endometrial cancer cell lines, but Wallbillich did observe decreased expression of fatty acid synthetase (FAS) in metformin-treated tumor tissue from a xenograft model (73). In endometrial cancer cell cultures, metformin treatment lowers secretion of insulin-like growth factor (IGF-1) (70), downregulates expression of insulin receptor (68) and IGF-1R (70, 75), inhibits phosphorylation of IGF-1R (68), and increases expression of insulin-like growth factor-binding protein 1 (IGFBP-1) (75). In other cancer types, this is associated with inhibitory phosphorylation of the signaling adapter, insulin receptor substrate-1 (IRS-1) and inhibition of the downstream PI3K/Akt/mTOR and mitogen-activated protein-kinase/extracellular signal-regulated kinase (MAPK/ERK) pathways (92–94). Inhibition of PI3K/Akt signaling has been observed in metformin-treated endometrial cancer cells (70), and both Akt and ERK1/2 are inhibited in endometrial cancer cells incubated with serum from women with PCOS who are receiving metformin treatment (79). The cumulative result is inhibition of individual cell growth and proliferation, decreased synthesis of proteins and fatty acids, as well as decreased paracrine and endocrine release of pro-proliferative systemic factors.
In addition to IGF-1-related signaling pathways, metformin treatment of endometrial cancer cells has been reported to affect the activity of the transcription factor signal transducer and activator of transcription 3 (STAT3), which is usually activated via signaling by various growth factors and cytokines to dimerize, translocate to the nucleus, and induce transcription of multiple pro-survival and pro-proliferative genes (95). STAT3 levels are elevated in endometrial cancer cells, in particular the serine-phosphorylated form, phospho-STAT3 Ser727 (96). High glucose concentrations induces transcription of STAT3 as well as its upstream regulators Janus kinases 1 and 2 (JAK1/2), while metformin treatment reduces total STAT3 protein as well as phospho-STAT3 Ser727 (73). This is associated with significantly decreased expression of multiple pro-survival downstream targets of STAT3, including c-Myc and B-cell lymphoma (Bcl)-2 and –XL, providing another possible mechanism for metformin's anti-cancer activity. The authors also examined the effect of metformin in a xenograft model of endometrioid endometrial cancer cells. While not statistically significant, metformin exposure was associated with a trend toward decreased tumor size, and analysis of tumor tissues demonstrated decreased expression of STAT3 and its targets (73).
Another recently-reported target of metformin in endometrial cancer is the transcription factor forkhead box protein 1 (FOXO1), which plays numerous roles in cellular function, including regulation of gluconeogenesis, adipogenesis, protection from oxidative stress, and tumor suppression (97). FOXO1 is negatively regulated via inhibitory phosphorylation by Akt, which causes it to translocate out of the nucleus to the cytoplasm where it is degraded (98). Conversely, AMPK activation leads to FOXO1 nuclear localization and activation (99). Zou demonstrated that endometrial cancer cells show decreased levels of phospho-AMPK and total FOXO1 protein, and endometrial cancer tissues show significantly less staining of activated AMPK and higher levels of phospho-Akt compared to controls (72). This is associated with a shift toward cytoplasmic (inactive) rather than nuclear (active) FOXO1 staining. In vitro, they reported that metformin treatment increased FOXO1 protein levels, decreased inhibitory FOXO1 phosphorylation, and increased FOXO1 nuclear accumulation in an AMPK-dependent manner, resulting in inhibition of endometrial cancer cell proliferation. Conversely, knockdown of FOXO1 expression using siRNA partially attenuates the antiproliferative effect of metformin on endometrial cancer cells. Metformin treatment inhibited growth of tumors in a xenograft mouse model, and tumor staining also showed increased phospho-AMPK and nuclear localization of FOXO1 as well as decreased staining of the proliferative marker Ki-67 (72). Decreased FOXO1 expression during metformin treatment has been reported by others as well (68).
Metformin treatment has also been shown to inhibit epithelial-to-mesenchymal transition (EMT) in endometrial cancer cell lines. Metformin increases epithelial markers such as E-cadherin and pan-keratin in endometrial cancer cells in vitro (76, 100) and decreases mesenchymal markers (e.g., N-cadherin, fibronectin, vimentin) (76, 100) and transcriptional drivers of EMT (e.g., Twist-1, snail-1, zinc finger E-box-binding homeobox-1 [ZEB-1]) (100). In cultured cells, metformin was able to attenuate the molecular and morphologic changes induced by EMT-inducing stimuli such as 17β-estradiol and transforming-growth factor-β (TGF-β) (76). Correspondingly, histologic staining for E-cadherin was significantly higher in endometrial carcinomas taken from patients with a history of metformin use (100).
Metformin has demonstrated the ability to synergize with other endometrial cancer therapies in vitro, leading to enhanced apoptosis of cultured endometrial cell lines in the presence of paclitaxel (74, 101), cisplatin (101, 102), or progestin (89). For the latter two, this effect is dependent on downregulation of glycoxylase I (GloI), a mediator of chemotherapy resistance (89, 101). Metformin treatment was also able to increase expression of the progesterone receptor in endometrial cancer cells (88) and sensitize progestin-resistant endometrial carcinoma cells to medroxyprogesterone-induced apoptosis (89). Conversely, metformin alters expression of the estrogen receptor (ER) in endometrial cancer cells, decreasing the ERα isoform while increasing expression of ERβ, with overall inhibition of estradiol-induced proliferation (90). In endometrial cancer patients with type 2 diabetes, metformin leads to decreased expression of the estrogen receptor in tumor tissue compared to insulin treatment (103).
Possible mutation-specific effects of metformin have also been explored in preclinical in vivo models of endometrial cancer. Oral metformin is capable of reducing in vitro cell proliferation as well as tumor size in xenograft models of multiple human and mouse endometrial cancer cell lines. (67). This effect occurred only in cell lines with activating K-Ras mutations but not wild-type K-Ras and could be partially attenuated by siRNA-based inhibition of K-Ras expression. Moreover, metformin treatment was shown to cause mislocalization of K-Ras to the cytoplasm in a protein kinase C (PKC)-dependent manner. No association was seen between metformin-responsiveness and PTEN mutations. Another study utilized a primary endometrioid endometrial carcinoma xenograft model using cells taken directly from patient biopsies for culture and inoculation into nude mice. The authors noted that one sample contained a K-Ras mutation while the other was wild-type; neither tumor was inhibited by metformin treatment, either alone or in combination with cisplatin (104). It should be noted that the dosage of metformin used in this study was lower than in the study by Iglesias (250 mg/kg/day for 21 days vs. 1 g/kg/day for 29-64 days). However, in both these studies, the tumors that displayed no susceptibility to metformin treatment also showed no changes in levels of activated AMPK or downstream mediators, emphasizing the likely importance of this pathway for mediating metformin-induced tumor suppression. Several studies have now shown that higher doses of metformin are required in mice to have antitumor effects than those administered in clinical trials and this may be a reflection of pharmacokinetic differences between rodents and humans (86, 105, 106).
Attention is now also being paid to the impact of metformin on other cellular components of the tumor microenvironment (TME) beyond the cancer cells themselves as described in Rivadeneira and Delgoffe (107). Metformin is capable of decreasing the rate of tumor cell oxygen consumption and thus is able to reduce hypoxia levels within the tumor. The reduction of hypoxia can enhance the activity of agents aimed at stimulating anti-tumor T-cells (108). Further effects of metformin on the immune microenvironment are hypothesized to be mediated through tumor-associated macrophage reprogramming from an M2 to M1-like phenotype (109). A review on the effects of metformin on other tumor cells is beyond the scope of this work. In sum, the pre-clinical data provide sufficient evidence for continued evaluation of metformin's antineoplastic potential. Nonetheless, they also highlight the likely complex interaction between tumor- and patient-specific factors dictating the efficacy of metformin as an anticancer treatment and underscore the need for ongoing human studies.
Metformin in Presurgical and Other Clinical Trials in Endometrial Cancer
The preponderance of preclinical data has prompted several early phase clinical trials of metformin in human endometrial cancers (Table 3). Many groups have utilized the pre-surgical window approach, in which patients with a biopsy-based histologic diagnosis of endometrial cancer receive metformin treatment during the period prior to hysterectomy. Compared to baseline levels or control patients, patients receiving metformin (between 850 and 2,250 mg daily) showed a post-treatment reduction in markers of DNA replication (topoisomerase IIα) (112) and cellular proliferation (Ki-67) (111, 114, 115, 119). No change in Ki-67 staining was observed by Soliman and colleagues, though notably the dose and duration of metformin exposure was the lowest among these studies (117). Metformin treatment was associated with histologic evidence for inhibition of key signaling pathways including PI3K/Akt/mTOR (111, 112, 114, 116, 117, 119) and MAPK/ERK (112, 117). Tumor immunohistochemistry in one trial also revealed decreased expression of estrogen receptor after metformin treatment (114). Plasma measures of a hyperinsulinemic state, including insulin, IGF-1, glucose, and leptin were significantly reduced post-metformin (111, 112, 116, 117). Furthermore, serum from metformin-treated patients showed decreased ability to stimulate DNA synthesis in cultured endometrial cancer cells (112), suggesting that systemic effects of metformin also play a role in its antiproliferative activity.
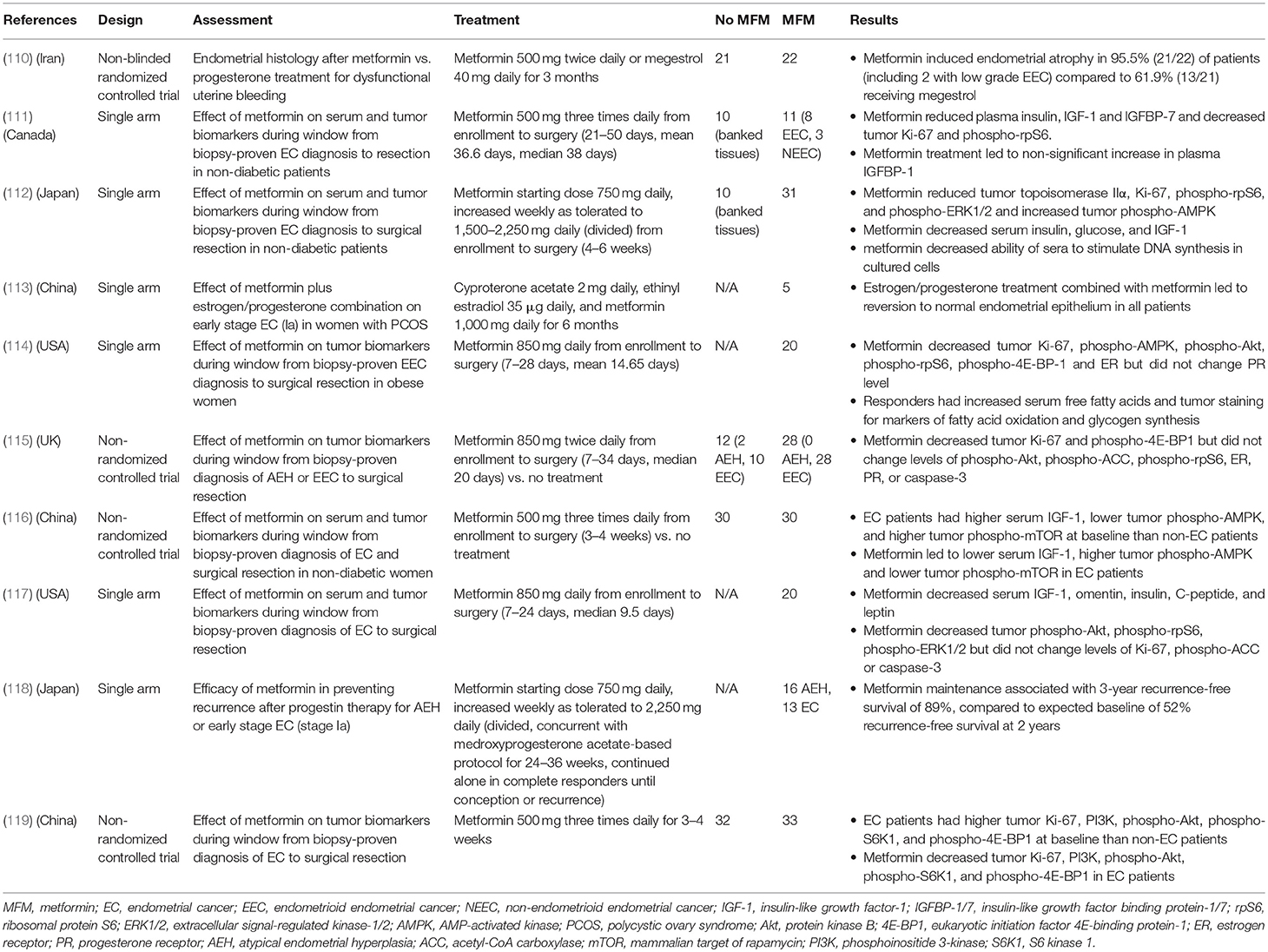
Table 3. Clinical trials of metformin in endometrial cancer.
In terms of clinical outcomes, a randomized study by Tabrizi examined the ability of metformin to reverse endometrial hyperplasia or disordered proliferative endometrium in patients with abnormal uterine bleeding compared to megestrol. Metformin was able to induce endometrial atrophy/restore endometrial histology in 95.5% of patients compared to 61.9% in the megestrol group. Significantly, this study included two patients with low grade endometrial carcinomas (stage Ia) who received metformin. After 3 months of treatment, repeat biopsy showed conversion to atrophic endometrium (110). A study of 5 PCOS patients with early stage endometrial carcinoma showed that co-treatment with the oral contraceptive Diane-35 (cyproterone and ethinyl estradiol) and metformin for 6 months led to reversion to normal epithelia on repeat biopsy in all patients (113). This included three patients who had previously been on megestrol treatment for 3 months with documented progesterone resistance. Mitsuhashi also used a combination approach of metformin with medroxyprogesterone to induce remission in patients with atypical endometrial hyperplasia (AEH) or early stage endometrial cancer, and further studied the use of maintenance metformin to prevent relapse. Metformin treatment led to a relapse-free survival of 89% at 3 years, which was higher than their projected baseline of 52% (118). These studies were also notable for their stated goal of fertility preservation, an important consideration for some endometrial cancer patients.
Larger trials are underway, including a phase 3 trial of metformin monotherapy as chemoprevention for endometrial cancer compared to placebo and lifestyle interventions in non-diabetic obese women (NCT01697566) and a phase 2/3 trial by the Gynecologic Oncology Group for advanced (stage III, IVA, IVB) or recurrent endometrial cancer that will compare the addition of metformin vs. placebo to combination paclitaxel/carboplatin as first-line therapy (NCT02065687). Several phase 2 or earlier studies will utilize metformin in combination with hormonal treatments such as megestrol acetate (NCT01968317) or levonorgestrel (as an intrauterine device) for early stage endometrial cancer or complex atypical hyperplasia in young women with the goal of fertility preservation or contraindications to surgery (NCT02990728, NCT02035787, NCT01686126). Also ongoing are a phase 2 study of metformin combined with letrozole and everolimus for advanced or recurrent endometrial cancer (NCT01797523) and a phase 1/2 study of metformin plus metronomic cyclophosphamide and olaparib for advanced or recurrent endometrial cancer (NCT02755844). These efforts will give valuable insight into the preventative and therapeutic value of metformin in endometrial malignancy.
The Metabolic Microenvironment as a Therapeutic Target in Endometrial Cancer
The role of glucose metabolism in endometrial cancer is also still being explored. Malignant tissues in general have been known to have high levels of glycolytic metabolism, even in the presence of oxygen, a phenomenon known as the Warburg effect after its discoverer, Otto Warburg (120). One theory was that cancer cells are less dependent on oxidative phosphorylation, allowing them to survive in the often relatively hypoxic tumor microenvironment. However, in recent years, a more nuanced model has emerged for some tumor types, known as the reverse Warburg theory (121). This is based on the recognition of heterogeneity in tumor composition; malignant tissues are composed of cancer cells surrounded by diverse types of stromal cells and adipocytes, each of which make contributions to the tumor microenvironment (122, 123). Our research in breast and prostate cancer cell models among others have demonstrated that glycolysis occurs not in the tumor cells themselves, but in the surrounding stromal cells (124–127). This relationship is thought to occur via oxidative stress in the cancer associated stroma (CAS), driven by tumor cell generation of reactive oxygen species and stromal cell loss of caveolin-1 (CAV1), an inhibitor of nitric oxide production (124–129). CAS cells undergo metabolic reprogramming associated with mitophagy (selective degradation of mitochondria by autophagy) and cell autophagy, and they generate high levels of energy-rich metabolites (including lactate and ketones) through glycolysis, which is then shuttled to cancer cells as substrates for oxidative phosphorylation. We have reported that lactate shuttling to tumor epithelial cells is dependent on transporters of the monocarboxylate transporter (MCT) family, with MCT4 being important for lactate efflux from stromal cells and MCT1 for lactate uptake in tumor epithelial cells in a breast cancer cell co-culture system (130). Others have observed similar lactate shuttling in prostate cancer and sarcoma models and have reported that lactate upload promotes tumor cell proliferation and angiogenesis (131, 132). Aside from lactate, Sousa and colleagues have described a similar two-compartment metabolic system in pancreatic ductal adenocarcinoma in which stroma-associated pancreatic stellate cells are stimulated by contact with pancreatic cancer cells to undergo autophagy and secrete primarily alanine which fuels the tricarboxylic acid (TCA) cycle and biosynthesis in the cancer cells themselves (133).
In this model, metformin treatment may play an important part in disrupting cancer cell metabolism via its direct inhibition of mitochondrial respiration in the cancer cells. In agreement with this, our previous results suggest that metformin treatment of cultured breast cancer cells inhibits their ability to induce loss of CAV1 (a marker of tumor-stromal metabolic coupling) in co-cultured fibroblasts (126). We have also observed that a short course of metformin was also able to increase stromal CAV1 expression in vivo in patients with head and neck squamous cell carcinoma in a presurgical window of opportunity trial (106).
The existence of such symbiotic metabolic reprogramming has not yet been investigated closely in endometrial cancer, but supportive evidence comes from a study by Latif et al. that showed differential histologic localization for MCT1 (tumor) vs. MCT4 (stroma) for some though not all endometrial cancer samples analyzed (134). High MCT1 expression was also a poor prognostic indicator for recurrence-free, cancer-free, and overall survival in the same study. Moreover, Zhao studied endometrial stromal-epithelial cell interactions in a non-cancer primary cell culture model and showed that epithelial cell proliferation and migration was enhanced when cultured with conditioned media from CAV1 depleted stromal cells (135). A phase 2 study currently underway at our institution utilizes a combination of metformin and doxycycline for the treatment of breast and uterine cancers, with outcomes including the measurement of biomarkers of tumor-stromal metabolic compartmentalization, such as stromal CAV1 and MCT4 and tumor MCT1 (NCT02874430). This will provide new insight into the effects of metformin treatment on the metabolic microenvironment in endometrial cancer.
The Reverse Warburg framework also opens up the possibility of multi-targeted therapies that simultaneously act on aberrant glucose metabolism in both cancer and stromal cells, such as metformin combined with inhibitors of glycolysis, autophagy, or transport of lactate and other energetic substrates. Combined metformin and glycolytic inhibitors have been utilized in xenograft models of breast cancer, gastric cancer, and glioblastoma with synergistic inhibition of tumor growth or prolongation of survival occurring at doses where each agent is ineffective alone (136, 137). Such approaches would exploit the vulnerabilities of tumor-stroma metabolic reprogramming that typically allow for cancer cell survival under a variety of energetic conditions. Overall, the metabolic microenvironment of endometrial cancer represents a promising therapeutic target, one which metformin and other biguanides may be uniquely poised to act upon.
Conclusions
Endometrial cancer is a disease with few effective treatments for advanced and metastatic disease. In addition, the need for fertility-sparing options for patients with early stage disease means there is a need for more primary or adjunctive treatment approaches. A large body of evidence links endometrial cancer incidence to metabolic conditions such as obesity and hyperglycemic states. Increasing rates of the latter has been mirrored by a rise in the former, particularly in developing countries, highlighting the need for a better understanding of the contribution of the metabolic microenvironment to endometrial cancer tumorigenesis. There is substantial evidence that the mechanisms of nutrient utilization and synthesis are significantly dysregulated in malignancy on both an intracellular and intercellular level. Models such as the reverse Warburg effect especially emphasize the importance of considering the interplay between cancer epithelial cells and their surrounding stroma. Dysregulation of metabolic pathways may represent adaptations that facilitate survival and proliferation in some scenarios (e.g., the idea of the parasitic cancer cell), but can also become liabilities for cancer cells particularly in times of nutrient or energy deprivation. Drugs such as metformin may be uniquely poised to exploit these defects, possibly in conjunction with other therapies that target glucose utilization. Indeed, preclinical and early clinical studies have shown promise for metformin as an adjunctive treatment for endometrial cancer, with effects on both cancer-specific as well as patient-specific metrics. Further study is needed to elucidate the role of metformin as a therapy for endometrial cancer, and several clinical trials are underway that will greatly expand our understanding of its potential benefits.
Author Contributions
JJ and UM-O contributed to the conception and design of the manuscript. TL wrote the first draft of the manuscript. UM-O, RS, CK, SR, NR, and JJ contributed to manuscript revision. All authors read and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Global Burden of Disease Cancer, Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol (2017) 3:524–48. doi: 10.1001/jamaoncol.2016.5688
2. Global Burden of Disease Cancer, Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, et al. The global burden of cancer 2013. JAMA Oncol (2015) 1:505–27. doi: 10.1001/jamaoncol.2015.0735
3. American Cancer Society. Cancer Facts & Figures 2018. Atlanta, GA: American Cancer Society (2018).
4. Duska LR, Garrett A, Rueda BR, Haas J, Chang Y, Fuller AF. Endometrial cancer in women 40 years old or younger. Gynecol Oncol. (2001) 83:388–93. doi: 10.1006/gyno.2001.6434
5. Moir-Meyer GL, Pearson JF, Lose F, Scott RJ, Mcevoy M, Attia J, et al. Rare germline copy number deletions of likely functional importance are implicated in endometrial cancer predisposition. Hum Genet. (2015) 134:269–78. doi: 10.1007/s00439-014-1507-4
6. Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol. (1983) 15:10–7. doi: 10.1016/0090-8258(83)90111-7
7. Del Carmen MG, Birrer M, Schorge JO. Uterine papillary serous cancer: a review of the literature. Gynecol Oncol. (2012) 127:651–61. doi: 10.1016/j.ygyno.2012.09.012
8. Kilgore LC, Partridge EE, Alvarez RD, Austin JM, Shingleton HM, Noojin F III, et al. Adenocarcinoma of the endometrium: survival comparisons of patients with and without pelvic node sampling. Gynecol Oncol. (1995) 56:29–33. doi: 10.1006/gyno.1995.1005
9. Nout RA, Smit VT, Putter H, Jurgenliemk-Schulz IM, Jobsen JJ, Lutgens LC, et al. Vaginal brachytherapy versus pelvic external beam radiotherapy for patients with endometrial cancer of high-intermediate risk (PORTEC-2): an open-label, non-inferiority, randomised trial. Lancet (2010) 375:816–23. doi: 10.1016/S0140-6736(09)62163-2
10. Kong A, Johnson N, Kitchener HC, Lawrie TA. Adjuvant radiotherapy for stage I endometrial cancer: an updated Cochrane systematic review and meta-analysis. J Natl Cancer Inst. (2012) 104:1625–34. doi: 10.1093/jnci/djs374
11. Morice P, Leary A, Creutzberg C, Abu-Rustum N, Darai E. Endometrial cancer. Lancet (2016) 387:1094–108. doi: 10.1016/S0140-6736(15)00130-0
12. Quinn MA. Hormonal treatment of endometrial cancer. Hematol Oncol Clin North Am. (1999) 13:163–87.ix. doi: 10.1016/S0889-8588(05)70159-3
13. Network NCC. Uterine Neoplasms Version 1.2018 (2018) Available online at: https://www.nccn.org/professionals/physician_gls/pdf/uterine.pdf (Accessed April 30, 2018).
14. Moore TD, Phillips PH, Nerenstone SR, Cheson BD. Systemic treatment of advanced and recurrent endometrial carcinoma: current status and future directions. J Clin Oncol. (1991) 9:1071–88. doi: 10.1200/JCO.1991.9.6.1071
15. Humber CE, Tierney JF, Symonds RP, Collingwood M, Kirwan J, Williams C, et al. Chemotherapy for advanced, recurrent or metastatic endometrial cancer: a systematic review of Cochrane collaboration. Ann Oncol. (2007) 18:409–20. doi: 10.1093/annonc/mdl417
16. Russo A, Autelitano M, Bisanti L. Metabolic syndrome and cancer risk. Eur J Cancer (2008) 44:293–7. doi: 10.1016/j.ejca.2007.11.005
17. Esposito K, Chiodini P, Colao A, Lenzi A, Giugliano D. Metabolic syndrome and risk of cancer: a systematic review and meta-analysis. Diabetes Care (2012) 35:2402–11. doi: 10.2337/dc12-0336
18. Stocks T, Bjorge T, Ulmer H, Manjer J, Haggstrom C, Nagel G, et al. Metabolic risk score and cancer risk: pooled analysis of seven cohorts. Int J Epidemiol. (2015) 44:1353–63. doi: 10.1093/ije/dyv001
19. Cust AE, Kaaks R, Friedenreich C, Bonnet F, Laville M, Tjonneland A, et al. Metabolic syndrome, plasma lipid, lipoprotein and glucose levels, and endometrial cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC). Endocr Relat Cancer (2007) 14:755–67. doi: 10.1677/ERC-07-0132
20. Bjorge T, Stocks T, Lukanova A, Tretli S, Selmer R, Manjer J, et al. Metabolic syndrome and endometrial carcinoma. Am J Epidemiol. (2010) 171:892–902. doi: 10.1093/aje/kwq006
21. Zhang Y, Liu Z, Yu X, Zhang X, Lu S, Chen X, et al. The association between metabolic abnormality and endometrial cancer: a large case-control study in China. Gynecol Oncol. (2010) 117:41–6. doi: 10.1016/j.ygyno.2009.12.029
22. Friedenreich CM, Biel RK, Lau DC, Csizmadi I, Courneya KS, Magliocco AM, et al. Case-control study of the metabolic syndrome and metabolic risk factors for endometrial cancer. Cancer Epidemiol Biomarkers Prev. (2011) 20:2384–95. doi: 10.1158/1055-9965.EPI-11-0715
23. Rosato V, Zucchetto A, Bosetti C, Dal Maso L, Montella M, Pelucchi C, et al. Metabolic syndrome and endometrial cancer risk. Ann Oncol. (2011) 22:884–9. doi: 10.1093/annonc/mdq464
24. Trabert B, Wentzensen N, Felix AS, Yang HP, Sherman ME, Brinton LA. Metabolic syndrome and risk of endometrial cancer in the united states: a study in the SEER-medicare linked database. Cancer Epidemiol Biomarkers Prev. (2015) 24:261–7. doi: 10.1158/1055-9965.EPI-14-0923
25. Esposito K, Chiodini P, Capuano A, Bellastella G, Maiorino MI, Giugliano D. Metabolic syndrome and endometrial cancer: a meta-analysis. Endocrine (2014) 45:28–36. doi: 10.1007/s12020-013-9973-3
26. Ni J, Zhu T, Zhao L, Che F, Chen Y, Shou H, et al. Metabolic syndrome is an independent prognostic factor for endometrial adenocarcinoma. Clin Transl Oncol. (2015) 17:835–9. doi: 10.1007/s12094-015-1309-8
27. Aune D, Navarro Rosenblatt DA, Chan DS, Vingeliene S, Abar L, Vieira AR, et al. Anthropometric factors and endometrial cancer risk: a systematic review and dose-response meta-analysis of prospective studies. Ann Oncol. (2015) 26:1635–48. doi: 10.1093/annonc/mdv142
28. Wise MR, Jordan V, Lagas A, Showell M, Wong N, Lensen S, et al. Obesity and endometrial hyperplasia and cancer in premenopausal women: a systematic review. Am J Obstet Gynecol. (2016) 214:689.e1–689.e17. doi: 10.1016/j.ajog.2016.01.175
29. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. (2003) 348:1625–38. doi: 10.1056/NEJMoa021423
30. Secord AA, Hasselblad V, Von Gruenigen VE, Gehrig PA, Modesitt SC, Bae-Jump V, et al. Body mass index and mortality in endometrial cancer: a systematic review and meta-analysis. Gynecol Oncol. (2016) 140:184–90. doi: 10.1016/j.ygyno.2015.10.020
31. Friberg E, Orsini N, Mantzoros CS, Wolk A. Diabetes mellitus and risk of endometrial cancer: a meta-analysis. Diabetologia (2007) 50:1365–74. doi: 10.1007/s00125-007-0681-5
32. Liao C, Zhang D, Mungo C, Tompkins DA, Zeidan AM. Is diabetes mellitus associated with increased incidence and disease-specific mortality in endometrial cancer? A systematic review and meta-analysis of cohort studies. Gynecol Oncol. (2014) 135:163–71. doi: 10.1016/j.ygyno.2014.07.095
33. Anderson KE, Anderson E, Mink PJ, Hong CP, Kushi LH, Sellers TA, et al. Diabetes and endometrial cancer in the Iowa women's health study. Cancer Epidemiol Biomarkers Prev (2001) 10:611–6. Available online at: http://cebp.aacrjournals.org/content/10/6/611.long
34. Luo J, Beresford S, Chen C, Chlebowski R, Garcia L, Kuller L, et al. Association between diabetes, diabetes treatment and risk of developing endometrial cancer. Br J Cancer (2014) 111:1432–9. doi: 10.1038/bjc.2014.407
35. Byers T, Sedjo RL. Body fatness as a cause of cancer: epidemiologic clues to biologic mechanisms. Endocr Relat Cancer (2015) 22:R125–34. doi: 10.1530/ERC-14-0580
36. Dossus L, Lukanova A, Rinaldi S, Allen N, Cust AE, Becker S, et al. Hormonal, metabolic, and inflammatory profiles and endometrial cancer risk within the EPIC cohort–a factor analysis. Am J Epidemiol. (2013) 177:787–99. doi: 10.1093/aje/kws309
37. Hernandez AV, Pasupuleti V, Benites-Zapata VA, Thota P, Deshpande A, Perez-Lopez FR. Insulin resistance and endometrial cancer risk: a systematic review and meta-analysis. Eur J Cancer (2015) 51:2747–58. doi: 10.1016/j.ejca.2015.08.031
38. Gupta SC, Sung B, Prasad S, Webb LJ, Aggarwal BB. Cancer drug discovery by repurposing: teaching new tricks to old dogs. Trends Pharmacol Sci. (2013) 34:508–17. doi: 10.1016/j.tips.2013.06.005
39. Wurth R, Thellung S, Bajetto A, Mazzanti M, Florio T, Barbieri F. Drug-repositioning opportunities for cancer therapy: novel molecular targets for known compounds. Drug Discov Today (2016) 21:190–9. doi: 10.1016/j.drudis.2015.09.017
40. Pryor R, Cabreiro F. Repurposing metformin: an old drug with new tricks in its binding pockets. Biochem J (2015) 471:307–22. doi: 10.1042/BJ20150497
41. Diabetes Prevention Program Research G, Knowler WC, Fowler SE, Hamman RF, Christophi CA, Hoffman HJ, et al. 10-year follow-up of diabetes incidence and weight loss in the diabetes prevention program outcomes study. Lancet (2009) 374:1677–86. doi: 10.1016/S0140-6736(09)61457-4
42. Becker C, Jick SS, Meier CR, Bodmer M. Metformin and the risk of endometrial cancer: a case-control analysis. Gynecol Oncol. (2013) 129:565–9. doi: 10.1016/j.ygyno.2013.03.009
43. Ko EM, Sturmer T, Hong JL, Castillo WC, Bae-Jump V, Funk MJ. Metformin and the risk of endometrial cancer: a population-based cohort study. Gynecol Oncol. (2015) 136:341–7. doi: 10.1016/j.ygyno.2014.12.001
44. Tseng CH. Metformin and endometrial cancer risk in Chinese women with type 2 diabetes mellitus in Taiwan. Gynecol Oncol. (2015) 138:147–53. doi: 10.1016/j.ygyno.2015.03.059
45. Franchi M, Asciutto R, Nicotra F, Merlino L, La Vecchia C, Corrao G, et al. Metformin, other antidiabetic drugs, and endometrial cancer risk: a nested case-control study within Italian healthcare utilization databases. Eur J Cancer Prev. (2016) 26:225–31. doi: 10.1097/CEJ.0000000000000235
46. Arima R, Hautakoski A, Marttila M, Arffman M, Sund R, Ilanne-Parikka P, et al. Cause-specific mortality in endometrioid endometrial cancer patients with type 2 diabetes using metformin or other types of antidiabetic medication. Gynecol Oncol. (2017) 147:678–83. doi: 10.1016/j.ygyno.2017.10.014
47. Pierotti MA, Berrino F, Gariboldi M, Melani C, Mogavero A, Negri T, et al. Targeting metabolism for cancer treatment and prevention: metformin, an old drug with multi-faceted effects. Oncogene (2013) 32:1475–87. doi: 10.1038/onc.2012.181
48. Febbraro T, Lengyel E, Romero IL. Old drug, new trick: repurposing metformin for gynecologic cancers? Gynecol Oncol. (2014) 135:614–21. doi: 10.1016/j.ygyno.2014.10.011
49. Coperchini F, Leporati P, Rotondi M, Chiovato L. Expanding the therapeutic spectrum of metformin: from diabetes to cancer. J Endocrinol Invest. (2015) 38:1047–55. doi: 10.1007/s40618-015-0370-z
50. Imai A, Ichigo S, Matsunami K, Takagi H, Yasuda K. Clinical benefits of metformin in gynecologic oncology. Oncol Lett. (2015) 10:577–82. doi: 10.3892/ol.2015.3262
51. Chae YK, Arya A, Malecek MK, Shin DS, Carneiro B, Chandra S, et al. Repurposing metformin for cancer treatment: current clinical studies. Oncotarget (2016) 7:40767–80. doi: 10.18632/oncotarget.8194
52. Gong J, Kelekar G, Shen J, Shen J, Kaur S, Mita M. The expanding role of metformin in cancer: an update on antitumor mechanisms and clinical development. Target Oncol. (2016) 11:447–67. doi: 10.1007/s11523-016-0423-z
53. Irie H, Banno K, Yanokura M, Iida M, Adachi M, Nakamura K, et al. Metformin: a candidate for the treatment of gynecological tumors based on drug repositioning. Oncol Lett. (2016) 11:1287–93. doi: 10.3892/ol.2016.4075
54. Tang YL, Zhu LY, Li Y, Yu J, Wang J, Zeng XX, et al. Metformin use is associated with reduced incidence and improved survival of endometrial cancer: a meta-analysis. Biomed Res Int. (2017) 2017:5905384. doi: 10.1155/2017/5905384
55. Nevadunsky NS, Van Arsdale A, Strickler HD, Moadel A, Kaur G, Frimer M, et al. Metformin use and endometrial cancer survival. Gynecol Oncol. (2014) 132:236–40. doi: 10.1016/j.ygyno.2013.10.026
56. Ko EM, Walter P, Jackson A, Clark L, Franasiak J, Bolac C, et al. Metformin is associated with improved survival in endometrial cancer. Gynecol Oncol. (2014) 132:438–42. doi: 10.1016/j.ygyno.2013.11.021
57. Lemanska A, Zaborowski M, Spaczynski M, Nowak-Markwitz E. Do endometrial cancer patients benefit from metformin intake? Ginekol Pol. (2015) 86:419–23. doi: 10.17772/gp/2397
58. Al Hilli MM, Bakkum-Gamez JN, Mariani A, Cliby WA, Mc Gree ME, Weaver AL, et al. The effect of diabetes and metformin on clinical outcomes is negligible in risk-adjusted endometrial cancer cohorts. Gynecol Oncol. (2016) 140:270–6. doi: 10.1016/j.ygyno.2015.11.019
59. Ezewuiro O, Grushko TA, Kocherginsky M, Habis M, Hurteau JA, Mills KA, et al. Association of metformin use with outcomes in advanced endometrial cancer treated with chemotherapy. PLoS ONE (2016) 11:e0147145. doi: 10.1371/journal.pone.0147145
60. Hall C, Stone RL, Gehlot A, Zorn KK, Burnett AF. Use of metformin in obese women with type I endometrial cancer is associated with a reduced incidence of cancer recurrence. Int J Gynecol Cancer (2016) 26:313–7. doi: 10.1097/IGC.0000000000000603
61. Seebacher V, Bergmeister B, Grimm C, Koelbl H, Reinthaller A, Polterauer S. The prognostic role of metformin in patients with endometrial cancer: a retrospective study. Eur J Obstet Gynecol Reprod Biol. (2016) 203:291–6. doi: 10.1016/j.ejogrb.2016.06.013
62. Meireles CG, Pereira SA, Valadares LP, Rego DF, Simeoni LA, Guerra ENS, et al. Effects of metformin on endometrial cancer: systematic review and meta-analysis. Gynecol Oncol. (2017) 147:167–80. doi: 10.1016/j.ygyno.2017.07.120
63. Zhang ZJ, Li S. The prognostic value of metformin for cancer patients with concurrent diabetes: a systematic review and meta-analysis. Diabetes Obes Metab. (2014) 16:707–10. doi: 10.1111/dom.12267
64. Byrne FL, Poon IK, Modesitt SC, Tomsig JL, Chow JD, Healy ME, et al. Metabolic vulnerabilities in endometrial cancer. Cancer Res. (2014) 74:5832–45. doi: 10.1158/0008-5472.CAN-14-0254
65. Han J, Zhang L, Guo H, Wysham WZ, Roque DR, Willson AK, et al. Glucose promotes cell proliferation, glucose uptake and invasion in endometrial cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol Oncol. (2015) 138:668–75. doi: 10.1016/j.ygyno.2015.06.036
66. Cantrell LA, Zhou C, Mendivil A, Malloy KM, Gehrig PA, Bae-Jump VL. Metformin is a potent inhibitor of endometrial cancer cell proliferation–implications for a novel treatment strategy. Gynecol Oncol. (2010) 116:92–8. doi: 10.1016/j.ygyno.2009.09.024
67. Iglesias DA, Yates MS, Van Der Hoeven D, Rodkey TL, Zhang Q, Co NN, et al. Another surprise from Metformin: novel mechanism of action via K-Ras influences endometrial cancer response to therapy. Mol Cancer Ther. (2013) 12:2847–56. doi: 10.1158/1535-7163.MCT-13-0439
68. Sarfstein R, Friedman Y, Attias-Geva Z, Fishman A, Bruchim I, Werner H. Metformin downregulates the insulin/IGF-I signaling pathway and inhibits different uterine serous carcinoma (USC) cells proliferation and migration in p53-dependent or -independent manners. PLoS ONE (2013) 8:e61537. doi: 10.1371/journal.pone.0061537
69. Takahashi A, Kimura F, Yamanaka A, Takebayashi A, Kita N, Takahashi K, et al. Metformin impairs growth of endometrial cancer cells via cell cycle arrest and concomitant autophagy and apoptosis. Cancer Cell Int. (2014) 14:53. doi: 10.1186/1475-2867-14-53
70. Zhang Y, Li MX, Wang H, Zeng Z, Li XM. Metformin down-regulates endometrial carcinoma cell secretion of IGF-1 and expression of IGF-1R. Asian Pac J Cancer Prev. (2015) 16:221–5. doi: 10.7314/APJCP.2015.16.1.221
71. De Barros Machado A, Dos Reis V, Weber S, Jauckus J, Brum IS, Von Eye Corleta H, et al. Proliferation and metastatic potential of endometrial cancer cells in response to metformin treatment in a high versus normal glucose environment. Oncol Lett. (2016) 12:3626–32. doi: 10.3892/ol.2016.5041
72. Zou J, Hong L, Luo C, Li Z, Zhu Y, Huang T, et al. Metformin inhibits estrogen-dependent endometrial cancer cell growth by activating the AMPK-FOXO1 signal pathway. Cancer Sci. (2016) 107:1806–17. doi: 10.1111/cas.13083
73. Wallbillich JJ, Josyula S, Saini U, Zingarelli RA, Dorayappan KD, Riley MK, et al. High Glucose-mediated STAT3 activation in endometrial cancer is inhibited by metformin: therapeutic implications for endometrial cancer. PLoS ONE (2017) 12:e0170318. doi: 10.1371/journal.pone.0170318
74. Hanna RK, Zhou C, Malloy KM, Sun L, Zhong Y, Gehrig PA, et al. Metformin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and modulation of the mTOR pathway. Gynecol Oncol. (2012) 125:458–69. doi: 10.1016/j.ygyno.2012.01.009
75. Xie Y, Wang JL, Ji M, Yuan ZF, Peng Z, Zhang Y, et al. Regulation of insulin-like growth factor signaling by metformin in endometrial cancer cells. Oncol Lett. (2014) 8:1993–9. doi: 10.3892/ol.2014.2466
76. Liu Z, Qi S, Zhao X, Li M, Ding S, Lu J, et al. Metformin inhibits 17beta-estradiol-induced epithelial-to-mesenchymal transition via betaKlotho-related ERK1/2 signaling and AMPKalpha signaling in endometrial adenocarcinoma cells. Oncotarget (2016) 7:21315–31. doi: 10.18632/oncotarget.7040
77. Zhuo Z, Wang A, Yu H. Metformin targeting autophagy overcomes progesterone resistance in endometrial carcinoma. Arch Gynecol Obstet. (2016) 294:1055–61. doi: 10.1007/s00404-016-4148-0
78. Barry JA, Azizia MM, Hardiman PJ. Risk of endometrial, ovarian and breast cancer in women with polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod Update (2014) 20:748–58. doi: 10.1093/humupd/dmu012
79. Tan BK, Adya R, Chen J, Lehnert H, Sant Cassia LJ, Randeva HS. Metformin treatment exerts antiinvasive and antimetastatic effects in human endometrial carcinoma cells. J Clin Endocrinol Metab. (2011) 96:808–16. doi: 10.1210/jc.2010-1803
80. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci. (2012) 122:253–70. doi: 10.1042/CS20110386
81. El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. (2000) 275:223–8. doi: 10.1074/jbc.275.1.223
82. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. (2000) 348(Pt. 3):607–14. doi: 10.1042/bj3480607
83. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. (2001) 108:1167–74. doi: 10.1172/JCI13505
84. Stephenne X, Foretz M, Taleux N, Van Der Zon GC, Sokal E, Hue L, et al. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia (2011) 54:3101–10. doi: 10.1007/s00125-011-2311-5
85. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science (2005) 310:1642–6. doi: 10.1126/science.1120781
86. Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. (2007) 67:10804–12. doi: 10.1158/0008-5472.CAN-07-2310
87. Wang Y, Xu W, Yan Z, Zhao W, Mi J, Li J, et al. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J Exp Clin Cancer Res. (2018) 37:63. doi: 10.1186/s13046-018-0731-5
88. Xie Y, Wang YL, Yu L, Hu Q, Ji L, Zhang Y, et al. Metformin promotes progesterone receptor expression via inhibition of mammalian target of rapamycin (mTOR) in endometrial cancer cells. J Steroid Biochem Mol Biol. (2011) 126:113–20. doi: 10.1016/j.jsbmb.2010.12.006
89. Zhang Z, Dong L, Sui L, Yang Y, Liu X, Yu Y, et al. Metformin reverses progestin resistance in endometrial cancer cells by downregulating GloI expression. Int J Gynecol Cancer (2011) 21:213–21. doi: 10.1097/IGC.0b013e318207dac7
90. Zhang J, Xu H, Zhou X, Li Y, Liu T, Yin X, et al. Role of metformin in inhibiting estrogen-induced proliferation and regulating ERalpha and ERbeta expression in human endometrial cancer cells. Oncol Lett. (2017) 14:4949–56. doi: 10.3892/ol.2017.6877
91. Harwood HJ Jr, Petras SF, Shelly LD, Zaccaro LM, Perry DA, Makowski MR, et al. Isozyme-nonselective N-substituted bipiperidylcarboxamide acetyl-CoA carboxylase inhibitors reduce tissue malonyl-CoA concentrations, inhibit fatty acid synthesis, and increase fatty acid oxidation in cultured cells and in experimental animals. J Biol Chem. (2003) 278:37099–111. doi: 10.1074/jbc.M304481200
92. Ning J, Clemmons DR. AMP-activated protein kinase inhibits IGF-I signaling and protein synthesis in vascular smooth muscle cells via stimulation of insulin receptor substrate 1 S794 and tuberous sclerosis 2 S1345 phosphorylation. Mol Endocrinol. (2010) 24:1218–29. doi: 10.1210/me.2009-0474
93. Karnevi E, Said K, Andersson R, Rosendahl AH. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer (2013) 13:235. doi: 10.1186/1471-2407-13-235
94. Quinn BJ, Dallos M, Kitagawa H, Kunnumakkara AB, Memmott RM, Hollander MC, et al. Inhibition of lung tumorigenesis by metformin is associated with decreased plasma IGF-I and diminished receptor tyrosine kinase signaling. Cancer Prev Res. (2013) 6:801–10. doi: 10.1158/1940-6207.CAPR-13-0058-T
95. Yuan J, Zhang F, Niu R. Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci Rep. (2015) 5:17663. doi: 10.1038/srep17663
96. Tierney BJ, Mccann GA, Naidu S, Rath KS, Saini U, Wanner R, et al. Aberrantly activated pSTAT3-Ser727 in human endometrial cancer is suppressed by HO-3867, a novel STAT3 inhibitor. Gynecol Oncol. (2014) 135:133–41. doi: 10.1016/j.ygyno.2014.07.087
97. Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell (2004) 117:421–6. doi: 10.1016/S0092-8674(04)00452-0
98. Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci USA. (2003) 100:11285–90. doi: 10.1073/pnas.1934283100
99. Yun H, Park S, Kim MJ, Yang WK, Im DU, Yang KR, et al. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J. (2014) 281:4421–38. doi: 10.1111/febs.12949
100. Laskov I, Abou-Nader P, Amin O, Philip CA, Beauchamp MC, Yasmeen A, et al. Metformin increases E-cadherin in tumors of diabetic patients with endometrial cancer and suppresses epithelial-mesenchymal transition in endometrial cancer cell lines. Int J Gynecol Cancer (2016) 26:1213–21. doi: 10.1097/IGC.0000000000000761
101. Dong L, Zhou Q, Zhang Z, Zhu Y, Duan T, Feng Y. Metformin sensitizes endometrial cancer cells to chemotherapy by repressing glyoxalase I expression. J Obstet Gynaecol Res. (2012) 38:1077–85. doi: 10.1111/j.1447-0756.2011.01839.x
102. Uehara T, Mitsuhashi A, Tsuruoka N, Shozu M. Metformin potentiates the anticancer effects of cisplatin under normoxic conditions in vitro. Oncol Rep. (2015) 33:744–50. doi: 10.3892/or.2014.3611
103. Markowska A, Pawalowska M, Filas V, Korski K, Grybos M, Sajdak S, et al. Does Metformin affect ER, PR, IGF-1R, beta-catenin and PAX-2 expression in women with diabetes mellitus and endometrial cancer? Diabetol Metab Syndr. (2013) 5:76. doi: 10.1186/1758-5996-5-76
104. Schrauwen S, Coenegrachts L, Cattaneo A, Hermans E, Lambrechts D, Amant F. The antitumor effect of metformin with and without carboplatin on primary endometrioid endometrial carcinoma in vivo. Gynecol Oncol. (2015) 138:378–82. doi: 10.1016/j.ygyno.2015.06.006
105. Chandel NS, Avizonis D, Reczek CR, Weinberg SE, Menz S, Neuhaus R, et al. Are metformin doses used in murine cancer models clinically relevant? Cell Metab. (2016) 23:569–70. doi: 10.1016/j.cmet.2016.03.010
106. Curry J, Johnson J, Tassone P, Vidal MD, Menezes DW, Sprandio J, et al. Metformin effects on head and neck squamous carcinoma microenvironment: window of opportunity trial. Laryngoscope (2017) 127:1808–15. doi: 10.1002/lary.26489
107. Rivadeneira DB, Delgoffe GM. Antitumor T-cell reconditioning: improving the metabolic fitness for optimal cancer immunotherapy. Clin Cancer Res. (2018) 24:2473–81. doi: 10.1158/1078-0432.CCR-17-0894
108. Sharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 blockake is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol Res. (2017) 5:9–16. doi: 10.1158/2326-6066.CIR-16-0103
109. Wang JC, Sun X, Ma Q, Fu GF, Cong LL, Zhang H, et al. Metformin's antitumor and anti-angiogenic activities are mediated by skewing macrophage polarization. J Cell Mol Med. (2018) 22:3825–936. doi: 10.1111/jcmm.13655
110. Tabrizi AD, Melli MS, Foroughi M, Ghojazadeh M, Bidadi S. Antiproliferative effect of metformin on the endometrium–a clinical trial. Asian Pac J Cancer Prev. (2014) 15:10067–70. doi: 10.7314/APJCP.2014.15.23.10067
111. Laskov I, Drudi L, Beauchamp MC, Yasmeen A, Ferenczy A, Pollak M, et al. Anti-diabetic doses of metformin decrease proliferation markers in tumors of patients with endometrial cancer. Gynecol Oncol. (2014) 134:607–14. doi: 10.1016/j.ygyno.2014.06.014
112. Mitsuhashi A, Kiyokawa T, Sato Y, Shozu M. Effects of metformin on endometrial cancer cell growth in vivo: a preoperative prospective trial. Cancer (2014) 120:2986–95. doi: 10.1002/cncr.28853
113. Li X, Guo YR, Lin JF, Feng Y, Billig H, Shao R. Combination of diane-35 and metformin to treat early endometrial carcinoma in PCOS women with insulin resistance. J Cancer (2014) 5:173–81. doi: 10.7150/jca.8009
114. Schuler KM, Rambally BS, Difurio MJ, Sampey BP, Gehrig PA, Makowski L, et al. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med. (2015) 4:161–73. doi: 10.1002/cam4.353
115. Sivalingam VN, Kitson S, Mcvey R, Roberts C, Pemberton P, Gilmour K, et al. Measuring the biological effect of presurgical metformin treatment in endometrial cancer. Br J Cancer (2016) 114:281–9. doi: 10.1038/bjc.2015.453
116. Cai D, Sun H, Qi Y, Zhao X, Feng M, Wu X. Insulin-like growth factor 1/mammalian target of rapamycin and AMP-activated protein kinase signaling involved in the effects of metformin in the human endometrial cancer. Int J Gynecol Cancer (2016) 26:1667–72. doi: 10.1097/IGC.0000000000000818
117. Soliman PT, Zhang Q, Broaddus RR, Westin SN, Iglesias D, Munsell MF, et al. Prospective evaluation of the molecular effects of metformin on the endometrium in women with newly diagnosed endometrial cancer: a window of opportunity study. Gynecol Oncol. (2016) 143:466–71. doi: 10.1016/j.ygyno.2016.10.011
118. Mitsuhashi A, Sato Y, Kiyokawa T, Koshizaka M, Hanaoka H, Shozu M. Phase II study of medroxyprogesterone acetate plus metformin as a fertility-sparing treatment for atypical endometrial hyperplasia and endometrial cancer. Ann Oncol. (2016) 27:262–6. doi: 10.1093/annonc/mdv539
119. Zhao Y, Sun H, Feng M, Zhao J, Zhao X, Wan Q, et al. Metformin is associated with reduced cell proliferation in human endometrial cancer by inbibiting PI3K/AKT/mTOR signaling. Gynecol Endocrinol. (2018) 34:428–32. doi: 10.1080/09513590.2017.1409714
120. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
121. Fu Y, Liu S, Yin S, Niu W, Xiong W, Tan M, et al. The reverse Warburg effect is likely to be an Achilles' heel of cancer that can be exploited for cancer therapy. Oncotarget (2017) 8:57813–25. doi: 10.18632/oncotarget.18175
122. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. (2011) 17:1498–503. doi: 10.1038/nm.2492
123. Romero IL, Mukherjee A, Kenny HA, Litchfield LM, Lengyel E. Molecular pathways: trafficking of metabolic resources in the tumor microenvironment. Clin Cancer Res. (2015) 21:680–6. doi: 10.1158/1078-0432.CCR-14-2198
124. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle (2009) 8:3984–4001. doi: 10.4161/cc.8.23.10238
125. Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: autophagy drives compartment-specific oncogenesis. Cell Cycle (2010) 9:3534–51. doi: 10.4161/cc.9.17.12908
126. Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: a new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle (2010) 9:3256–76. doi: 10.4161/cc.9.16.12553
127. Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer's disease, and “Neuron-Glia Metabolic Coupling”. Aging (2010) 2:185–99. doi: 10.18632/aging.100134
128. Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, et al. The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle (2010) 9:1960–71. doi: 10.4161/cc.9.10.11601
129. Ko YH, Lin Z, Flomenberg N, Pestell RG, Howell A, Sotgia F, et al. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: implications for preventing chemotherapy resistance. Cancer Biol Ther. (2011) 12:1085–97. doi: 10.4161/cbt.12.12.18671
130. Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle (2011) 10:1772–83. doi: 10.4161/cc.10.11.15659
131. Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. (2012) 72:5130–40. doi: 10.1158/0008-5472.CAN-12-1949
132. Goodwin ML, Jin H, Straessler K, Smith-Fry K, Zhu JF, Monument MJ, et al. Modeling alveolar soft part sarcomagenesis in the mouse: a role for lactate in the tumor microenvironment. Cancer Cell (2014) 26:851–62. doi: 10.1016/j.ccell.2014.10.003
133. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature (2016) 536:479–83. doi: 10.1038/nature19084
134. Latif A, Chadwick AL, Kitson SJ, Gregson HJ, Sivalingam VN, Bolton J, et al. Monocarboxylate transporter 1 (MCT1) is an independent prognostic biomarker in endometrial cancer. BMC Clin Pathol. (2017) 17:27. doi: 10.1186/s12907-017-0067-7
135. Zhao L, Zhou S, Zou L, Zhao X. The expression and functionality of stromal caveolin 1 in human adenomyosis. Hum Reprod. (2013) 28:1324–38. doi: 10.1093/humrep/det042
136. Cheong JH, Park ES, Liang J, Dennison JB, Tsavachidou D, Nguyen-Charles C, et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol Cancer Ther. (2011) 10:2350–62. doi: 10.1158/1535-7163.MCT-11-0497
Keywords: tumor microenvironment, metabolism, metformin, endometrial cancer, reverse Warburg
Citation: Lee TY, Martinez-Outschoorn UE, Schilder RJ, Kim CH, Richard SD, Rosenblum NG and Johnson JM (2018) Metformin as a Therapeutic Target in Endometrial Cancers. Front. Oncol. 8:341. doi: 10.3389/fonc.2018.00341
Received: 31 May 2018; Accepted: 06 August 2018;
Published: 28 August 2018.
Edited by:
Simona Pisanti, Università degli Studi di Salerno, ItalyReviewed by:
Ronca Roberto, Università degli Studi di Brescia, ItalyRaquel Aloyz, Lady Davis Institute (LDI), Canada
Copyright © 2018 Lee, Martinez-Outschoorn, Schilder, Kim, Richard, Rosenblum and Johnson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer M. Johnson, jennifer.m.johnson@jefferson.edu