





Pheochromocytomas and paragangliomas have a highly diverse genetic background, with a third of the cases carrying a germline mutation in 1 of 14 identified genes.
This study aimed to evaluate next-generation sequencing for more efficient genetic testing of pheochromocytoma and paraganglioma and to establish germline and somatic mutation frequencies for all known susceptibility genes.
A targeted next-generation sequencing approach on an Illumina MiSeq instrument was used for a mutation analysis in 86 unselected pheochromocytoma and paraganglioma tumor samples. The study included the genes EGLN1, EPAS1, KIF1Bβ, MAX, MEN1, NF1, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2, TMEM127, and VHL. Results were verified in tumor and constitutional DNA with Sanger sequencing.
In all cases with clinical syndromes or known germline mutations, a mutation was detected in the expected gene. Among 68 nonfamilial tumors, 32 mutations were identified in 28 of the samples (41%), including germline mutations in EGLN1, KIF1Bβ, SDHA, SDHB, and TMEM127 and somatic mutations in EPAS1, KIF1Bβ, MAX, NF1, RET, and VHL, including one double monoallelic EPAS1 mutation.
Targeted next-generation sequencing proved to be fast and cost effective for the genetic analysis of pheochromocytoma and paraganglioma. More than half of the tumors harbored mutations in the investigated genes. Notably, 7% of the apparently sporadic cases carried germline mutations, highlighting the importance of comprehensive genetic testing. KIF1Bβ, which previously has not been investigated in a large cohort, appears to be an equally important tumor suppressor as MAX and TMEM127 and could be considered for genetic testing of these patients.
Pheochromocytomas (PCCs) and abdominal paragangliomas (PGLs) are catecholamine-producing tumors that arise from neural crest-derived cells of the adrenal medulla and the extraadrenal paraganglia, respectively. More than 30% of the cases have been reported to carry germline mutations in a growing list of susceptibility genes: EGLN1/PHD2, EPAS1/HIF2A, KIF1Bβ, MAX, MEN1, NF1, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2/SDH5, TMEM127, and VHL, which are involved in diverse but interconnecting pathways (1, 2). For several of the genes, germline mutations cause hereditary tumor syndromes in which PCC and/or PGL can be among the manifestations. Activating RET mutations cause multiple endocrine neoplasia (MEN) type 2 (MEN2), VHL mutations cause von Hippel-Lindau disease (VHL), NF1 mutations cause neurofibromatosis type 1 (NF1), and mutations in the different SDHx genes are associated with familial pheochromocytoma-paraganglioma syndromes (PGL1–5). Although germline mutations in TMEM127 (3, 4) and MAX (5, 6) have been demonstrated to occur in 1%–3% of PCCs without other germline mutations, genetic alterations in the genes EGLN1 and KIF1Bβ have so far been described in only a few isolated cases, and their prevalence in PCC and PGL, as well as associated phenotypic features, is still unknown (7–10). Furthermore, PCC has been reported in a few patients with MEN type 1 (MEN1), caused by mutations in the MEN1 gene (2, 11), and most recently a MEN1 patient with PGL was reported for the first time (12), but MEN1 mutations have so far not been identified in sporadic PCCs or PGLs (13).
Somatic mutations in the genes known to be associated with familial disease have been considered rare (2, 14) until recently when somatic NF1 mutations were discovered in 20%–25% of sporadic PCCs (15, 16). In addition, recent work identified that the gene EPAS1, encoding hypoxia-inducible factor (HIF)-2α, is involved in PCC and PGL development (17). Somatic EPAS1 mutations have now been identified in several tumors (17–20), and a germline, predisposing mutation has been reported in one patient (21). Despite this recent progress, most of the sporadic PCCs and PGLs remain genetically unexplained. It is also unknown whether tumors may have mutations in more than one of the susceptibility genes because comprehensive investigations of all the genes in the same cohort are lacking.
Genetic testing can aid in decisions about treatment and screening in PCC/PGL patients and family members (22, 23). However, traditional Sanger sequencing of all the associated genes, in a clinical or a research setting, is expensive and time consuming. In this study, we used a targeted next-generation sequencing (NGS) approach to perform a mutation analysis in all the genes that have been shown to be associated with PCCs/PGLs. For the first time, all the genes were investigated in large cohort of unselected tumors, with the aim of further elucidating the biology of PCCs/PGLs and to evaluate the usefulness of NGS in the genetic testing.
Materials and Methods
Patients and samples
Tumor samples (72 PCCs and 14 PGLs) were obtained from 86 unselected patients operated in Sweden (Supplemental Table 1). Eighteen of the tumors were from patients with a known syndromic form of the disease (nine MEN2A, two VHL, three NF1) or a known mutation [four with SDHB mutation (24)]. The remaining tumors (58 PCCs and 10 PGLs) were from apparently sporadic cases, ie, nonfamilial and without syndromic features. As additional sensitivity controls, three PCCs with known NF1 mutation status from a previous study (15) were included in the NGS analysis. All samples were collected and studied with informed consent and approval from the local ethic committees. Tumor DNA was quantified with a Quant-iT PicoGreen double-stranded DNA assay (Invitrogen), and 250 ng of DNA from each sample was used for library preparation.
Next-generation sequencing
Probes for targeted sequencing with the TruSeq custom amplicon kit (Illumina) were designed to cover the genes EGLN1, KIF1Bβ, MAX, MEN1, NF1, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2, TMEM127, and VHL (Supplemental Tables 2 and 3) including exon-intron boundaries, using the online DesignStudio software (Illumina). The DNA library was prepared according to the manufacturer's protocol, and two library pools of 48 samples each were sequenced in two separate runs on a MiSeq sequencer (Illumina), using 2 × 150 bp paired-end reads. The alignment of the NGS data to the human reference genome and the variant calling were performed using software provided by Illumina. Silent sequence variants and missense variants reported in the HapMap or 1000 Genomes projects were excluded, except for very rare (allele frequency ≤ 0.02) missense variants, which are listed separately (Supplemental Table 4). The remaining variants were classified as mutations, and their potential effects were predicted with two different algorithms: PolyPhen-2 (40) and MutationTaster (41). The deletions or insertions were verified by inspection in Integrative Genomics Viewer (25).
Sanger sequencing
All mutations were confirmed by Sanger sequencing, and mutational status was also investigated in corresponding constitutional DNA. Because the involvement of the EPAS1 gene was not known when we designed our NGS assay, the mutation-prone exons 9 and 12 of EPAS1 were analyzed separately with Sanger sequencing. In one case with two different EPAS1 mutations, cloning and sequencing of EPAS1 cDNA was performed to determine whether the mutations occurred in cis or trans.
Materials and methods are described in more detail in the Supplemental Information of this paper.
Results
A next-generation sequencing panel for PCC/PGL susceptibility genes
In this NGS design, 13 PCC/PGL susceptibility genes were covered by 272 amplicons (Supplemental Table 3). Of these, 265 amplicons (97%) yielded sequence reads, with a mean depth of 915× per amplicon and sample. The amplicons that, for some or for all samples, did not yield sequencing results contained parts of EGLN1 exon 1, KIF1Bβ exons 1 and 6, NF1 exons 1 and 8, SDHC exon 3, and TMEM127 exon 2 and also MEN1 exons 9 and 10 for which design was not possible. Because NF1 was frequently mutated in the material, Sanger sequencing was performed in most samples to cover the two missing NF1 exons (Supplemental Table 5).
Mutations in PCCs and PGLs with known syndromes
NGS screening of 18 samples with previously known syndromes and/or germline mutations revealed a mutation in the expected gene in all cases (Figure 1, Supplemental Table 6, and Supplemental Figure 1), but one NF1 case also had a somatic mutation in KIF1Bβ. The three sensitivity control samples with previously defined NF1 alterations also showed expected mutations (Supplemental Table 7). The number of sequence reads for each detected mutation is provided in Supplemental Table 8.
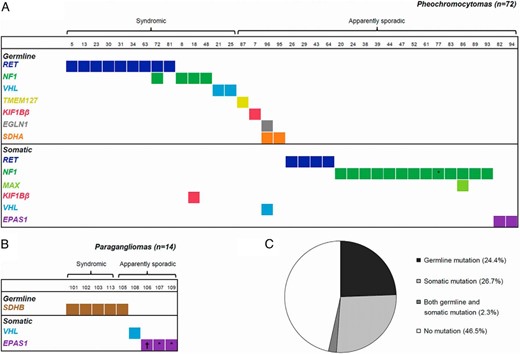
Mutations detected in an unselected cohort of 86 pheochromocytomas and paragangliomas. Tested genes included EGLN1, EPAS1, KIF1Bβ, MAX, MEN1, NF1, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2, TMEM127, and VHL. Mutations were classified as germline or somatic based on their presence or absence in constitutional DNA (blood or normal tissue). A, Distribution of mutations among pheochromocytomas. Thirty-five samples (49%) without any mutation are not shown. B, Distribution of mutations among paragangliomas. Five samples (36%) without any mutation are not shown. C, Mutation status in the entire cohort. *, The mutation could not be analyzed in constitutional DNA and is thus not known with certainty to be somatic; †, the sample had two different somatic EPAS1 mutations.
Novel mutations in apparently sporadic PCCs and PGLs
In 68 apparently sporadic tumors (58 PCCs and 10 PGLs), 32 protein-altering mutations were uncovered (Table 1 and Figure 1), which were all verified by Sanger sequencing (Supplemental Figure 1). Analysis of constitutional DNA revealed that 6 of 32 of the mutations (19%) were germline, whereas 23 (72%) were somatic (Table 1 and Supplemental Figure 1). Three mutations (9%) could not be investigated in constitutional DNA but were hypothesized to be somatic because they comprise one NF1 mutation in a patient without any features of NF1 disease and two EPAS1 mutations in a hot spot site in which all the previously identified mutations have been shown to be somatic (17–19).
Mutations Identified Among 58 Pheochromocytomas and 10 Paragangliomas With Apparently Sporadic Presentation
| Case Identification | Gene | Mutation | Protein Alteration | Presence in Normal DNA | Predicted Mutation Impact | |
|---|---|---|---|---|---|---|
| PolyPhen-2a | MutationTasterb | |||||
| 7, PCC | KIF1Bβ | c.2504A>G | p.Tyr835Cys | Yes | Probably damaging (0.999) | Disease causing |
| 20, PCC | NF1 | c.1413_1440del | p.Lys471fs | No | Truncating (frameshift) | Not analyzable |
| 24, PCC | NF1 | c.935delG | p.Gly312fs | No | Truncating (frameshift) | Disease causing |
| 26, PCC | RET | c.2753T>C | p.Met918Thr | No | Probably damaging (0.999) | Disease causing |
| 29, PCC | RET | c.2753T>C | p.Met918Thr | No | Probably damaging (0.999) | Disease causing |
| 38, PCC | NF1 | c.879delC | p.Asn293fs | No | Truncating (frameshift) | Disease causing |
| 39, PCC | NF1 | c.3638_ 3700delinsA | p.Thr1213fs | No | Truncating (frameshift) | Not analyzable |
| 43, PCC | RET | c.2372A>T | p.Tyr791Phe | No | Possibly damaging (0.563) | Disease causing |
| 44, PCC | NF1 | c.1340T>C | p.Leu447Pro | No | Probably damaging (0.999) | Disease causing |
| 47, PCC | NF1 | c.7901_7924del | p.Phe2634_Glu2641del | No | Inframe deletion of 8 aa | Not analyzable |
| 52, PCC | NF1 | c.5327_5375dup | p.Val1777X | No | Truncating (frameshift) | Not analyzable |
| 61, PCC | NF1 | c.2806A>T | p.Lys936X | No | Truncating (nonsense) | Disease causing |
| 64, PCC | RET | c.2694_2705del | p.Asp898_Glu901del | No | Unknown (inframe deletion) | Disease causing |
| 77, PCC | NF1 | c.5123delT | p.Phe1708fs | n.d. | Truncating (frameshift) | Disease causing |
| 82, PCC | EPAS1 | c.1592C>G | p.Pro531Arg | No | Probably damaging (1.000) | Disease causing |
| 83, PCC | NF1 | c.3354_3375del | p.Glu1119fs | No | Truncating (frameshift) | Not analyzable |
| 86, PCC | MAX | c.97C>T | p.Arg33X | No | Truncating (nonsense) | Disease causing |
| NF1 | c.4174–2A>T | p.Val1392_Lys1444del | No | Inframe splice errorc | Not analyzable | |
| 87, PCC | TMEM127 | c.665C>T | p.Ala222Val | Yes | Possibly damaging (0.695) | Disease causing |
| 89, PCC | NF1 | c.3232delT | p.Ser1078fs | No | Truncating (frameshift) | Disease causing |
| 93, PCC | NF1 | c.5991G>A | p.Trp1997X | No | Truncating (nonsense) | Disease causing |
| 94, PCC | EPAS1 | c.1591C>T | p.Pro531Ser | No | Probably damaging (1.000) | Disease causing |
| 95, PCC | SDHA | c.629G>A | p.Arg210Gln | Yes | Possibly damaging (0.644) | Disease causing |
| 96, PCC | EGLN1 | c.799G>A | p.Glu267Lys | Yes | Probably benign (0.031) | Disease causing |
| SDHA | c.223C>T | p.Arg75X | Yes | Truncating (nonsense) | Disease causing | |
| VHL | c.386T>C | p.Leu129Pro | No | Probably damaging (1.000) | Disease causing | |
| 105, PGL | SDHB | c.683_684delAG | p.Glu228fs | Yes | Truncating (frameshift) | Disease causing |
| 106, PGL | EPAS1 | c.1208T>C | p.Leu403Pro | No | Probably damaging (1.000) | Disease causing |
| EPAS1 | c.1589C>T | p.Ala530Val | No | Probably damaging (1.000) | Disease causing | |
| 107, PGL | EPAS1 | c.1595A>G | p.Tyr532Cys | n.d. | Probably damaging (1.000) | Disease causing |
| 108, PGL | VHL | c.593T>G | p.Leu198Arg | No | Probably damaging (0.999) | Disease causing |
| 109, PGL | EPAS1 | c.1591C>G | p.Pro531Ala | n.d. | Probably damaging (1.000) | Disease causing |
| Case Identification | Gene | Mutation | Protein Alteration | Presence in Normal DNA | Predicted Mutation Impact | |
|---|---|---|---|---|---|---|
| PolyPhen-2a | MutationTasterb | |||||
| 7, PCC | KIF1Bβ | c.2504A>G | p.Tyr835Cys | Yes | Probably damaging (0.999) | Disease causing |
| 20, PCC | NF1 | c.1413_1440del | p.Lys471fs | No | Truncating (frameshift) | Not analyzable |
| 24, PCC | NF1 | c.935delG | p.Gly312fs | No | Truncating (frameshift) | Disease causing |
| 26, PCC | RET | c.2753T>C | p.Met918Thr | No | Probably damaging (0.999) | Disease causing |
| 29, PCC | RET | c.2753T>C | p.Met918Thr | No | Probably damaging (0.999) | Disease causing |
| 38, PCC | NF1 | c.879delC | p.Asn293fs | No | Truncating (frameshift) | Disease causing |
| 39, PCC | NF1 | c.3638_ 3700delinsA | p.Thr1213fs | No | Truncating (frameshift) | Not analyzable |
| 43, PCC | RET | c.2372A>T | p.Tyr791Phe | No | Possibly damaging (0.563) | Disease causing |
| 44, PCC | NF1 | c.1340T>C | p.Leu447Pro | No | Probably damaging (0.999) | Disease causing |
| 47, PCC | NF1 | c.7901_7924del | p.Phe2634_Glu2641del | No | Inframe deletion of 8 aa | Not analyzable |
| 52, PCC | NF1 | c.5327_5375dup | p.Val1777X | No | Truncating (frameshift) | Not analyzable |
| 61, PCC | NF1 | c.2806A>T | p.Lys936X | No | Truncating (nonsense) | Disease causing |
| 64, PCC | RET | c.2694_2705del | p.Asp898_Glu901del | No | Unknown (inframe deletion) | Disease causing |
| 77, PCC | NF1 | c.5123delT | p.Phe1708fs | n.d. | Truncating (frameshift) | Disease causing |
| 82, PCC | EPAS1 | c.1592C>G | p.Pro531Arg | No | Probably damaging (1.000) | Disease causing |
| 83, PCC | NF1 | c.3354_3375del | p.Glu1119fs | No | Truncating (frameshift) | Not analyzable |
| 86, PCC | MAX | c.97C>T | p.Arg33X | No | Truncating (nonsense) | Disease causing |
| NF1 | c.4174–2A>T | p.Val1392_Lys1444del | No | Inframe splice errorc | Not analyzable | |
| 87, PCC | TMEM127 | c.665C>T | p.Ala222Val | Yes | Possibly damaging (0.695) | Disease causing |
| 89, PCC | NF1 | c.3232delT | p.Ser1078fs | No | Truncating (frameshift) | Disease causing |
| 93, PCC | NF1 | c.5991G>A | p.Trp1997X | No | Truncating (nonsense) | Disease causing |
| 94, PCC | EPAS1 | c.1591C>T | p.Pro531Ser | No | Probably damaging (1.000) | Disease causing |
| 95, PCC | SDHA | c.629G>A | p.Arg210Gln | Yes | Possibly damaging (0.644) | Disease causing |
| 96, PCC | EGLN1 | c.799G>A | p.Glu267Lys | Yes | Probably benign (0.031) | Disease causing |
| SDHA | c.223C>T | p.Arg75X | Yes | Truncating (nonsense) | Disease causing | |
| VHL | c.386T>C | p.Leu129Pro | No | Probably damaging (1.000) | Disease causing | |
| 105, PGL | SDHB | c.683_684delAG | p.Glu228fs | Yes | Truncating (frameshift) | Disease causing |
| 106, PGL | EPAS1 | c.1208T>C | p.Leu403Pro | No | Probably damaging (1.000) | Disease causing |
| EPAS1 | c.1589C>T | p.Ala530Val | No | Probably damaging (1.000) | Disease causing | |
| 107, PGL | EPAS1 | c.1595A>G | p.Tyr532Cys | n.d. | Probably damaging (1.000) | Disease causing |
| 108, PGL | VHL | c.593T>G | p.Leu198Arg | No | Probably damaging (0.999) | Disease causing |
| 109, PGL | EPAS1 | c.1591C>G | p.Pro531Ala | n.d. | Probably damaging (1.000) | Disease causing |
Abbreviation: aa, amino acids; n.d., no data (but probably somatic; see text). Nucleotide and protein nomenclature were based on the following Ensembl transcript identifiers: RET: ENST00000355710; VHL: ENST00000256474; NF1: ENST00000358273; SDHA: ENST00000264932; SDHB: ENST00000375499; TMEM127: ENST00000258439; MAX: ENST00000358664; KIF1Bβ: ENST00000263934; EGLN1: ENST00000366641; EPAS1: ENST00000263734.
Analyzes missense mutations/variants.
Analyzes nucleotide substitutions and insertions/deletions up to 12 bp in the coding sequence.
Causing skipping of exon 32.
Mutations Identified Among 58 Pheochromocytomas and 10 Paragangliomas With Apparently Sporadic Presentation
| Case Identification | Gene | Mutation | Protein Alteration | Presence in Normal DNA | Predicted Mutation Impact | |
|---|---|---|---|---|---|---|
| PolyPhen-2a | MutationTasterb | |||||
| 7, PCC | KIF1Bβ | c.2504A>G | p.Tyr835Cys | Yes | Probably damaging (0.999) | Disease causing |
| 20, PCC | NF1 | c.1413_1440del | p.Lys471fs | No | Truncating (frameshift) | Not analyzable |
| 24, PCC | NF1 | c.935delG | p.Gly312fs | No | Truncating (frameshift) | Disease causing |
| 26, PCC | RET | c.2753T>C | p.Met918Thr | No | Probably damaging (0.999) | Disease causing |
| 29, PCC | RET | c.2753T>C | p.Met918Thr | No | Probably damaging (0.999) | Disease causing |
| 38, PCC | NF1 | c.879delC | p.Asn293fs | No | Truncating (frameshift) | Disease causing |
| 39, PCC | NF1 | c.3638_ 3700delinsA | p.Thr1213fs | No | Truncating (frameshift) | Not analyzable |
| 43, PCC | RET | c.2372A>T | p.Tyr791Phe | No | Possibly damaging (0.563) | Disease causing |
| 44, PCC | NF1 | c.1340T>C | p.Leu447Pro | No | Probably damaging (0.999) | Disease causing |
| 47, PCC | NF1 | c.7901_7924del | p.Phe2634_Glu2641del | No | Inframe deletion of 8 aa | Not analyzable |
| 52, PCC | NF1 | c.5327_5375dup | p.Val1777X | No | Truncating (frameshift) | Not analyzable |
| 61, PCC | NF1 | c.2806A>T | p.Lys936X | No | Truncating (nonsense) | Disease causing |
| 64, PCC | RET | c.2694_2705del | p.Asp898_Glu901del | No | Unknown (inframe deletion) | Disease causing |
| 77, PCC | NF1 | c.5123delT | p.Phe1708fs | n.d. | Truncating (frameshift) | Disease causing |
| 82, PCC | EPAS1 | c.1592C>G | p.Pro531Arg | No | Probably damaging (1.000) | Disease causing |
| 83, PCC | NF1 | c.3354_3375del | p.Glu1119fs | No | Truncating (frameshift) | Not analyzable |
| 86, PCC | MAX | c.97C>T | p.Arg33X | No | Truncating (nonsense) | Disease causing |
| NF1 | c.4174–2A>T | p.Val1392_Lys1444del | No | Inframe splice errorc | Not analyzable | |
| 87, PCC | TMEM127 | c.665C>T | p.Ala222Val | Yes | Possibly damaging (0.695) | Disease causing |
| 89, PCC | NF1 | c.3232delT | p.Ser1078fs | No | Truncating (frameshift) | Disease causing |
| 93, PCC | NF1 | c.5991G>A | p.Trp1997X | No | Truncating (nonsense) | Disease causing |
| 94, PCC | EPAS1 | c.1591C>T | p.Pro531Ser | No | Probably damaging (1.000) | Disease causing |
| 95, PCC | SDHA | c.629G>A | p.Arg210Gln | Yes | Possibly damaging (0.644) | Disease causing |
| 96, PCC | EGLN1 | c.799G>A | p.Glu267Lys | Yes | Probably benign (0.031) | Disease causing |
| SDHA | c.223C>T | p.Arg75X | Yes | Truncating (nonsense) | Disease causing | |
| VHL | c.386T>C | p.Leu129Pro | No | Probably damaging (1.000) | Disease causing | |
| 105, PGL | SDHB | c.683_684delAG | p.Glu228fs | Yes | Truncating (frameshift) | Disease causing |
| 106, PGL | EPAS1 | c.1208T>C | p.Leu403Pro | No | Probably damaging (1.000) | Disease causing |
| EPAS1 | c.1589C>T | p.Ala530Val | No | Probably damaging (1.000) | Disease causing | |
| 107, PGL | EPAS1 | c.1595A>G | p.Tyr532Cys | n.d. | Probably damaging (1.000) | Disease causing |
| 108, PGL | VHL | c.593T>G | p.Leu198Arg | No | Probably damaging (0.999) | Disease causing |
| 109, PGL | EPAS1 | c.1591C>G | p.Pro531Ala | n.d. | Probably damaging (1.000) | Disease causing |
| Case Identification | Gene | Mutation | Protein Alteration | Presence in Normal DNA | Predicted Mutation Impact | |
|---|---|---|---|---|---|---|
| PolyPhen-2a | MutationTasterb | |||||
| 7, PCC | KIF1Bβ | c.2504A>G | p.Tyr835Cys | Yes | Probably damaging (0.999) | Disease causing |
| 20, PCC | NF1 | c.1413_1440del | p.Lys471fs | No | Truncating (frameshift) | Not analyzable |
| 24, PCC | NF1 | c.935delG | p.Gly312fs | No | Truncating (frameshift) | Disease causing |
| 26, PCC | RET | c.2753T>C | p.Met918Thr | No | Probably damaging (0.999) | Disease causing |
| 29, PCC | RET | c.2753T>C | p.Met918Thr | No | Probably damaging (0.999) | Disease causing |
| 38, PCC | NF1 | c.879delC | p.Asn293fs | No | Truncating (frameshift) | Disease causing |
| 39, PCC | NF1 | c.3638_ 3700delinsA | p.Thr1213fs | No | Truncating (frameshift) | Not analyzable |
| 43, PCC | RET | c.2372A>T | p.Tyr791Phe | No | Possibly damaging (0.563) | Disease causing |
| 44, PCC | NF1 | c.1340T>C | p.Leu447Pro | No | Probably damaging (0.999) | Disease causing |
| 47, PCC | NF1 | c.7901_7924del | p.Phe2634_Glu2641del | No | Inframe deletion of 8 aa | Not analyzable |
| 52, PCC | NF1 | c.5327_5375dup | p.Val1777X | No | Truncating (frameshift) | Not analyzable |
| 61, PCC | NF1 | c.2806A>T | p.Lys936X | No | Truncating (nonsense) | Disease causing |
| 64, PCC | RET | c.2694_2705del | p.Asp898_Glu901del | No | Unknown (inframe deletion) | Disease causing |
| 77, PCC | NF1 | c.5123delT | p.Phe1708fs | n.d. | Truncating (frameshift) | Disease causing |
| 82, PCC | EPAS1 | c.1592C>G | p.Pro531Arg | No | Probably damaging (1.000) | Disease causing |
| 83, PCC | NF1 | c.3354_3375del | p.Glu1119fs | No | Truncating (frameshift) | Not analyzable |
| 86, PCC | MAX | c.97C>T | p.Arg33X | No | Truncating (nonsense) | Disease causing |
| NF1 | c.4174–2A>T | p.Val1392_Lys1444del | No | Inframe splice errorc | Not analyzable | |
| 87, PCC | TMEM127 | c.665C>T | p.Ala222Val | Yes | Possibly damaging (0.695) | Disease causing |
| 89, PCC | NF1 | c.3232delT | p.Ser1078fs | No | Truncating (frameshift) | Disease causing |
| 93, PCC | NF1 | c.5991G>A | p.Trp1997X | No | Truncating (nonsense) | Disease causing |
| 94, PCC | EPAS1 | c.1591C>T | p.Pro531Ser | No | Probably damaging (1.000) | Disease causing |
| 95, PCC | SDHA | c.629G>A | p.Arg210Gln | Yes | Possibly damaging (0.644) | Disease causing |
| 96, PCC | EGLN1 | c.799G>A | p.Glu267Lys | Yes | Probably benign (0.031) | Disease causing |
| SDHA | c.223C>T | p.Arg75X | Yes | Truncating (nonsense) | Disease causing | |
| VHL | c.386T>C | p.Leu129Pro | No | Probably damaging (1.000) | Disease causing | |
| 105, PGL | SDHB | c.683_684delAG | p.Glu228fs | Yes | Truncating (frameshift) | Disease causing |
| 106, PGL | EPAS1 | c.1208T>C | p.Leu403Pro | No | Probably damaging (1.000) | Disease causing |
| EPAS1 | c.1589C>T | p.Ala530Val | No | Probably damaging (1.000) | Disease causing | |
| 107, PGL | EPAS1 | c.1595A>G | p.Tyr532Cys | n.d. | Probably damaging (1.000) | Disease causing |
| 108, PGL | VHL | c.593T>G | p.Leu198Arg | No | Probably damaging (0.999) | Disease causing |
| 109, PGL | EPAS1 | c.1591C>G | p.Pro531Ala | n.d. | Probably damaging (1.000) | Disease causing |
Abbreviation: aa, amino acids; n.d., no data (but probably somatic; see text). Nucleotide and protein nomenclature were based on the following Ensembl transcript identifiers: RET: ENST00000355710; VHL: ENST00000256474; NF1: ENST00000358273; SDHA: ENST00000264932; SDHB: ENST00000375499; TMEM127: ENST00000258439; MAX: ENST00000358664; KIF1Bβ: ENST00000263934; EGLN1: ENST00000366641; EPAS1: ENST00000263734.
Analyzes missense mutations/variants.
Analyzes nucleotide substitutions and insertions/deletions up to 12 bp in the coding sequence.
Causing skipping of exon 32.
Four PCCs had somatic RET mutations, one of which was a 12-bp deletion in exon 15 that has previously been reported in a medullary thyroid carcinoma (26). The other three involved codon 791 and the hot spot codon 918 frequently altered in medullary thyroid carcinoma. One PCC and one PGL had somatic missense VHL mutations, and one PGL had a germline frameshift mutation in SDHB. Thirteen PCCs had somatic mutations in the NF1 gene, of which only one has been reported before (Trp1997X, reported in Catalogue Of Somatic Mutations In Cancer in a neurofibroma). In the cases of splice-site mutations, the erroneous splicing was verified in cDNA derived from the same tumor tissue (Supplemental Figure 2).
Among the less studied susceptibility genes, one PCC had a somatic nonsense mutation in MAX in a previously reported hot spot for germline mutations (5, 6), and another PCC revealed a germline missense mutation in TMEM127 (reported only once with allele frequency of 0.0005 in African Americans analyzed by the National Heart, Lung, and Blood Institute Exome Sequencing Project, release 6500). Two PCCs had germline mutations in SDHA, one missense and one nonsense, of which the latter was recently reported in another Swedish patient with PGL (27). Furthermore, one PCC had a novel germline KIF1Bβ mutation, and another PCC displayed a novel germline mutation in EGLN1.
Six EPAS1 mutations were identified in two PCCs and three PGLs. One PGL sample had two somatic EPAS1 mutations: one in exon 9, only two residues from the hydroxylation site Pro405, and the other in exon 12, one residue from the hydroxylation site Pro531. Cloning and sequencing of cDNA revealed that the two mutations were in cis (Supplemental Figure 3). Apart from the above-mentioned mutation in exon 9, all EPAS1 mutations occurred in or near the primary hydroxylation site in exon 12.
Bioinformatic analysis suggests that all detected mutations are probably disease causing, with the exception of the one in EGLN1, which was predicted as benign by one algorithm and damaging by the other (Table 1). For some of the identified mutations, the ratio between the numbers of reads may suggest a tumor heterogeneity and/or loss of the normal allele (Supplemental Table 8). Five different rare missense polymorphisms of unknown pathogenic significance were also detected among the investigated tumors (Supplemental Table 4). Prediction algorithms indicated that some of the variants may be pathogenic, but because the frequencies of the variants in the tumor material were similar to previously reported population frequencies (Supplemental Table 4), there is no indication of their involvement in PCC/PGL disease at the present, and they were therefore not included in mutation counts or statistical analysis. Reanalysis of medical records showed that the patients carrying the MEN1 polymorphism did not suffer from any MEN1-related disease.
Clinical features of cases with mutations
The entire cohort consisted of 52% females and 48% males, with a mean age at surgery of 51 years. The tumors included 84% PCCs and 16% PGLs, 21% malignant according to Armed Forces Institute of Pathology criteria (9% according to World Health Organization criteria). Patients with germline mutations were younger than patients with somatic or no mutations (mean 40 y vs 55 y, P = .00012), with VHL and SDHB patients being the youngest (mean 22 y and 28 y, respectively) (Supplemental Figure 4). No significant differences in tumor size or hormone levels were observed between different genotype groups, but when considering the previously known gene expression clusters (1), RET/NF1/TMEM127/KIF1Bβ cases had higher epinephrine levels than VHL/SDHx/EPAS1 cases (P = .026). Both SDHB mutations (P < .0001) and EPAS1 mutations (P = .029) were associated with PGL. SDHB mutations were also associated with malignancy (according to the World Health Organization, P = .0050 and according to Armed Forces Institute of Pathology, P = .0078), whereas all EPAS1 cases were benign. NF1 mutations occurred only in PCCs, but the association was not significant. Three cases of PCCs were bilateral (all MEN2A), and two cases presented with multiple PGLs (one with a germline SDHB mutation and one with somatic double mutation in EPAS1). Upon reinvestigation of medical records, two patients with somatic EPAS1 mutations were found to have polycythemia vera. The clinical data of patients with apparently sporadic PCC/PGL who carried germline mutations are summarized in Table 2.
Clinical and Genetic Data for Cases With Apparently Sporadic Presentation Who Carried Germline Mutations
| Case Identification | Mutation | Age, y | Gender | Tumor Type | Tumor Size, Weight | Malignancy | Hormonal Profile EPI/NE/DA | Other Cancers |
|---|---|---|---|---|---|---|---|---|
| 7 | KIF1Bβ p.Tyr835Cys | 54 | Female | PCC | 3 × 3 × 2.5 cm, 16 g | Benign | N/E/− | Endometrial carcinoma |
| 87 | TMEM127 p.Ala222Val | 55 | Female | PCC | 5.5 × 4 × 2.5 cm, 48 g | Benign | N/E/− | Not found |
| 95 | SDHA p.Arg210Gln | 64 | Male | PCC | 6 × 5 × 3 cm, 66 g | Benign | N/E/− | Not found |
| 96 | SDHA p.Arg75X | 47 | Male | PCC | 3.6 × 2.5 × 1.6 cm, 23 g | Benign | −/−/− | Not found |
| EGLN1 p.Glu267Lys | ||||||||
| 105 | SDHB p.Glu228fs | 35 | Female | PGL | 3 × 2 × 3 cm, − | Malignant (metastasis after 12 y) | N/N/N | 3 Cholesteatomas |
| Case Identification | Mutation | Age, y | Gender | Tumor Type | Tumor Size, Weight | Malignancy | Hormonal Profile EPI/NE/DA | Other Cancers |
|---|---|---|---|---|---|---|---|---|
| 7 | KIF1Bβ p.Tyr835Cys | 54 | Female | PCC | 3 × 3 × 2.5 cm, 16 g | Benign | N/E/− | Endometrial carcinoma |
| 87 | TMEM127 p.Ala222Val | 55 | Female | PCC | 5.5 × 4 × 2.5 cm, 48 g | Benign | N/E/− | Not found |
| 95 | SDHA p.Arg210Gln | 64 | Male | PCC | 6 × 5 × 3 cm, 66 g | Benign | N/E/− | Not found |
| 96 | SDHA p.Arg75X | 47 | Male | PCC | 3.6 × 2.5 × 1.6 cm, 23 g | Benign | −/−/− | Not found |
| EGLN1 p.Glu267Lys | ||||||||
| 105 | SDHB p.Glu228fs | 35 | Female | PGL | 3 × 2 × 3 cm, − | Malignant (metastasis after 12 y) | N/N/N | 3 Cholesteatomas |
Abbreviations: DA, dopamine levels in urine before surgery; E, elevated; EPI, epinephrine levels in plasma or urine before surgery; N, normal; NE, norepinephrine levels in plasma or urine before surgery; −, missing data.
Clinical and Genetic Data for Cases With Apparently Sporadic Presentation Who Carried Germline Mutations
| Case Identification | Mutation | Age, y | Gender | Tumor Type | Tumor Size, Weight | Malignancy | Hormonal Profile EPI/NE/DA | Other Cancers |
|---|---|---|---|---|---|---|---|---|
| 7 | KIF1Bβ p.Tyr835Cys | 54 | Female | PCC | 3 × 3 × 2.5 cm, 16 g | Benign | N/E/− | Endometrial carcinoma |
| 87 | TMEM127 p.Ala222Val | 55 | Female | PCC | 5.5 × 4 × 2.5 cm, 48 g | Benign | N/E/− | Not found |
| 95 | SDHA p.Arg210Gln | 64 | Male | PCC | 6 × 5 × 3 cm, 66 g | Benign | N/E/− | Not found |
| 96 | SDHA p.Arg75X | 47 | Male | PCC | 3.6 × 2.5 × 1.6 cm, 23 g | Benign | −/−/− | Not found |
| EGLN1 p.Glu267Lys | ||||||||
| 105 | SDHB p.Glu228fs | 35 | Female | PGL | 3 × 2 × 3 cm, − | Malignant (metastasis after 12 y) | N/N/N | 3 Cholesteatomas |
| Case Identification | Mutation | Age, y | Gender | Tumor Type | Tumor Size, Weight | Malignancy | Hormonal Profile EPI/NE/DA | Other Cancers |
|---|---|---|---|---|---|---|---|---|
| 7 | KIF1Bβ p.Tyr835Cys | 54 | Female | PCC | 3 × 3 × 2.5 cm, 16 g | Benign | N/E/− | Endometrial carcinoma |
| 87 | TMEM127 p.Ala222Val | 55 | Female | PCC | 5.5 × 4 × 2.5 cm, 48 g | Benign | N/E/− | Not found |
| 95 | SDHA p.Arg210Gln | 64 | Male | PCC | 6 × 5 × 3 cm, 66 g | Benign | N/E/− | Not found |
| 96 | SDHA p.Arg75X | 47 | Male | PCC | 3.6 × 2.5 × 1.6 cm, 23 g | Benign | −/−/− | Not found |
| EGLN1 p.Glu267Lys | ||||||||
| 105 | SDHB p.Glu228fs | 35 | Female | PGL | 3 × 2 × 3 cm, − | Malignant (metastasis after 12 y) | N/N/N | 3 Cholesteatomas |
Abbreviations: DA, dopamine levels in urine before surgery; E, elevated; EPI, epinephrine levels in plasma or urine before surgery; N, normal; NE, norepinephrine levels in plasma or urine before surgery; −, missing data.
Discussion
In this study, we designed a targeted NGS assay for all so far known susceptibility genes in pheochromocytoma and paraganglioma and used it for mutation analysis in a cohort of unselected tumors. In addition to the most commonly tested genes, our resequencing effort included KIF1Bβ, which has previously not been investigated in large cohorts, NF1, in which somatic alterations were recently shown to be of importance, and MEN1, which has had a disputed involvement.
Mutations in KIF1Bβ have previously only been reported in a few, isolated cases. Patients with germline KIF1Bβ mutations have been reported with other tumors, such as lung adenocarcinomas, colorectal carcinomas, and neuroblastomas (9). Here we report a pheochromocytoma patient with a germline missense mutation in KIF1Bβ who later presented with an endometrial carcinoma. The mutation affects a highly conserved region of the protein close to the site of the functionally relevant T827I mutation identified in neuroblastoma (8, 28). Another PCC had a somatic missense mutation in KIF1Bβ in combination with a germline NF1 mutation. Even though a germline KIF1Bβ mutation was detected in only one case, the frequency in apparently sporadic pheochromocytomas (1.7%) is similar to previously reported frequencies of germline MAX and TMEM127 mutations (4, 6). This suggests that KIF1Bβ may be an equally important tumor suppressor gene that warrants further studies. Germline SDHA mutations have, so far, mainly been associated with PGLs and rarely reported in PCC (29), but here we report two mutations in PCC patients. Mutations in MAX, TMEM127, RET, VHL, and SDHB were found in expected frequencies (2, 4, 6, 14), whereas no mutations were detected in SDHC, SDHD, or SDHAF2. Furthermore, we did not detect any mutations in the MEN1 gene, suggesting that it is of no or minor importance in sporadic PCC/PGL. One PCC patient carried a germline mutation in EGLN1, causing a substitution of glutamic acid against lysine. Two different prediction algorithms gave inconclusive results regarding its pathogenicity, and the importance in PCC is uncertain.
More than 22% of the apparently sporadic PCCs harbored somatic mutations in NF1, which was the most frequently mutated gene in the cohort. This is in agreement with the recent discovery of somatic NF1 mutations (30) and confirms the role of NF1 as an important tumor suppressor in nonfamilial PCC. In agreement with recent findings (17–19), we identified somatic EPAS1 mutations in 7.4% of sporadic tumors but observed that they were 10 times more prevalent in PGLs, of which as many as 30% had mutations, compared with PCCs (3.4%). Two of the patients with EPAS1 mutations also had polycythemia, which is in agreement with previous findings (17–20). Interestingly, one PGL displayed two somatic EPAS1 mutations, in cis, that were close to the Pro405 and the Pro531 hydroxylation sites, respectively. The finding of a double mutation is in agreement with our previous observation of two different EPAS1 mutations in a PCC, which were also in cis and in proximity to each of the hydroxylation sites (42). Speculatively, the inactivation of both hydroxylation sites may lead to an even higher stability and accumulation of the HIF-2α protein, and thus a greater cell survival advantage, than one mutation may cause alone, as previously has been demonstrated for the HIF-1α protein (31).
Taken together, the identified mutations (Figure 1) support that PCCs and abdominal PGLs have somewhat different genetic backgrounds despite their common origin in chromaffin cells. Interestingly, some PCCs harbored deleterious mutations in more than one of the investigated genes, for example a splice-site NF1 mutation in combination with a nonsense MAX mutation. These results, as well as two recently reported cases with mutations in two different susceptibility genes (16), suggest that genetic alterations in two or more susceptibility genes may contribute to the tumor development. However, further studies will be required to identify which events are drivers in the tumorigenesis. In previous studies of subsets of the cases we have shown a CpG island methylator phenotype (CIMP) in association with SDHB mutation (24, 32), in agreement with the recent demonstration of a hypermethylator phenotype in SDHx-related tumors (33). In the present cohort, CIMP was associated with SDHB mutation but not with other mutations, and CIMP was also observed in one wild-type malignant paraganglioma (Supplemental Table 1).
In total, our results showed that 26.7% of all cases carried germline mutations. Notably, as many as 7% of the apparently sporadic cases in our cohort carried germline mutations, even though no family history or other signs of hereditary disease were known based on the patient's medical records. The clinical follow-up of such patients should include renewed family history and, upon establishment of the mutation as germline, follow-up of family members should be considered. In addition, mutation carriers may benefit from screening for other tumors because mutations in many of the genes are associated with different cancer types (1) and patients with SDHB mutations have a well-known high risk of PCC/PGL metastasis (23). In such cases, genetic testing could also be offered to family members. For KIF1Bβ mutations the clinical phenotype is not established due to the low number of cases so far, but family investigation and surveillance to discover other tumors could be justified (9). The lack of family history for the cases reported here would indicate that the penetrance is variable and needs further family investigations. Explanations may include de novo mutations or mechanisms such as copy number variations or epigenetic regulation of gene expression that may contribute to the development of PCC/PGL.
Concerning the methodology, the targeted NGS approach allowed simultaneous analysis of the known PCC/PGL susceptibility genes with a high sequencing depth (mean 915× when multiplexing 48 samples), 100% sensitivity in samples with known syndromes and/or mutations and a high specificity since all mutations detected with the method could be validated with Sanger sequencing. Of the intended targets, 3% did not yield sequence results. It may be possible to obtain better results with optimization, for example, a redesign of the problematic enrichment probes or potentially by using other commercially available amplicon enrichment assays. However, very GC-rich regions may remain particularly difficult to target because this is a known weakness of available capture techniques (34). For diagnostic procedures, awareness of such not captured regions will be essential to estimate the risk of false-negative results, although it should not hinder the use of NGS for efficient mutation analysis of the vast majority of exons. The laboratory procedure for the library preparation and NGS used in this study required three days of hands-on time for 48 samples and 174 exons (272 amplicons). In our hands, reagent costs were approximately 1 € per exon and sample (184 € for all genes in one sample), which corresponds to about one eighth of the reagent cost for standard Sanger sequencing (8 € per exon and sample) in our laboratory. The bioinformatic pipeline for alignment and variant calling provided by Illumina was straightforward, but required editing of the default thresholds to enable the detection of insertions and deletions larger than 25 bp. This was essential because three cases had exonic insertions or deletions of 28, 49, and 63 nucleotides.
As for many other tumor forms, whole-exome sequencing has proved to be a valuable tool for discovering new genes involved in PCC and PGL (5, 19, 33, 35). Two studies, limited to three and 11 samples, have also attempted whole-exome sequencing for clinical genetic screening in PCC and PGL (36, 37). Here we show that NGS on a smaller scale offers an opportunity for efficient targeted genetic analysis. For potential diagnostic use, the targeted approach is less costly and likely to raise less ethical concerns regarding patient integrity and incidental findings. In a recent study, genetic testing in PCC and PGL was performed with Roche 454 technology (38). The authors reported the technique as rapid and cost-effective compared with Sanger sequencing but experienced similar drawbacks as reported here with some exons not yielding results. However, the study was limited to nine PCC/PGL susceptibility genes and did, for example, not include the large NF1 and KIF1Bβ genes, which are especially difficult and costly to sequence manually. After the completion of this resequencing effort, the genetic landscape of PCCs and PGLs has been further characterized through the discovery of somatic HRAS mutations reported elsewhere (35) and germline FH (fumarate hydratase) mutations (39). This further supports the role of RAS/RAF/MAPK activation and tricarboxylic acid cycle dysfunction, respectively, in subsets of the tumors and highlights the need for high throughput sequencing to uncover their genetic complexity.
In conclusion, our results show that targeted NGS on a bench-top instrument is a fast and cost-effective method for genetic analysis of PCC and PGL, but whereas the most commonly analyzed genes were covered, optimization would be required to cover the small proportion of targets that did not yield results. The usefulness in a clinical setting was demonstrated when we used the same gene panel and method for genetic testing of a young PGL patient, who was found to carry a nonsense SDHA mutation (27). In addition, the results show that the identified familial susceptibility genes are of importance in a large proportion of the nonfamilial tumors: 41% were found to have either germline or somatic mutations, strengthening the role of the so far known genes in the tumor development. However, more than half of the sporadic tumors still remain without any known molecular marker for the origin of their disease. Further research to discover additional driver events and markers of malignancy is warranted and could involve genome-wide search for coding mutations and epigenetic modifications as well as alterations of regulatory regions.
Apart from those genes known for many years (eg, RET, VHL), our knowledge regarding the prevalence of PCCs/PGLs, and the complete phenotype of patients with mutations in the less commonly involved genes (eg, KIF1Bβ) is limited. The main reason is that comprehensive mutation analysis has been time consuming and costly, and genetic testing has therefore often been based on incomplete clinical and/or histopathological parameters. This has resulted in an underestimation especially of the rarely affected genes and an incomplete knowledge of the associated phenotypes. This study shows that the costs for analyzing all known candidate genes simultaneously, independently of other parameters, can be kept low due to new techniques. By applying such techniques in all cases with PCCs/PGLs it is expected that we can broaden our knowledge as described above, which may facilitate a personalized management of the patients in terms of surveillance and potential target-directed therapies.
Acknowledgments
We thank Ms Lisa Ånfalk (Karolinska University Hospital) for excellent blood and tissue collection and handling.
This work was supported by grants from the University of Linköping (to O.G.), the LiU Cancer Network (to O.G. and P.S.), the Swedish Research Council (to P.S. and C.L.), the Cancer Society in Stockholm (to M.B., A.H., and C.L.), the Swedish Cancer Society (to C.L.), and StratCan at Karolinska Institutet (to A.A.). Support for the Illumina sequencing was provided by the Genetics Services Unit at the Wisconsin National Primate Research Center, Grant P51RR000167/P51OD011106.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- CIMP
CpG island methylator phenotype
- HIF
hypoxia-inducible factor
- MEN
multiple endocrine neoplasia
- MEN1
MEN type 1
- MEN2
MEN type 2
- NF1
neurofibromatosis type 1
- NGS
next-generation sequencing
- PCC
pheochromocytoma
- PGL
paraganglioma.
References
Author notes
J.W. and A.A. contributed equally to this work. O.G. and P.S. contributed equally to this work.

{kind=link}