





Abstract
Whereas human neutrophils are effective and efficient killers of bacteria, macrophages such as those derived from monocytes are almost devoid of killing activity. Nevertheless, monocytes can be transformed into effective killers of mycobacteria or staphylococci when exposed to clinical concentrations of a phenothiazine or to inhibitors of efflux pumps (reserpine and verapamil), or to ouabain, an inhibitor of K+ transport. Because the rates of multidrug-resistant Mycobacterium tuberculosis (MDR-TB) continue to escalate globally, and because no new effective drug has been made available for almost 40 years, compounds that enhance the killing activity of monocytes against MDR-TB are obviously needed. This review covers the specific characteristics of MDR-TB, identifies a variety of agents that address these characteristics and therefore have potential for managing MDR-TB. Because the mechanism by which these agents enhance the killing of intracellular bacteria is important for the intelligent design of new anti-tubercular agents, the review correlates the mechanisms by which these agents manifest their effects. Lastly, a model is presented which describes the mechanisms by which distinct efflux pumps of the phagosome–lysosome complex are inhibited by agents that are known to inhibit K+ flux. The model also predicts the existence of a K+ activated exchange (pump) that is probably located in the membrane that delineates the lysosome. This putative pump, which is immune to inhibitors of K+ flux, is identified as being the cause for the acidification of the lysosome thereby activating its hydrolytic enzymes. Because the non-killer macrophage can be transformed into an effective killer by a variety of compounds that inhibit K+ transport, perhaps it would be wise to develop drugs that enhance the killing activity of these cells inasmuch as this approach would not be subject to any resistance, as is the eventual case for conventional antibiotics.
Introduction
Mycobacterium tuberculosis has infected man since the birth of civilization.1,2 This long association has in all probability been the selection process by which this organism evolved into a steadfast human pathogen. The steadfast nature of this human infection has been the basis for its eventual elimination as a human pathogen inasmuch as the effectiveness of therapy with antibiotics available in the 1950s was readily proven by the steady decline of new cases of pulmonary tuberculosis throughout the Western World.3,4 With respect to Third World countries, infections continued to increase due to man-made conditions such as famine, war and over-crowding, as well as to the cost of anti-tubercular drugs, which for many countries was beyond their affordability.5 The movement of large numbers of people (refugees) from Third World countries that had experienced war, political strife or famine, to parts of the West, contributed decades later, to significant increases of new cases of pulmonary tuberculosis. Although these new cases of pulmonary tuberculosis were initially restricted to these immigrants,6–8 the infection began to spread rapidly among the indigenous population of the cities where the immigrants had settled.8 Despite the availability of effective anti-tubercular compounds, new cases of pulmonary tuberculosis continued to escalate, and in some cities such as New York, the rate of new cases was extremely alarming.9,10 One of the reasons for this escalation involved the appearance of large numbers of cases that were infected with M. tuberculosis that was resistant to the two most effective anti-tubercular drugs, isoniazid and rifampicin;11 hence, patients infected with multidrug-resistant M. tuberculosis (MDR-TB) strains were sources for new transmissions of infections, which rapidly manifested themselves as active disease in patients co-infected with HIV.12 Moreover, because of the availability of anti-tubercular drugs and ineffective therapy, strains initially susceptible to isoniazid and rifampicin could infect another patient who, due to the selection of a spontaneous mutation that resulted in resistance to one antibiotic, would be managed poorly with these agents, and a second spontaneous mutation that caused resistance to the second antibiotic would be selected.13 Therapy of MDR-TB is extremely problematic, regardless of whether one uses all five first-line drugs of defence (isoniazid, rifampicin, streptomycin, ethambutol, pyrazinamide), or second line of defence compounds.14 Furthermore the use of these therapeutic modalities produces significant morbidity in view of the problem of non-compliance.15 Hence new cases of MDR-TB have continued to increase in many of the urban centres of Western and Third World countries.16
Tuberculosis: the disease and requirements for an effective anti-TB drug
Mycobacterium tuberculosis infects the lung parenchyma intracellularly and generally remains silent for 90% of those infected, and for many of these cases, the infection is resolved, and for still fewer cases, evidence of infection is noted only at autopsy. When the organism breaks free of its intracellular prison in large numbers it manifests its presence by dissemination to other sites of the lung, thereby increasing the size of the area infected. At this time the organism may be expelled via micro-droplets of sputum to the environment when the patient coughs. When this occurs the infection has progressed to one of active disease. At this stage the patient is infectious. The percentage of infected patients that progress to active disease can be drastically increased by conditions that promote immuno-incompetence.17–24
An effective anti-tubercular drug must satisfy two conditions—namely, it must be able to inhibit the replication of M. tuberculosis at its intracellular location and/or kill the organism directly at that site.15,25,26 Surprisingly, with few exceptions, almost all studies that report on the anti-tubercular activity of old or new drugs have been conducted in vitro. Subsequent to these studies, the compounds are then tested in animal models for their ability to cure or reduce a tuberculosis infection. Because very few, if any, show any effectiveness on the infected mouse, it is not surprising that few of these compounds reach clinical trials, and even fewer may make it to the market place. For this reason the last effective anti-TB compound that reached the market was rifampicin, almost 40 years ago.27 Why is there such a discrepancy between the many drugs that demonstrate activity in vitro and fail to cure a TB infection in vivo? This is probably due to their inability to reach the intracellular organism or maintain activity at that site.28,29
The phenothiazines: compounds that satisfy the two essential aspects of an effective anti-tubercular drug
The antimicrobial activity of phenothiazines has been known for over a century.26 The first phenothiazine to be examined for antibacterial properties was the dye methylene blue.26,30 This dye (Figure 1) could render mobile bacteria immobile31,32 as well as inhibit the in vitro growth of some Gram-positive bacteria.33–35 However, soon after the demonstration of its antibacterial properties, the dye was shown to cause cats to become lethargic.36 Interest in the neuroleptic properties of the dye overshadowed its antimicrobial properties—hence, the dye was used as a lead compound for the synthesis of the first neuroleptic, chlorpromazine, which is a colourless phenothiazine (Figure 1). The availability of chlorpromazine in 1957 resulted in its worldwide use for the therapy of psychoses and severe neuroses, through which it soon became clear that chlorpromazine had antibacterial properties, including those of an anti-tubercular drug.37 Nevertheless, little interest in chlorpromazine, or any other phenothiazine, as an antibacterial drug (antibiotic) was generated, since it was the ‘Golden Age of Antibiotics’38 and the drugs available at that time (1960s) were shown to be very effective at controlling tuberculosis, given the rapid and steady decline of the disease.39 However, with the extensive use of antibiotics, the problem of resistance began to appear in the early 1960s40 and escalated to such significant levels that for many bacterial infections, treatment became problematic.41–44 Although the response of the pharmaceutical industry kept pace initially with the problem, by rapidly making available new antibiotics, the time between the introduction of a given antibiotic and ensuing significant rates of resistance grew shorter.45–48 By the 1990s, antibiotic resistance became the ‘norm’ for many bacterial infections; with respect to M. tuberculosis this was to rifampicin and isoniazid, the two most effective anti-tubercular compounds. MDR-TB was significant in many areas of the world, including urban centres in Western Europe (e.g. Lisbon, Barcelona), and in Northern Europe (e.g. Riga).49 The failure of conventional anti-tubercular therapy experienced early in the 1990s spurred a search for new anti-tubercular drugs—a search that has thus far resulted in no new compounds that are as effective as those to which resistance had developed. Because the problem of MDR-TB primarily took place in countries that were economically disadvantaged,27,28 the required incentive was not present for the creation of new and effective compounds, given the high cost of drug development and the poor market conditions present in the countries affected. Prior to and during the emergence of MDR-TB, phenothiazines had been observed to have potential for the therapy of tuberculosis.25 The studies described in that review indicated that the administration of chlorpromazine to patients presenting with tuberculosis resulted in cures.25 The ability of chlorpromazine to cure tuberculosis resulted in a series of in vitro studies that not only showed that chlorpromazine was an effective anti-mycobacterial compound,50–54 but also that other phenothiazines (see Figure 1) were equally effective.51,55–62 Nevertheless, the concentrations of chlorpromazine and other phenothiazines required for in vitro inhibition of mycobacterial growth were well beyond those that could be achieved in the patient.25,26 However, because phenothiazines were known to be concentrated by tissues and organs containing large populations of macrophages,63–65 Crowle et al. demonstrated that physiological concentrations of chlorpromazine present in the medium could enhance the killing of M. tuberculosis that had been phagocytosed by macrophages.50 Because chronic administration of chlorpromazine is known to produce a wide range of mild-to-severe side effects, the use of this compound for the therapy of tuberculosis was not seriously considered.25,26 Given the fact that phenothiazines are concentrated by macrophages and other cells rich in lysosomes,66–72 thioridazine (Figure 1), being equivalent to chlorpromazine in all of its anti-mycobacterial properties,25,26,53,54 was studied for its ability to enhance the killing of phagocytosed bacteria, including M. tuberculosis.73–75 This series of ex vivo studies demonstrated what had been previously observed for chlorpromazine,50 namely that concentrations in the medium that were below those present in the plasma of patients chronically treated with thioridazine could result in the killing of intracellular mycobacteria.73 These results led the way for studies that would determine whether phenothiazines could reduce bacterial load of the lung in mice infected with M. tuberculosis.
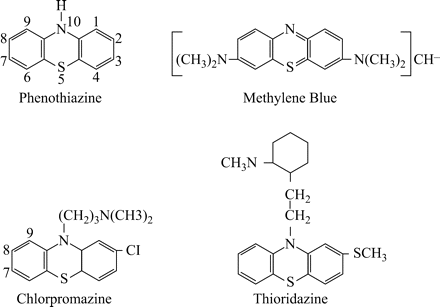
Methylene blue, chlorpromazine, thioridazine and general phenothiazine structures.
That thioridazine can effectively reduce that load is shown by the preliminary study summarized in Figure 2. However, because the manner of infection involved massive intra-peritoneal doses of M tuberculosis, whereas the number of organisms of the pulmonary system could be effectively reduced, those present in the spleen and liver were relatively unaffected. Hence, the possibility of vertical transmission of M. tuberculosis to the lungs or other organs remained a distinct possibility. Since that demonstration, others have shown that derivatives of chlorpromazine could also reduce the number of organisms recovered from the lungs of the infected mouse.76,77 Other phenothiazines have also been suggested for the management of tuberculosis.57,58,61
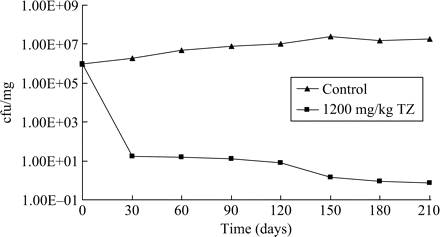
Effect of thioridazine (TZ) on the recovery of M. tuberculosis from infected mice. Four groups, each consisting of eight female mice were infected intraperitoneally with approx. 106 cfu of M. tuberculosis H37Rv ATCC 27294 and treated 3 days later with daily doses of thioridazine (100, 400 or 1200 mg/kg). The fourth group received no drug. At intervals of 30 days one mouse from each of the four groups was sacrificed, their lungs removed, weighed and homogenized, and mycobacteria were freed from cellular debris with the use of NaOH/sodium citrate/cysteine. Following centrifugation to remove cell debris, the supernatant was used for the preparation of serial dilutions required for colony-forming unit determination. Because treatment with daily doses of 100 and 400 mg/kg produced marginal results, these data are not shown. Controls, filled triangles; 1200 mg/kg, filled squares.
How does a phenothiazine enhance the killing of intracellular bacteria including mycobacteria?
Phenothiazines have been shown to inhibit the transport of K+ from external to internal cellular compartments such as transport channels of cardiac Kir2.1 cells and red blood cells78–80 and between intracellular compartments (e.g. diencephalic neurons, rat liver mitochondria).81,82 They also inhibit the binding of calcium to calmodulin, the calcium binding protein of mammalian cells.83 The binding of calcium to calmodulin-like proteins of bacteria has been amply demonstrated.84–93 The inhibition of calcium access to Ca2+-dependent ATPase inhibits transport processes such as those performed by influx and efflux pumps.94–96 Because phenothiazines inhibit access to calcium, they inhibit the activity of calcium-dependent ATPase, and hence the transport processes.94–101
Bacteria as well as mammalian cells contain efflux pumps that extrude noxious agents from the periplasm and cytoplasm of the former102 and from the cytoplasm of the latter.103 Understanding the effects of a phenothiazine on calcium-dependent transport processes of the bacterium or the mammalian cell predict that their respective efflux pumps will be affected by a phenothiazine. This prediction has been shown to be correct whenever studied.94–101 Since the first contact of phenothiazine with the bacterium takes place at the surface of the cell envelope, it would be expected that although the phenothiazine could penetrate this structure as a consequence of a concentration gradient and its amphipathic structure, one of the many efflux pumps of the bacterium would recognize this molecule and extrude it once it reached the periplasm of the bacterium. Although this expectation has yet to be fully studied, the effect of phenothiazine on the efflux system of the bacterial cell has been shown to be one of inhibition.96–101 Phenothiazines readily intercalate between nucleic bases of the DNA helix.104–108 The degree of intercalation is dependent upon the number of guanosine-cytosine residues.105 When phenothiazines intercalate into DNA they inhibit all DNA-based processes as well as the degree of coiling and uncoiling of DNA promoted by gyrases.109 The inhibition of the efflux pump by a phenothiazine would result in large numbers of phenothiazine molecules entering the cell, reaching their intercalative sites of the DNA and thereby inhibiting the replication of the bacterium.
The concentrations of the phenothiazine in the medium required for the inhibition of bacterial replication vary greatly depending on the type of bacterium: the MIC of phenothiazines for Gram-negative bacteria ranges from 100 to 200 mg/L, whereas for Gram-positive bacilli or cocci the MIC ranges from 10 to 40 mg/L.26 Although not proven, there seems to be a strong correlation between bacteria that contain a highly effective system of efflux pumps and the concentration of the phenothiazine required for the in vitro inhibition of replication. As an example, as much as 35% of the genome of Gram-negative bacteria codes for influx/efflux pumps (transporters) and, with respect to Escherichia coli, as many as 20 MDR efflux pumps have been genetically characterized.102 Although this organism has an obvious redundancy of efflux pumps, the major efflux pump that accounts for MDR in this organism is coded by the acrAB-TolC operon.97 When this operon is deleted, the organism becomes extremely susceptible to antibiotics, and to chlorpromazine and thioridazine.97 When both the acrAB-intact and acrAB-deleted strains are induced to high levels of resistance to tetracycline, the genetic expression of the AcrAB efflux pump of the former has been increased 6-fold whereas the AcrEF efflux pump of the latter strain has been increased 80-fold.97 The increased genetic expression of these pumps renders both strains significantly more resistant to chlorpromazine and thioridazine than their parents. These results support the contention that chlorpromazine and thioridazine are substrates of the AcrAB and AcrEF efflux pump systems and that when the ability of the pumps to extrude the agents is exceeded, the agents reach their intercalative targets and bacterial replication is inhibited. Because the concentration of either phenothiazine in the medium that enhances the killing of intracellular bacteria is about a 100th of that needed to kill the bacterium in vitro, 73–77 the killing activity appears to be related to the phenothiazine being concentrated by the non-killing macrophage to levels comparable to those required for killing in vitro.50 Electron microscopic studies of the effects of phenothiazines on phagocytosed Staphylococcus aureus demonstrate that the in vitro effect of these agents on the bacterium's morphology is reproduced when the bacterium is phagocytosed by macrophages that are subsequently exposed to concentrations of the agent (see Figure 3) which produce neither an inhibition of replication nor a change in the morphology of the organism.110
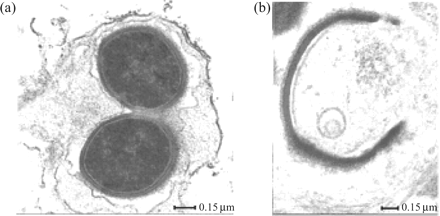
The ex vivo effects of thioridazine on the ultrastructure of Staphylococcus aureus phagocytosed by monocyte-derived macrophages. (a) Control; (b) thioridazine-treated 6 h post-phagocytosis; thioridazine (0.1 mg/L) was added to the medium 30 min after phagocytosis.110
Requirement of K+ for intracellular killing
The killing activity of neutrophils, although highly complex, has been shown by a series of elegant studies conducted by Segal's group111 to depend upon the availability of K+ to the phagolysosome112 and the dependence of this process on active Ca2+ channels of the phagolysosome unit. The essential need for Ca2+ required for the availability of K+ involves a Ca2+-dependent ATPase that is employed for the generation of energy required in K+ transport. Because the K+ concentration needed to trigger the acidification process required for the activation of hydrolases113 is higher than that present in the cytoplasm11,112 one would have to assume that the membrane of the phagolysosome complex would contain the required energy-driven efflux pumps. Because phenothiazines do transform non-killing macrophages into effective killers50,73–75 and because phenothiazines are potent inhibitors of K+ transport processes that are dependent upon Ca2+-dependent ATPase, it seems probable that killing is enhanced by the phenothiazine's inhibition of K+ efflux from the phagolysosome. If this hypothesis is correct, then one would predict that inhibitors of K+ transport would also enhance the killing activity of non-killing macrophages. As is evident from Figure 4, ouabain, verapamil and reserpine, which are inhibitors of K+ transport processes, also enhance the killing activity of non-killer macrophages.114 The question of whether the K+ efflux pump units pre-exist in the lysosome or are part of the phagosome unit, and hence have their origins in the plasma membrane of the macrophage, has not yet been addressed. Nevertheless, with the exception of smooth muscle,115 the plasma membrane of most mammalian cells has a plethora of K+ transport units.116 In all likelihood the plasma membrane that delimits the phagosome probably contains many K+ transport units whose origins are from the plasma membrane of the macrophage. Due to the invagination process that results in the phagosome, the plasma membrane would pump K+ from the lumen of the phagosome to the cytoplasm, a region of the cell that is known to have a high concentration of the ion.117 It is at these K+ transport sites of the phagosome where ouabain, verapamil, reserpine and phenothiazines inhibit K+ efflux; and thereby the concentration of K+ within the phagosome would be maintained to levels needed for the acidification of the phagolysosome and activation of the hydrolases.111,112 However, recent studies have shown that the killing activity of monocyte-derived macrophages, which are identical to those used in the experiments described in Figure 4, is dependent upon extracellular calcium and extracellular ATP,118,119 both of which are under transport control mediated by Ca2+-dependent ATPase. As is the case for the K+ transport systems, the transport process for Ca2+ would be reversed in the phagosome if this system were to be retained in the plasma membrane that delimits the phagosome unit. Phagocytosis of the bacterium by macrophages should result in a phagosome that contains K+ and Ca2+ transport systems that pump out these ions to the cytoplasm of the cell. It is hypothesized that failure to kill bacteria present in the phagosome–lysosome is due to the low concentration of these ions in the complex that results from their extrusion to the cytoplasm. The inhibition of either one or both of these processes by a phenothiazine may establish conditions by which the concentration of these ions can be increased to a point where other ATP-driven membrane exchange systems involved in the cytosolic homeostasis are activated, leading to an intense hydrolytic activity of the phagosome–lysosome ATPases, in particular the V-ATPases.113,118,119 The import of H+ thereby creating the acidification of the lysosome component that had fused with the phagosome and the activation of the hydrolases needed for the killing of the bacterium. These ATP-driven membrane exchange systems must also be immune to ouabain, phenothiazines, etc, Figure 5 presents a hypothetical model which describes the aforementioned events that ultimately transform non-killer macrophages into effective killers. The phagocytosis of the bacterium (Figure 5a) results in a phagolysosome whose pumps extrude K+ and Ca2+ from the vacuole to the cytoplasm (IV). When a phenothiazine is presented to the macrophage that has phagocytosed the mycobacterium (Figure 5b), the efflux of K+ and Ca2+ from the phagolysosome is prevented, the build up of H+ takes place, the hydrolases are now activated and the bacterium is digested (VII).
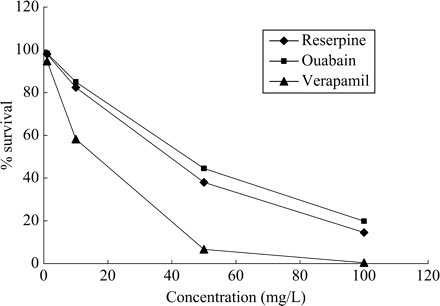
Effects of ouabain, verapamil and reserpine on the killing activity of human monocyte-derived macrophages.114
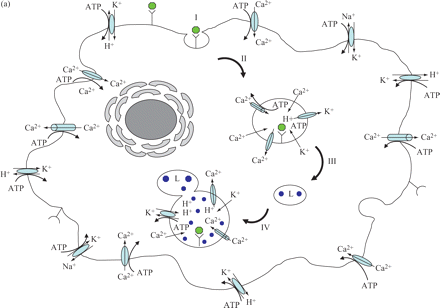
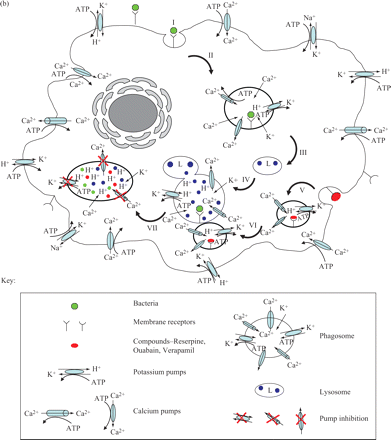
Hypothetical model suggested for the enhancement of killing of human monocyte-derived macrophages by phenothiazines and other efflux pumps. (a) Infected and untreated macrophage. (b) Infected and treated macrophage. Sequence of events described in the model: I, binding of bacteria to plasma membrane; II, invagination and formation of phagosome; III and IV, maturation of phagosome and fusion of the lysosome with the phagosome; V, binding of inhibitor of efflux pump and formation of vesicle; VI, fusion of vesicle containing the inhibitor of efflux pump with matured phagolysosome; VII, inhibition of Ca2+ and K+ efflux by inhibitor of efflux pump; K+ leaks into phagolysosome with cytosolic homeostasis mechanisms activated leading to an increased activity of the ATPases; acidification of the phagolysosome; activation of hydrolases; killing of bacteria. Note: hydrolysis of ATP to ADP by ATPases is not shown in the diagram.
The search for inhibitors of efflux pumps that may also enhance the killing activity of non-killing macrophages
Our previous studies demonstrated unusual properties of an extract made from the Portuguese nuisance plant Carpobrotus edulis.120,121 Among these were: (i) the ability to invoke Th1 responses of a variety of T-cell subsets as well as to stimulate the production of cytokines that are involved in the immune response to an infectious agent; (ii) the ability to render highly resistant mouse lymphoma cells that contain the MDR efflux pump transport pg1 completely susceptible to cytotoxic agents; (iii) the ability to inhibit the MDR efflux pump of these mouse lymphoma cells as evident from the increased retention of the efflux pump substrate rhodamine 1,2,3; and (iv) the ability to enhance the killing of intracellular (phagocytosed) M. tuberculosis as well as Mycobacterium avium,122 whereas the highest concentration of the extract was devoid of any in vitro activity against these mycobacteria. Although it is not yet known whether the activities noted for the C. edulis extract are due to one or more substances [the extract and the inhibitors of K+ transport (ouabain, reserpine and verapamil) enhance the killing of intracellular bacteria by non-killing macrophages whereas they do not have any in vitro activity against these bacteria], it seems possible that the activity noted for the extract is due to one agent, and that this agent manifests its effects via the inhibition of K+ transport. Other plant extracts as well as plant-derived compounds that have been shown to inhibit efflux pumps of cancer cells also have activity against phagocytosed bacteria. The suggestion that inhibitors of the P-glycoprotein of mammalian cells may also inhibit the efflux pump of bacteria receives support from the studies that used piperidine, an alkaloid isolated from the fruits of Piper longum123 and its derivative piperine, to inhibit the P-glycoprotein of Caco-2 cells,124 as well to increase the retention of ethidium bromide in S. aureus by the inhibition of efflux activity.125 Polyphenols obtained from plant sources have been shown to inhibit efflux pumps of Caco-2 cells126 and have been shown to enhance the killing of intracellular M. tuberculosis.127
Other plant-derived agents that have activity against efflux pumps of cancer cells may also enhance the killing activity of macrophages against bacteria, perhaps even mycobacteria. Voacamine, a bisindolic alkaloid from Peschiera fuchsiaefolia, induces a significant increase of drug retention in cancer cells by its ability to inhibit the MDR transporter protein, P-glycoprotein.128 Irofulven, a novel anticancer agent derived from the mushroom, reverses the resistance of cancer cells to cytotoxic agents by inhibiting the MDR efflux pump responsible for this resistance.129 Curcumin mixture and three major curcuminoids purified from turmeric (curcumin I, II and III) have been shown to modulate the function of MDR protein 1 (MRP1) of HEK293 cells stably transfected with MRP1-pcDNA3.1.130 These and many other plant-derived compounds that are active against efflux pumps of cancer cells may serve as lead compounds for the synthesis of new agents that have activity against intracellular bacteria.
Concluding remarks
MDR-TB is an intracellular infection of the non-killing macrophage of the lung, so any drug that is to be effective must have activity at this site. Conventionally, anti-tubercular drugs are designed to have direct activity against intracellular MDR-TB and, as has been the case for all other antibiotics, resistance ensues with usage. Because the non-killer macrophage can be transformed into an effective killer by a variety of compounds that inhibit K+ transport, perhaps it would be wiser to develop drugs that enhance the killing activity of these cells inasmuch as this approach would not be subject to any resistance as is inevitably the case for conventional antibiotics.
Acknowledgements
This work was partially supported by grants EU-FSE/FEDER-POCTI-37579/FCB/2001 and EU-FSE/FEDER-POCI/SAU-MMO/59370/2004 provided by the Fundação para a Ciência e a Tecnologia (FCT) of Portugal. M. Martins was supported by grant SFRH/BD/14319/2003 (FCT, Portugal).
Transparency declarations
None to declare.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}