




Abstract
Sulforaphane (SFN), an isothiocyanate first isolated from broccoli, exhibits chemopreventive properties in prostate cancer cells through mechanisms that are poorly understood. We recently reported on a novel mechanism of chemoprotection by SFN in human colon cancer cells, namely the inhibition of histone deacetylase (HDAC). Here, we show that addition of 15 µM SFN also inhibited HDAC activity by 40, 30 and 40% in BPH-1, LnCaP and PC-3 prostate epithelial cells, respectively. The inhibition of HDAC was accompanied by a 50–100% increase in acetylated histones in all three prostate cell lines, and in BPH-1 cells treated with SFN there was enhanced interaction of acetylated histone H4 with the promoter region of the P21 gene and the bax gene. A corresponding 1.5- to 2-fold increase was seen for p21 Cip1/Waf1 and Bax protein expression, consistent with previous studies using HDAC inhibitors, such as trichostatin A. The downstream events included cell cycle arrest and activation of apoptosis, as evidenced by changes in cell cycle kinetics and induction of multi-caspase activity. These findings provide new insight into the mechanisms of SFN action in benign prostate hyperplasia, androgen-dependent prostate cancer and androgen-independent prostate cancer cells, and they suggest a novel approach to chemoprotection and chemotherapy of prostate cancer through the inhibition of HDAC.
Introduction
Prostate cancer is the second leading cause of cancer-related death in men. With over 230 000 men estimated to be diagnosed with prostate cancer in 2004, preventive measures that target the various steps involved in cancer initiation and progression could significantly decrease the incidence and mortality of prostate cancer. Epidemiological studies suggest that cruciferous vegetable intake may lower overall risk of prostate cancer, particularly during the early stages ( 1 – 4 ), and there is growing interest in identifying the specific chemoprotective constituents and their mechanisms of action.
Sulforaphane (SFN) is an isothiocyanate found in cruciferous vegetables and is especially high in broccoli and broccoli sprouts ( 5 ). SFN is an effective chemoprotective agent in carcinogen-induced animal models ( 6 – 8 ), as well as in xenograft models of prostate cancer ( 9 ). Recent work has clearly implicated multiple mechanisms of SFN action, with the majority of studies focusing on SFN as a potent Phase-2 enzyme inducer and effects on cell cycle arrest and apoptosis ( 9 – 18 ).
We reported on a novel mechanism of chemoprotection by SFN in human colon cancer cells, namely the inhibition of histone deacetylase (HDAC) ( 19 ). SFN was shown to (i) inhibit nuclear HDAC activity, (ii) augment the levels of acetylated histones, (iii) increase acetylated histone H4 interactions with the P21 promoter and (iv) elevate the expression of p21 Cip1/Waf1 in HCT116 cells.
Several clinical trials are currently ongoing aimed at establishing the chemotherapeutic efficacy of HDAC inhibitors, based on evidence that cancer cells undergo cell cycle arrest, differentiation and apoptosis in vitro , and that tumor volume and/or tumor number may be reduced in animal models ( 20 – 22 ).
Inhibitors of HDAC have generated considerable recent interest as novel chemoprotective agents because they target epigenetic events that can occur at various stages of cancer development ( 23 , 24 ). Moreover, since epigenetic mechanisms exist in a wide array of target tissues, HDAC inhibitors have the potential for broad applicability. In the case of prostate cancer, for example, it has been shown that HDAC activity increases in metastatic cells compared with prostate hyperplasia ( 25 ) and over-expression of HDAC1 in PC-3 cells results in an increase in cell proliferation and an overall decrease in cell differentiation ( 26 ). This may be of particular importance in the progression to androgen independence. Importantly, inhibitors of HDAC, including suberoylanilide hydroxamic acid (SAHA), valproic acid, depsipeptide and sodium butyrate, have been demonstrated to be effective against prostate cell lines and xenograft models ( 27 – 29 ).
Based on the studies with HDAC inhibitors in prostate cancer cell lines and our recent work in human colon cancer cells, we sought to test the broad applicability of HDAC inhibition by SFN using various prostate cell lines, including consideration of the downstream consequences of HDAC inhibition on apoptosis and cell cycle arrest. We report here, for the first time, that SFN inhibits HDAC activity in BPH-1, LnCaP and PC-3 cells, and provide support for SFN as an effective chemopreventive agent for prostate hyperplasia and/or cancer.
Materials and methods
Cell culture
Human benign prostate hyperplasia epithelial cells (BPH-1), androgen-independent prostate cancer epithelial cells (PC-3) and androgen-dependent prostate cancer epithelial cells (LNCaP) were obtained from American Type Tissue Collection (Manassas, VA). Cells were grown and maintained in RPMI 1640 with glutamine plus 10% FBS and treated with test agents at ∼50–70% confluency. Unless stated otherwise, cells were harvested 48 h after treatment with 3–15 µM D,L-SFN (LKT Labs, St Louis, MN) or 8 h after treatment with 100 ng/ml trichostatin A (TSA, Biomol, Plymouth Meeting, PA). The final concentration of vehicle, DMSO, did not exceed 0.1%. Dose and treatment times for TSA and SFN were identical to our recent study ( 19 ), and based on a prior investigation by Billin et al . ( 30 ) using TSA, and another by Hu et al . ( 31 ) which reported plasma concentrations for SFN in the rat on the order of 20 µM. Pilot experiments identified the peak of TSA inhibitory activity to occur 8 h after treatment (M.C. Myzak and R.H. Dashwood, unpublished data).
Western blotting
Protein concentrations were determined by Lowry assay from attached cell populations. Proteins (10–20 µg) were separated by SDS–PAGE on a 4–12% bis-Tris gel (Novex, San Diego, CA) and were transferred to nitrocellulose membrane (Invitrogen, Carlsbad, CA). Equal protein loading was confirmed with Amido Black staining and β-actin levels. The membrane was blocked for 1 h with 2% BSA, followed by either overnight incubation with primary antibody at 4°C or 1 h incubation with primary antibody at room temperature, and was finally incubated for 1 h with secondary antibody conjugated with horseradish peroxidase (Bio-Rad, Hercules, CA). Antibody dilutions were as follows: acetylated histone H3, 1:100 (Upstate, Charlottesville, VA); acetylated histone H4, 1:100 (Upstate); p21, 1.5 mg/ml (Neomarkers, Fremont, CA); caspase-3, 1:2000 (Calbiochem, San Diego, CA); Bax, 1:500 (BD Pharmingen, San Diego, CA), Bcl-2 (Santa Cruz Biotechnologies, Santa Cruz, CA), p53 (Santa Cruz Biotechnologies) and β-actin, 1:5000 (Sigma, St Louis, MO). Detection was by Western Lightning Chemiluminescence Reagent Plus (PE Life Sciences, Boston, MA) with image analysis on an AlphaInnotech photodocumentation system. Densitometry was performed by NIH Image J.
HDAC activity assay
The Fluor-de-Lys HDAC activity assay kit (Biomol) was used, as reported previously ( 19 ). Attached cell lysates (15 µg total protein) from cells treated with SFN or TSA were incubated with Fluor-de-Lys substrate for 10 min at 37°C to initiate the HDAC reaction. Fluor-de-Lys Developer was then added, and the mixture was incubated for another 10 min at room temperature. Fluorescence was measured using a Spectra Max Gemini XS fluorescent plate reader (Molecular Devices), with excitation 360 nm and emission 460 nm.
Chromatin immunoprecipitation
BPH-1 cells were treated with SFN as above. Formaldehyde (Sigma) was added directly to media to a final concentration of 1% and incubated for 10 min at 37°C. Attached cell lysates were sonicated eight times for 15 s using a Heat Systems-Ultrasonics Sonicator (Model W-225R) on setting 5. A Chromatin Immunoprecipitation (ChIP) kit (Upstate) was used according to the manufacturer's instructions, with anti-acetylated histone H4 antibody. Primers and PCR conditions used for amplification of the P21 promoter were exactly as reported previously ( 19 ). Primers and PCR conditions used for amplification of the bax promoter were as follows: F, 5′-TAATCTCAGCACTTTGGGAGG; R, 5′-GACAGGGTCTCACTGTGTTGC. PCR products were detected after 30 cycles of the following cycling conditions: 94°C for 30 s, 52°C for 30 s and 72°C for 30 s.
Real-time PCR
Total RNA was extracted using an RNeasy kit (Qiagen, Valencia, CA) from BPH-1 cells 12 h after treatment with 0 or 15 µM SFN, and first-strand cDNA was prepared from 4 µg total RNA using an Omniscript Reverse Transcriptase Kit (Qiagen). Levels of bax mRNA were quantified by real-time PCR and normalized to glyceraldehyde-3-phosphate dehydrogenase ( GAPDH ), using the following primers: bax forward primer 5′ TGC TTC AGG GTT TCA TCC AG 3′ and reverse primer 5′ GGC GGC AAT CAT CCT CTG 3′; GAPDH forward primer 5′ GAA GGT GAA GGT CGG AGT C 3′ and reverse primer 5′GAA GAT GGT GAT GGG ATT TC3′. PCR was conducted over 40 cycles (95°C for 10 s, 56°C for 20 s and 72°C for 20 s) using an Opticon Monitor 2 system (Finnzymes, Finland), in 50 µl containing cDNA, SYBR Green I dye (DyNAmo master solution, Finnzymes) and the corresponding primers.
Multi-caspase activity
Multi-caspase activity was assessed using a flow-cytometry based Multi-caspase assay kit (Guava Technologies, Hayward, CA.). Cells were trypsinized, washed in D-PBS and stained with a fluorochrome-conjugated caspase inhibitor, sulforhodamine-valyl-alanyl-aspartyl-fluoromethylketone (SR-VAD-FMK) according to the manufacturer's instructions. This inhibitor readily crosses cell membranes and covalently binds to the active forms of multiple caspases, which are cleaved from inactive precursors (procaspases) during apoptosis induction. Subsequently, cells were washed three times and stained with 7-amino-actinomycin-D (7-AAD) for 15 min prior to analysis on a Guava Personal Cell Analyzer (PCA) (Guava Technologies).
Cell cycle analysis
A flow cytometric assay was also performed to assess effects of SFN on cell cycle. BPH-1 and PC-3 cells were treated with SFN and cells (attached and floating) were harvested by trypsinization at 24 or 48 h post-treatment. One million cells were fixed by slow, dropwise addition of 2 ml cold 70% ethanol, followed by storage at 4°C for 24 h. After fixation, cells were washed, pelleted and resuspended in 0.04 mg/ml propidium iodide and 100 mg/ml RNase in PBS. The sample was incubated at room temperature for 30 min and analyzed on the Guava PCA. Multi-Cycle analysis software (Phoenix Flow Systems, San Diego, CA) was used to generate histograms and determine number of cells in each phase of the cell cycle.
Statistics
One-way analysis of variance (ANOVA) was performed to assess the differences between groups. Differences in means among treatments were tested by Dunnett's test, and the level of significance was designated as follows: *P < 0.05, **P < 0.01 and ***P < 0.001.
Results
We recently identified SFN as an HDAC inhibitor in human colorectal cancer cells ( 19 ), and the present study sought to verify that SFN also acts as an HDAC inhibitor in benign prostate hyperplasia and prostate cancer cells. The addition of 15 µM SFN for 48 h inhibited HDAC activity in BPH-1, LnCaP and PC-3 cell lysates by 40, 30 and 40%, respectively, compared with the corresponding controls ( Figure 1 ). The addition of the known HDAC inhibitor, TSA, lowered HDAC activity by 30% in BPH-1 and PC-3 cells, but had no significant effect in LnCaP cells.
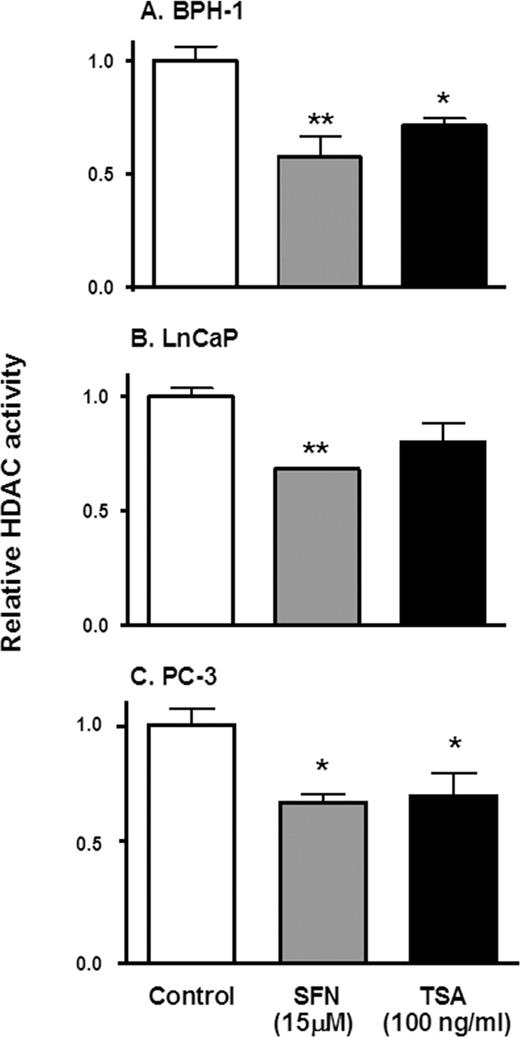
SFN inhibits HDAC activity in prostate epithelial cells. BPH-1 ( A ), LnCaP ( B ) and PC-3 cells ( C ) were harvested 48 h after treatment with SFN (15 µM), or 8 h after TSA exposure (100 ng/ml), and cell lysates were analyzed for HDAC activity, as reported ( 19 ). Results = normalized mean ± SD, n = 3. *P < 0.05, ** P < 0.01. Average baseline HDAC activity for BPH-1, LnCaP and PC-3 cells were 7.7, 8.4 and 12.2 arbitrary florescence units (AFU), respectively.
Immunoblotting confirmed that SFN increased the amount of acetylated histone H3 and acetylated H4 in all three prostate cell lines (ranging from 50 to 100% increase), compared with untreated controls ( Figure 2 ). An increase in acetylated histones was also detected in cells treated with TSA, most notably in PC-3 cells ( Figure 2C ).
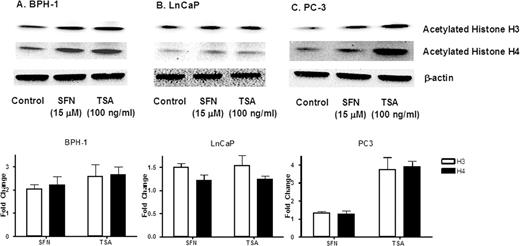
SFN increases acetylated histone levels in prostate cell lines. Cells were treated with SFN or TSA as described in the legend to Figure 1 , and acetylated histone H3 or acetylated histone H4 levels were assessed by immunoblotting. Equal protein loading was confirmed using β-actin. Results are representative of two or more separate experiments. Fold change was calculated using density values normalized to β-actin and expressed as relative change between SFN/TSA treated and control.
Because the SFN-mediated increase in acetylated histones H3 and H4 was most striking in BPH-1 cells ( Figure 2 ), these cells were examined for changes in the acetylation status of histone H4 associated with the promoter region of the P21 gene. Using anti-acetylated histone H4 antibody followed by PCR with primers specific for the P21 promoter, ChIP assays showed a concentration-dependent increase in the amount of acetylated histone H4 associated with the P21 promoter ( Figure 3A ). Expression of the p21 protein, a well known target of HDAC inhibitors ( 32 – 41 ), was increased 50–100% by SFN in all three prostate cell lines, compared with untreated controls ( Figure 3B ).
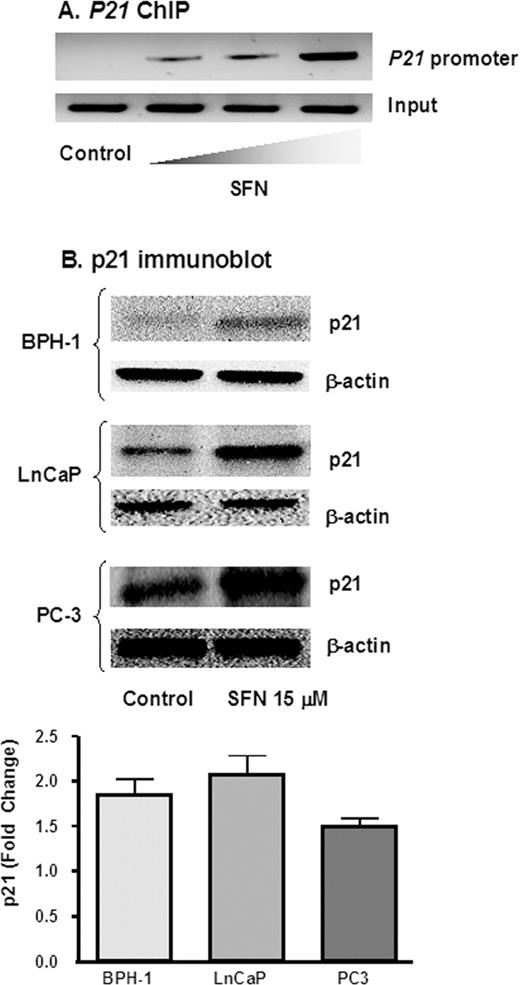
SFN increases p21 expression in prostate cells. ( A ), BPH-1 cells were treated with SFN and 48 h later DNA was cross-linked to proteins, ChIP was performed using acetylated histone H4, and following reversal of cross-linking and isolation of DNA, PCR was performed with primers to the P21 promoter. Wedge symbol, 3, 9, 15 µM SFN; control, 0 µM SFN (vehicle alone). ( B ), Cells were treated with 0 or 15 µM SFN and 48 h later the attached cells were isolated and cell lysates were immunoblotted for p21 protein. Equal protein loading was confirmed using β-actin. Results are representative of two or more separate experiments. Fold change was calculated using density values normalized to β-actin and expressed as relative change between 0 and 15 µM SFN.
To determine the downstream effects of HDAC inhibition by SFN, pro- and anti-apoptotic protein expression was examined by immunoblotting. A 50% increase in the pro-apoptotic protein Bax was detected in all three prostate cell lines ( Figure 4A–C ), and SFN also caused an increase in p53 protein levels in BPH-1 and LnCaP cells, compared with controls ( Figure 4A and B ). As expected, p53 was not detected in p53-null PC-3 cells ( Figure 4C ). The increased expression of pro-apoptotic proteins was coupled with a 30 and 40% decrease in the anti-apoptotic protein Bcl-2 in SFN-treated BPH-1 and PC-3 cells, respectively ( Figure 4A and C ), but not in LnCaP cells ( Figure 4B ), which express low levels of Bcl-2 protein. To establish the association between the histone acetylation status and expression of specific apoptosis-related genes, we performed ChIP using primers specific for the bax promoter. There was a marked increase in acetylated histone H4 associated with the bax promoter ( Figure 4D ) and a significant increase in bax mRNA expression, 12 h after SFN treatment ( Figure 4E ).
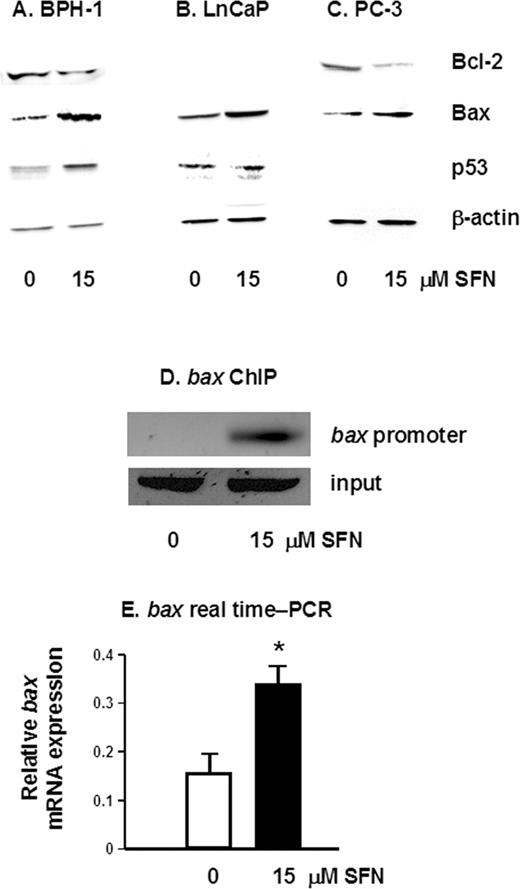
SFN alters the expression of pro- and anti-apoptotic proteins. Attached cells were harvested 48 h after treatment of BPH-1 ( A ), LnCaP ( B ) and PC-3 cells ( C ) with 0 or 15 µM SFN, and cell lysates were immunoblotted for Bcl-2, Bax and p53, as indicated. Equal protein loading was confirmed using β-actin. Results are representative of two or more separate experiments. ( D ) BPH-1 cells were treated with 0 or 15 µM SFN and ChIP was performed as described in the legend to Figure 3 , except that PCR was performed with primers to the bax promoter. ( E ) Real-time PCR results for bax mRNA expression in BPH-1 cells 12 h after treatment with 0 or 15 µM SFN; mean ± SD, n = 3; *P < 0.05.
Changes induced by SFN in Bcl-2 and Bax expression were associated with an increase in multi-caspase activity ( Figure 5 ). Cells stained positive in this assay contain one or more active (cleaved) caspases, which is indicative of apoptosis. Treatment of both BPH-1 and PC-3 cells with 15 µM SFN resulted in a significant increase in multi-caspase activity at 48 h ( Figure 5A and B ). In addition, there was a loss of procaspase-3 in both BPH-1 and PC-3 cells after treatment with 15 µM at 48 h by immunoblotting ( Figure 5C and D ), suggesting cleavage of pro-caspase-3 and subsequent activation of caspase-3.
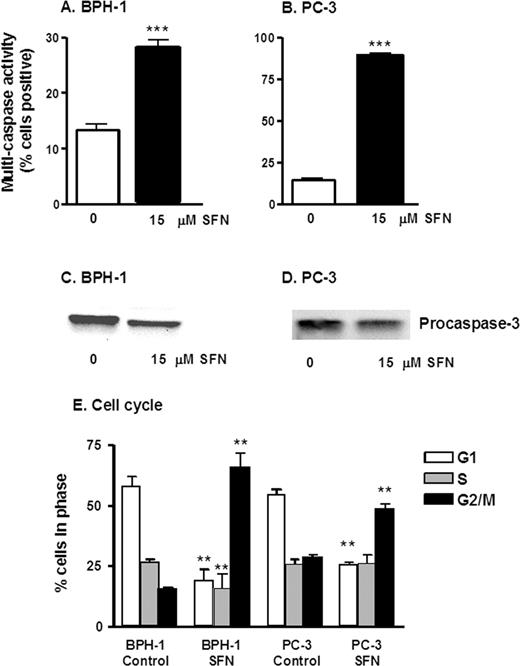
SFN increases multi-caspase activity and causes cell cycle arrest in prostate epithelial cells. BPH-1 cells ( A ) and PC-3 cells ( B ) were harvested 48 h after treatment with 0 or 15 µM SFN, as indicated, and the attached cells were examined for multi-caspase activity using a Guava PCA. Results represent mean ± SD, n = 3, from experiments conducted on two or more separate occasions. BPH-1 ( C ) and PC-3 ( D ) cells were treated as above, and cell lysates were immunoblotted for procaspase-3. In ( E ) BPH-1 and PC-3 were harvested, fixed and stained for cell cycle analysis. Attached and floating cells were fixed in 70% ethanol and stained with propidium iodide, and cell cycle kinetics was examined using the Guava PCA, followed by data analysis with Multi-Cycle software. Results indicate mean ± SD, n = 3; **P < 0.01, ***P < 0.001.
The increase in multi-caspase activity induced by SFN in BPH-1 cells ( Figure 5A and B ) was paralleled by an increase in the number of floating cells and a concomitant decrease in the attached cell population ( Figure 6A and B ). In PC-3 cells, a concentration-dependent increase in floating cells was observed ( Figure 6C ), coupled with a concentration-dependent decrease in the attached cell population ( Figure 6D ). Under the microscope, the floating cells had morphological hallmarks clearly distinct from the attached cells, consistent with the cells detaching and undergoing apoptosis.
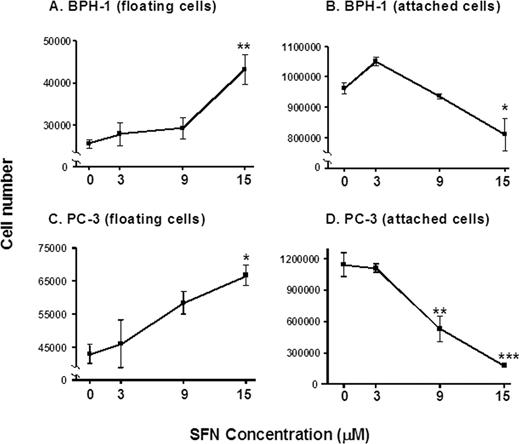
SFN increases the proportion of prostate cells in the floating population. BPH-1 ( A and B ) and PC-3 cells ( C and D ) were treated with 0, 3, 9, or 15 µM SFN and 48 h later the floating and attached cells were counted. Results are given as mean ± SD, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001.
The induction of apoptosis, and the detachment of cells, is frequently preceded by changes in cell cycle kinetics ( 42 ). In BPH-1 cells, there was a significant loss of cells in the G 1 phase of the cell cycle and an increase in G 2 /M cells, 24 h after SFN treatment ( Figure 5E ). In the comparison of 0 versus 15 µM SFN, the proportion of BPH-1 cells in G 2 /M increased from 15.31 to 65.34%, implying the presence of a G 2 /M arrest. In PC-3 cells, there was a concentration-dependent decrease in G 1 and an increase in G 2 /M indicating a G 2 /M arrest 48 h following SFN treatment ( Figure 5E ).
Discussion
We recently provided the first evidence for SFN as an HDAC inhibitor in human embryonic kidney 293 cells and HCT116 human colorectal cancer cells ( 19 ). The present investigation has confirmed that SFN acts in a similar manner in human prostate cell lines, and we extend these findings by showing associations between HDAC inhibition, alterations in acetylated histone status, enhanced expression of pro-apoptotic proteins, and corresponding increases in markers of cell cycle arrest and apoptosis (for scheme, Figure 7 ). Previous reports have described cell cycle arrest in LnCaP, PC-3, and other prostate cell lines treated with SFN ( 9 , 10 , 18 ), but not in the context of HDAC inhibition, and to our knowledge this is the first study with SFN in BPH-1 cells.
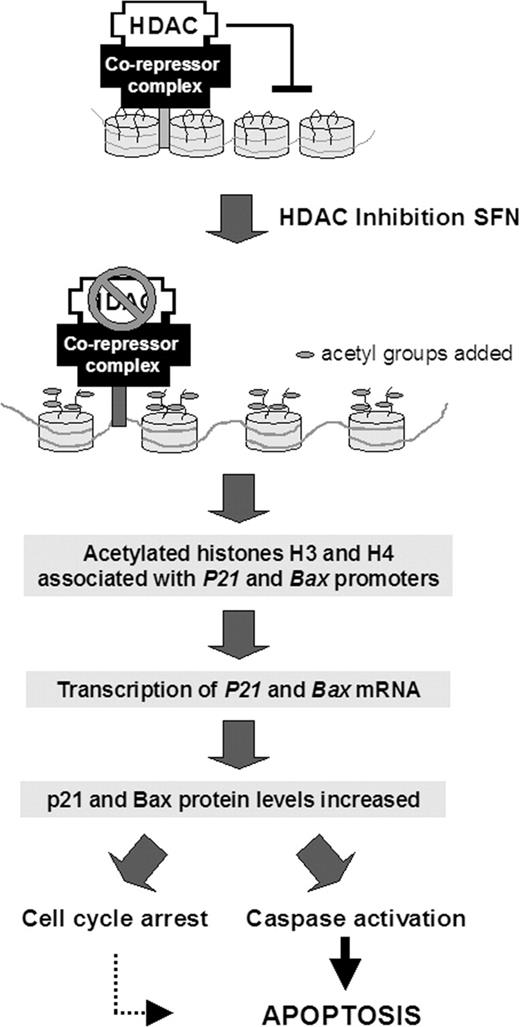
Model for the mechanism of SFN-induced apoptosis through HDAC inhibition in prostate cells. SFN inhibits HDAC activity, which results in an increase in acetylated histones associated with the promoter region of genes such as P21 and bax . Interactions between the histones and DNA are loosened, facilitating transcription factor access to P21 and bax . The corresponding de-repression of these genes activates transcription, leading to increased mRNA and protein expression, enabling cycle arrest, caspase activation, and apoptosis induction in prostate cancer cells.
In vivo studies have established that SFN is an effective cancer chemopreventive agent in several animal models ( 6–8 ), and it is thought to induce Phase 2 detoxification enzymes through interaction of Nrf-2 with the antioxidant response element (ARE) ( 43–45 ). However, SFN also has a marked effect on cell cycle checkpoint controls and cell survival/apoptosis in various cancer cell lines, through molecular mechanisms that remain poorly understood ( 9–18 ). The present investigation provides possible insight into this question by showing that SFN is an inhibitor of HDAC activity in prostate epithelial cells. Whereas previous reports described downstream changes in apoptosis markers in prostate cells after SFN treatment ( 9 , 10 , 18 ), we show for the first time a connection between (i) HDAC inhibition, (ii) increased acetylated histones on the bax promoter, (iii) elevated bax mRNA expression, (iv) increased Bax protein levels and (v) activation of caspases ( Figure 7 ).
In all three prostate cell lines, HDAC inhibition was significant at a concentration of 15 µM SFN. Several previous studies used SFN doses in the range 20–100 µM and observed oxidative stress and apoptosis in vitro . Recently, it was reported that plasma concentrations reached 20 µM SFN in rats after a single oral dose of 50 µmol ( 31 ). However, in humans, after consumption of broccoli sprouts, peak plasma levels were reported at 2 µM ( 46 ). To date, actual tissue levels of SFN have not been reported; however, cells in culture can accumulate millimolar levels of SFN ( 47 ). It is feasible that SFN may reach sufficient concentrations in tissues to act as an HDAC inhibitor in vivo . We have found in other studies that in wild-type and Apcmin mice, consumption of ∼6 µmol SFN/day for 10 weeks significantly inhibited HDAC activity and increased acetylated histones in the prostate. HDAC inhibition was also apparent in colonic mucosa and peripheral blood mononuclear cells, suggesting that long-term administration of SFN in the diet can inhibit HDAC activity in vivo ( 48 ).
In general, late-stage androgen-independent PC-3 cells often require higher doses of agents or do not respond as well in terms of undergoing apoptosis or cell cycle arrest compared with androgen-dependent or benign hyperplastic cells. Thus, it is noteworthy that androgen-independent PC-3 cells responded well to SFN. This may be attributed to the fact that PC-3 cells have higher HDAC activity compared with normal prostate and BPH cells ( 25 ), and that in general, HDAC inhibitors exhibit greater effects toward cancer cells ( 22 ).
Previously, we identified SFN-Cys, a metabolite of SFN generated via the mercapturic acid pathway, as the likely ultimate HDAC inhibitor in colon cancer cells ( 19 ). The working hypothesis is that SFN-Cys is the active HDAC inhibitor and may be formed in two ways; (i) directly from SFN as it is metabolized via the mercapturic acid pathway, with the initial step involving glutathione (GSH) conjugation catalyzed by glutathione- S -transferases (GST) ( 49–52 ) or (ii) indirectly from SFN-NAC, where the HDAC enzyme recognizes SFN-NAC and deacetylates it to form SFN-Cys. In prostate cancer, GSTP1 is frequently turned off by epigenetic modifications through methylation of its promoter ( 53 , 54 ). According to a recent study, CpG island hypermethylation in the promoter region of GSTP1 was observed in several prostate cancer cell lines. PC-3 cells express GSTP1, but contain a mixture of both methylated and unmethylated alleles, while LnCaP cells do not express GSTP1 ( 53 ). Thus, differences in sensitivity between LnCaP and PC-3 cells may also be related to differences in GST activity. Although GSTP1 can be silenced in LnCaP and PC-3 cell lines (the status is unknown in BPH-1), addition of 15 µM SFN nonetheless exhibited significant HDAC inhibitory activity ( Figure 1 ). It has been demonstrated that conjugation of SFN to GSH can occur as a facile reaction, without catalysis by GST ( 50 ), and it might be that slower conjugation of GSH enhances HDAC inhibition such that the SFN-Cys metabolite is generated more slowly, and available for a more sustained period prior to clearance. To explore this hypothesis, future studies should examine the kinetics of SFN-Cys generation and turnover in cell lines with altered GST expression.
The degree of HDAC inhibition and downstream gene expression with SFN was similar to that of known pharmacological inhibitors of HDAC. As with other HDAC inhibitors ( 32 – 41 ), such as TSA, SAHA and sodium butyrate, SFN increased p21 protein expression in prostate cells, and ChIP assays confirmed an increase in the expression of acetylated histone H4 associated with the P21 promoter. The P21 promoter lacks an ARE, suggesting an Nrf-2-independent mechanism for the induction of p21 by SFN, but downstream effectors of Nrf-signaling might act on P21 gene expression. Future studies should employ siRNA to knockdown Nrf-2, or Nrf-2 deficient cell lines, to examine the inter-relationships between Nrf2 signaling and HDAC inhibition.
Importantly, 15 µM SFN also increased acetylated histone H4 association with the bax promoter and augmented bax mRNA expression ( Figure 4D and E ), indicating that SFN might directly mediate Bax induction through chromatin remodeling. Further studies are in progress to determine whether SFN-induced changes in Bcl-2, Bax and multi-caspase activity necessarily result from HDAC inhibition rather than other mechanisms, such as modulation of cell cycle control proteins, tubulin polymerization and MAPK [reviewed in ( 55 )]. It is noteworthy that PC-3 cells, which lack p53 ( Figure 4C ), had strong induction of p21 ( Figure 3B ), supporting a p53-independent mechanism for SFN in these cells.
Apoptosis induction and cell cycle alterations have been reported for HDAC inhibitors ( 20–22 ), including dietary agents ( 56 ). In previous studies with SFN by others, a G 2 /M arrest was noted in PC-3 cells ( 57 ), whereas a G 1 arrest was observed in DU-145 and LnCaP cells ( 10 , 18 ). Similarly, we demonstrated that SFN treatment in BPH-1 and PC-3 cells induced pro-apoptotic markers ( Figure 4–6 ) and a G 2 /M cell cycle arrest ( Figure 5E ). As is characteristic of other HDAC inhibitors, SFN induced expression of p21, but cells underwent G 2 /M arrest ( 22 ). Further mechanistic studies are required, especially in the context of other known targets of HDAC inhibitors, such as the cyclins ( 58 ). Nonetheless, one important implication of these findings is that both benign prostate hyperplasia and androgen-independent prostate cancer might respond favorably to SFN treatment, via the induction of cell cycle arrest and apoptosis.
In summary, the present investigation has established an association between inhibition of HDAC activity, increased histone acetylation on bax and P21 promoters, elevated bax RNA and protein expression, and concomitant increase in cell cycle arrest and apoptotic markers after SFN treatment ( Figure 7 ). These results suggest that SFN may be effective against benign prostate hyperplasia, androgen-dependent prostate cancer and androgen-independent prostate cancer potentially through the inhibition of HDAC activity. The present work focused on p21 and Bax, which are well-established targets of HDAC inhibitors and key regulators of cell cycle kinetics and apoptosis, but additional targets of HDAC inhibition warrant further investigation. Collectively, the results suggest that, in addition to its well-recognized effects on Phase 2 pathways, an alterative mechanism by which SFN can modulate gene expression is through the inhibition of HDAC activity, leading to apoptosis induction in cancer cells. The convergence of multiple mechanisms has many advantages; the present results and published data clearly indicate that SFN should be useful both as a chemopreventive agent and as a chemotherapeutic agent in the prostate.
The BPH-1 cell line was obtained from Dr Simon W. Hayward (Vanderbilt University, Nashville, TN). The authors thank Anna Hsu of the Department of Nutrition and Exercise Sciences, and Julie Oughton of the Flow Cytometry Facility Core of the Environmental Health Sciences Center (Oregon State University), for assistance with flow cytometry and cell cycle analyses. The Hagen and Tanguay laboratories are also gratefully acknowledged for providing access to real-time PCR instruments. This work was supported in part by NIH grants CA66525 (RHD), CA80176 (RHD), CA90890 (RHD) and CA107693 (EH) and by the National Institute of Environmental Health Sciences Center grant P30 ES00210. Additional support was from the Oregon Agricultural Experiment Station.
Conflict of Interest Statement : None declared.
References
Kolonel,L.N., Hankin,J.H., Whittemore,A.S. et al . (
Cohen,J.H., Kristal,A.R. and Stanford,J.L. (
Kristal,A.R. and Lampe,J.W. (
Giovannucci,E., Rimm,E.B., Liu,Y., Stampfer,M.J. and Willett,W.C. (
Zhang,Y., Talalay,P., Cho,C.G. and Posner,G.H. (
Chung,F.L., Conaway,C.C., Rao,C.V. and Reddy,B.S. (
Fahey,J.W., Zhang,Y. and Talalay,P. (
Zhang,Y., Kensler,T.W., Cho,C.G., Posner,G.H. and Talalay,P. (
Singh,A.V., Xiao,D., Lew,K.L., Dhir,R. and Singh,S.V. (
Chiao,J.W., Chung,F.L., Kancherla,R., Ahmed,T., Mittelman,A. and Conaway,C.C. (
Gingras,D., Gendron,M., Boivin,D., Moghrabi,A., Theoret,Y. and Beliveau,R. (
Bonnesen,C., Eggleston,I.M. and Hayes,J.D. (
Gamet-Payrastre,L., Li,P., Lumeau,S., Cassar,G., Dupont,M.A., Chevolleau,S., Gasc,N., Tulliez,J. and Terce,F. (
Fimognari,C., Nusse,M., Cesari,R., Iori,R., Cantelli-Forti,G. and Hrelia,P. (
Fimognari,C., Nusse,M., Berti,F., Iori,R., Cantelli-Forti,G. and Hrelia,P. (
Jackson,S.J. and Singletary,K.W. (
Gamet-Payrastre,L., Lumeau,S., Gasc,N., Cassar,G., Rollin,P. and Tulliez,J. (
Wang,L., Liu,D., Ahmed,T., Chung,F.L., Conaway,C. and Chiao,J.W. (
Myzak,M.C., Karplus,P.A., Chung,F.-L. and Dashwood,R.H. (
Rosato,R.R. and Grant,S. (
Marks,P., Rifkind,R.A., Richon,V.M., Breslow,R., Miller,T. and Kelly,W.K. (
Marks,P.A., Richon,V.M. and Rifkind,R.A. (
Oh,H.J., CHung,E.J., Lee,S., Loaiza-Perez,A., Sausville,E.A. and Trepel,J.B. (
Kopelovich,L., Crowell,J.A. and Fay,J.R. (
Patra,S.K., Patra,A. and Dahiya,R. (
Halkidou,K., Gaughan,L., Cook,S., Leung,H.Y., Neal,D.E. and Robson,C.N. (
Fronsdal,K. and Saatcioglu,F. (
Thelen,P., Schweyer,S., Hemmerlein,B., Wuttke,W., Seseke,F. and Ringert,R.H. (
Butler,L.M., Agus,D.B., Scher,H.I., Higgins,B., Rose,A., Cordon-Cardo,C., Thaler,H.T., Rifkind,R.A., Marks,P.A. and Richon,V.M. (
Billin,A.N., Thirlwell,H. and Ayer,D.E. (
Hu,R., Hebbar,V., Kim,B.R., Chen,C., Winnik,B., Buckley,B., Soteropoulos,P., Tolias,P., Hart,R.P. and Kong,A.N. (
Druesne,N., Pagniez,A., Mayeur,C., Thomas,M., Cherbuy,C., Duee,P.H., Martel,P. and Chaumontet,C. (
Hinnebusch,B.F., Meng,S., Wu,J.T., Archer,S.Y. and Hodin,R.A. (
Remiszewski,S.W., Sambucetti,L.C., Atadja,P. et al . (
Woo,S.H., Frechette,S., Abou Khalil,E. et al . (
Della Ragione,F., Criniti,V., Della Pietra,V., Borriello,A., Oliva,A., Indaco,S., Yamamoto,T. and Zappia,V. (
Richon,V.M., Sandhoff,T.W., Rifkind,R.A. and Marks,P.A. (
Lavelle,D., Chen,Y.H., Hankewych,M. and DeSimone,J. (
Eickhoff,B., Ruller,S., Laue,T., Kohler,G., Stahl,C., Schlaak,M. and van der Bosch,J. (
Finzer,P., Kuntzen,C., Soto,U., zur Hausen,H. and Rosl,F. (
Kobayashi,H., Tan,E.M. and Fleming,S.E. (
Diaz,G.D., Li,Q. and Dashwood,R.H. (
Dinkova-Kostova,A.T., Holtzclaw,W.D., Cole,R.N., Itoh,K., Wakabayashi,N., Katoh,Y., Yamamoto,M. and Talalay,P. (
Thimmulappa,R.K., Mai,K.H., Srisuma,S., Kensler,T.W., Yamamoto,M. and Biswal,S. (
Kwak,M.K., Egner,P.A., Dolan,P.M., Ramos-Gomez,M., Groopman,J.D., Itoh,K., Yamamoto,M. and Kensler,T.W. (
Ye,L., Dinkova-Kostova,A.T., Wade,K.L., Zhang,Y., Shapiro,T.A. and Talalay,P. (
Zhang,Y. (
Myzak,M.C., Dashwood,W.M., Orner,G.A., Dashwood,R.H. and Ho,E. (
Zhang,Y., Kolm,R.H., Mannervik,B. and Talalay,P. (
Kassahun,K., Davis,M., Hu,P., Martin,B. and Baillie,T. (
Conaway,C.C., Krzeminski,J., Amin,S. and Chung,F.L. (
Conaway,C.C., Yang,Y.M. and Chung,F.L. (
Yegnasubramanian,S., Kowalski,J., Gonzalgo,M.L., Zahurak,M., Piantadosi,S., Walsh,P.C., Bova,G.S., De Marzo,A.M., Isaacs,W.B. and Nelson,W.G. (
Millar,D.S., Ow,K.K., Paul,C.L., Russell,P.J., Molloy,P.L. and Clark,S.J. (
Myzak,M.C. and Dashwood,R.H. (
Myzak,M.C. and Dashwood,R.H. (
Singh,S.V., Herman-Antosiewicz,A., Singh,A.V., Lew,K.L., Srivastava,S.K., Kamath,R., Brown,K.D., Zhang,L. and Baskaran,R. (
Author notes
1Linus Pauling Institute, 2Molecular and Cellular Biology Program, 3Department of Nutrition and Exercise Sciences and 4Department of Environmental and Molecular Toxicology, Oregon State University, Corvallis, OR 97331, USA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}