




Abstract
Breast cancer resistance protein (BCRP/ABCG2) is known to actively transport various anticancer drugs and to restrict the uptake of the food carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5- b ]pyridine from the gut lumen. The present study reveals that BCRP is involved in the transport of phase-2 metabolites of the carcinogen benzo[ a ]pyrene (BP) in the human intestinal cell line Caco-2. Treatment with the selective BCRP inhibitor Ko 143 (5 μM) inhibited the apical transport of BP-3-sulfate (BP3S) to 83% of control levels in TC7 cells and to 64% of control levels in Caco-2 cells. The apical transport of BP-3-glucuronide was inhibited by Ko 143 to 76% of control levels in TC7 cells. Furthermore, the expression of BCRP is most likely aryl hydrocarbon receptor (AhR) dependent, as treatment of Caco-2 cells with known AhR agonists including 2,3,7,8-tetrachlorodibenzo- p -dioxin, BP, indolo[3,2- b ]carbazole and benzo[ k ]fluoranthene increased both mRNA and protein levels of BCRP. Induced BCRP protein was found to be functionally active, since pre-treatment of TC7 cells with oltipraz, indolo[3,2- b ]carbazole or benzo[ k ]fluoranthene increased the amount of apically transported BP3S to as much as 180% of that in the controls. The induction of BCRP (mRNA and protein expression) by indolo[3,2- b ]carbazole was inhibited in Caco-2 cells by co-incubation with the AhR antagonist PD98059 (2′-amino-3′-methoxyflavone). In summary, this study provides strong evidence that BCRP is an important part of the intestinal barrier protecting the body from food-associated contaminants such as the carcinogen BP.
Introduction
Breast cancer resistance protein (BCRP/MXR/ABCG2) is a relatively recently discovered member of the ATP-binding cassette (ABC) superfamily that has been associated with the phenomenon of multidrug resistance. As a 72 kDa half-transporter encoded by the ABCG2 gene, BCRP contains only six transmembrane domains with a single ABC element and is known to form homodimers in order to be functional ( 1 ). High levels of BCRP expression in human normal tissues are found in colon, small intestine, placenta and liver ( 2 ), indicating that BCRP contributes to the barrier function of these tissues. In polarized cells of the gut, kidney and liver epithelium, BCRP is localized to the apical membrane ( 2 ) where it mediates a unidirectional transport of its substrates to the luminal side of the organ acting as an efflux pump.
As shown in Bcrp1(−/−) mice, this ABC transport protein also plays an important role in the protection of the body against dietary toxins such as the heterocyclic aromatic amine and cooked food mutagen, 2-amino-1-methyl-6-phenylimidazo[4,5- b ]pyridine (PhIP) ( 3 ).
Another group of food-associated carcinogens are polycyclic aromatic hydrocarbons (PAH) ( 4 ). In the present study, benzo[ a ]pyrene (BP) was used as a model PAH as it exhibits high carcinogenic potential, and because its metabolism has been studied most intensively in recent decades. As a product of incomplete combustion of organic material it occurs ubiquitously in the environment and has been identified in numerous food items, particularly in grilled and broiled meat ( 5 ). Despite the fact that tobacco smoke and occupational exposure are well known sources of BP, PAH-contaminated food is thought to be the main source of exposure of the general population to BP ( 6 ). BP is regarded as one of the most potent carcinogens to which humans are exposed daily, but the toxic potential of BP seems to depend on the route of administration, as has been shown in several studies with experimental animals. Oral administration appears to lead to less toxicity than intraperitoneal injection, an effect that can be related to the high metabolic turnover of BP in the gastrointestinal tract ( 7 ).
Overall, the detoxification process of lipophilic compounds such as PAH involves phase-1 [e.g. cytochrome P450 (CYP) monooxygenases CYP1A1, CYP1A2 and CYP1B1] and phase-2 xenobiotic-metabolizing enzymes (XME) [e.g. sulfotransferases (SULT), UDP-glucuronosyl-transferases (UGT), and glutathione S -transferases (GST)]. Due to the strong hydrophilic nature of the generated phase-2 metabolites, they cannot cross the cell membrane passively. For the prevention of adverse effects of an intracellular accumulation of these compounds, they need to be transported actively out of the cell. This transport can be mediated by members of the ABC family of drug transporters, which are expressed on the apical [P-glycoprotein (P-gp), multidrug resistance-associated protein 2 (MRP2), and BCRP] or on the basolateral (MRP1, MRP3) domain of intestinal cells.
The human colon adenocarcinoma cell line Caco-2 is the most common in vitro model used to investigate biotransformation and transport processes for the prediction of the oral bioavailability of xenobiotics. In Caco-2 cells, BCRP has been found to be localized to the apical membrane. Furthermore, the transport of some well-known substrates of BCRP (e.g. estrone-3-sulfate) has been shown to be inhibited by BCRP inhibitors such as Ko 143 ( 8 ). Therefore, the Caco-2 cell line may be used as a model cell line for the investigation of the characteristics of BCRP.
We have previously reported ( 9 , 10 ) that the major phase-2 metabolites of BP formed by Caco-2 cells are sulfate conjugates [BP-1-sulfate (BP1S) and BP-3-sulfate (BP3S)], which are transported actively out of the cells. In these studies no contribution of P-gp and/or MRP2 to the apically directed transport of these BP conjugates could be demonstrated, and the transport protein involved thus remained to be identified.
Since many known BCRP substrates, e.g. flavopiridol, sulfasalazine, PhIP, anthracyclines and indolocarbazole derivatives ( 11 , 12 ), share some common structural features such as planar and aromatic entities, and since BCRP has been reported to be involved in the transport of conjugated organic compounds such as sulfates and glucuronides ( 13 ), we investigated the possible involvement of BCRP in the transport of the phase-2 metabolites of BP [BP3S and BP-3-glucuronide (BP3G)] out of the human intestinal cell line Caco-2 in this study.
Both XME and some ABC-transport proteins show an adaptive response upon exposure to xenobiotics, forming a biochemical barrier against their systemic uptake after ingestion ( 9 , 10 , 33 ). Thus, it is likely that there are common regulatory mechanisms for both XME and transport proteins because both components of the biochemical barrier are critical for efficient detoxification of ingested xenobiotics.
To the best of our knowledge it is not known if PAH affect the expression of BCRP. Thus, we hypothesize that aryl hydrocarbon receptor (AhR) agonists such as PAH may not only modulate phase-1 and phase-2 enzymes but also the expression of BCRP, and we address the question of whether the AhR is involved in the regulation of BCRP.
Materials and methods
Chemicals
Reserpine, BP, PD98059 (2′-amino-3′-methoxyflavone) ( 14 ), α-naphthoflavone (α-NF) and 3-methylcholanthrene (3-MC) were purchased from Sigma (Deisenhofen, Germany). Oltipraz was obtained from McKesson Bioservices (San Francisco, CA, USA). 2,3,7,8-Tetrachlorodibenzo- p -dioxin (TCDD) was purchased from Promochem (Wesel, Germany). Benzo[ k ]fluoranthene (B[ k ]F), indeno-[1,2,3- cd ]-fluoranthene (INF), and the phase-1 metabolite of BP, 3-hydroxy-benzo[ a ]pyrene (BP-3-OH), were synthesized at the Biochemical Institute for Environmental Carcinogens (Grosshansdorf, Germany). The purity of the compounds was >99% as indicated by GC-MSD and high-performance liquid chromatography (HPLC) analyses. Indolo[3,2- b ]carbazole (ICZ) was a kind gift from Prof J.Bergman (Karolinska Institute, Stockholm, Sweden) and 3′-methoxy-4′-nitroflavone was kindly provided by Prof Abel, University of Düsseldorf, Germany.
The BCRP inhibitor Ko 143 was generously provided by Professor A.H.Schinkel (The Netherlands Cancer Institute, Amsterdam, NL). S3025, a chlorogenic acid derivative and specific MRP2 inhibitor ( 15 ), was a kind gift from Dr A.W.Herling (Aventis Pharma Germany, Frankfurt, Germany). Stock solutions of all test compounds were prepared in dimethylsulfoxide (DMSO) and stored at −20°C until used.
Cell culture
The human colon adenocarcinoma cell line Caco-2 was obtained from the European Collection of Cell Cultures (ECACC, Porton Down, UK) and maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco-Invitrogen, Karlsruhe, Germany) supplemented with 10% fetal calf serum (FCS), 100 IU/ml penicillin and 100 μg/ml streptomycin in a humidified atmosphere of 5% CO 2 in air at 37°C. The cells were used at passages 56 through 66 for all experiments. TC7 cells [a subclone of Caco-2 ( 16 )] were cultured in DMEM supplemented with 20% FCS, 1% non-essential amino acids, 100 IU/ml penicillin and 100 μg/ml streptomycin. TC7 cells were used at passages 26 through 30. The BCRP-overexpressing human colon carcinoma cell line HCT 116 NRI was a generous gift from Dr H.Komatani [Merck Research Laboratories, Ibaraki, Japan ( 17 )]. The cells were cultured in the same medium as Caco-2 cells.
Transport of BP3S in the presence of inhibitors of BCRP and MRP2
Caco-2 cells were seeded onto Transwell™ inserts (4.71 cm 2 growth area, 0.4 μm pore size, polycarbonate membranes; Corning Costar, Cambridge, MA, USA) and cultured for 17 days after seeding. The medium was changed every other day. Before starting the experiments, we checked the cell monolayer integrity by measuring the transepithelial electrical resistance (TEER) (voltohmmeter with chopstick electrode; EVOM, World Precision Instruments, Sarasota, FL, USA). The net value had to exceed 300 Ω/cm 2 . The experiment was started by exchanging the culture medium from both chambers of the Transwell™ plate for medium containing the inhibitors Ko 143 (5 μM), reserpine (25 μM), S 3025 (50 μM) or vehicle (DMSO, 0.1%). After pre-incubation of the cells with the inhibitors for 1 h, the medium from both chambers of the Transwell™ plate was exchanged again for medium supplemented with the substrate BP-3-OH (5 μM) and the respective inhibitor (in the same concentration used for pre-incubation). The plates were placed on an orbital shaker during the experiment. At the indicated times, medium samples from both chambers of the Transwell™ plate were collected and stored at −20°C until HPLC analysis.
Transport of BP3S and BP3G in TC7 cells in the presence of Ko 143
In a similar manner, TC7 cells were cultured on Transwell™ plates for 12 days and subsequently pre-treated with 50 μM of oltipraz for 48 h to induce UGT1A6 ( 18 ). The experiment was started by pre-incubation of the cells with 5 μM of Ko 143, and the experiment was continued as described above. All treatments were carried out on both sides of the Transwell™ plates.
Transport of BP3S by HCT 116 NRI cells
The BCRP-overexpressing cell line HCT 116 NRI [generated by selection with NB-506, an indolocarbazole derivative and anticancer agent ( 17 )] and parental HCT 116 WT cells were seeded onto Transwell™ plates as described above. The cells were grown for 4 days and the medium was replaced every day. The experiment was started by exchanging the culture medium from both chambers of the Transwell™ plate for medium containing 2.5 μM of the substrate BP-3-OH. After 3, 6 and 9 h of incubation, medium samples were taken and stored at −20°C until HPLC analysis for BP3S.
Transport of BP3S by TC7 cells treated with AhR agonists
In order to test whether treatment with different AhR agonists would induce functionally active BCRP, TC7 cells were treated with the following: (i) oltipraz (4-methyl-5-[2-pyrazinyl]-1,2-dithiole-3-thione], a synthetic dithiole-1,2-thione and antischistosomal agent ( 19 ) which acts as a bi-functional inducer ( 40 ) and has AhR-agonistic properties ( 18 ); (ii) ICZ, which is formed from the natural compound glucobrassicin through condensation of indole-3-carbinol under acidic conditions in the stomach and is considered one of the most potent natural AhR agonists ( 37 ); and (iii) B[ k ]F, which is a PAH also found in food products and is a well-known strong AhR agonist.
TC7 cells were cultured on Transwell™ plates for 6 days and subsequently treated with oltipraz (50 μM), ICZ (2.5 μM), B[ k ]F (5 μM) or DMSO (0.1%, v/v) for 3 days. The medium was replaced daily. Preliminary experiments showed that the induction of CYP1A1 and CYP1B1 would lead to a decrease in the amount of substrate available for phase-2 enzymes (SULT and UGT) in comparison with untreated cells. We, therefore, blocked phase-1 enzymes by incubation with α-NF, a well established CYP 1A1 and 1B1 inhibitor ( 20 ). Cells were pre-incubated with α-NF (150 μM) for 1 h, and the incubation experiment was subsequently started by replacing the medium with medium containing 5 μM of the substrate BP-3-OH and α-NF (50 μM final concentration). All treatment compounds were added to both chambers of the Transwell™ plate. Medium samples were collected from both chambers of the Transwell™ plate and analyzed for BP3S, as described above.
Expression of BCRP and CYP1A1 ( mRNA ) in the presence of ICZ and AhR antagonist PD98059 or 3′-methoxy-4′-nitroflavone ( NF )
After reaching confluency, Caco-2 cells were cultured in small culture flasks (25 cm 2 ) for 14 days, pre-treated for 1 h with 10 μM of PD98059 ( 14 ) and then incubated with the ICZ (2.5 μM) and PD98059 (10 μM) mixture. After 8 and 24 h of incubation, the medium was removed, the cell monolayer was washed with ice-cold phosphate-buffered saline (PBS), and cells were harvested for RNA isolation. Treatment with 10 μM NF was done in the same manner.
Western blotting
Caco-2 cells were cultured in large culture flasks (75 cm 2 ) for 9 days and treated for 3 days with different test compounds: oltipraz (50 μM), ICZ (2.5 μM), B[ k ]F (5 μM), 3-MC (5 μM), BP (10 μM), TCDD (50 nM) or DMSO (0.1%, v/v). For harvesting the cells, the cell monolayer was washed with ice-cold PBS, and cells were scraped into ice-cold lysis buffer (2 mM Tris, 50 mM mannitol, pH 7.0) supplemented with 10 μM phenylmethylsulfonyl fluoride.
Cells were pelleted by brief centrifugation (5 min, 10000× g , 4°C) and homogenized in a small volume of lysis buffer by ultrasonication on ice. The protein content of the whole cell lysate was determined by the bicinchoninic acid method ( 21 ). Samples were diluted with 2× loading buffer [100 mM Tris, 4% (w/v) SDS, 20% (v/v) glycerol, 0.2% (w/v) bromophenolblue, pH 6.8; supplemented with 10 mM DTT] and heated at 95°C for 5 min. The samples (80 μg of protein) were loaded onto a 10% SDS–polyacrylamide gel and subjected to electrophoresis. The separated proteins were immobilized by electrotransfer onto nitrocellulose membranes (Amersham Biosciences, Braunschweig, Germany) by semi-dry blotting. Subsequently, blots were blocked with 5% (w/v) non-fat dry milk in PBS-T [PBS containing 0.07% (v/v) Tween-20] for 1 h at room temperature. Probing was performed overnight at 4°C with anti-BCRP antibody BXP-21 (anti-mouse IgG 2a , Calbiochem, Darmstadt, Germany; dilution 1:2000). The horseradish peroxidase-conjugated anti-mouse secondary antibody was then applied for 1.5 h at room temperature (Amersham Biosciences, Braunschweig, Germany; dilution 1:10000). The detection was carried out with an enhanced chemiluminescence detection kit (ECL advance™) following the manufacturer's instructions (Amersham Biosciences, Braunschweig, Germany).
Preparation of mRNA and real-time quantitative PCR analysis
Total mRNA was prepared from freshly isolated cells, and reverse transcription (RT) was performed as described in Lampen et al . ( 22 ). One hundred nanograms of mRNA was used as a template in a cDNA synthesis reaction using 100 pmol of oligodT 18 primer in the presence of MMLV reverse transcriptase (Promega, Heidelberg, Germany). The reaction was performed at 37°C for 1 h. Real-time quantitative PCR was performed in an Mx3000 cycler (Stratagene, Amsterdam, The Netherlands) using SybrGreen dye (Molecular Probes, Leiden, The Netherlands). One of the two primers was placed at the junction between two exons to avoid amplification of contaminating genomic DNA as far as possible. Primer concentrations were between 200 and 600 nM. Quantitative values were obtained from the threshold PCR cycle number (Ct) at which the increase in signal associated with an exponential growth for PCR product can first be detected. The relative mRNA levels in each sample were normalized to its β-actin content. Fold inductions in comparision to β-actin were calculated using Mx3000 Software, which used the 2 −(ΔΔCt) formular method by which ΔΔCt is the ΔCt (treated) − ΔCt (DMSO), ΔCt is Ct (gene X) − Ct (β-actin) and Ct is the cycle at which the threshold is crossed.
Primers for human CYP1A1 were 5′-ACATCACCTACGCCAGTCGC-3′ for sense and 5′-GCTGCTGCCTACCCTAGTCTCA-3′ for antisense (GenBank accession no. X02612). Primers for human BCRP were 5′-CAGGTCTGTTGGTCAATCTCACA-3′ for sense and 5′-TCCATATCGTGGAATGCTGAAG for antisense (GenBank accession no. AF098951). Primers for human β-actin were 5′-CGTCCACCGCAAATGCTT-3′ for sense and 5′-GTTTTCTGCGCAAGTTAGGTTTTGT-3′ for antisense (GenBank accession no. NM 01101). The thermal cycling comprised a denaturation step at 95°C for 5 min, 40 cycles at (95°C for 30 s, 59°C for 30 s, 72°C for 30 s). Consequently, at the end of the PCR cycles, the PCR products were analyzed using the heat dissociation protocol to confirm that a single PCR product was detected by SYBR Green dye. In prevalidation experiments the conditions for the PCR cycles were optimized and the PCR slope and PCR efficiency were calculated for each gene.
HPLC analysis
The sample clean-up and HPLC analysis of the medium samples so obtained were performed as described in detail by Buesen et al . ( 10 ). The BP metabolites were separated employing HPLC (HP 1100 series, Agilent Technologies, Waldbronn, Germany) on a reversed-phase C18 column (PAH 16 plus, 5-μm particle size, J. T. Baker, Griesheim, Germany). The detection was carried out using a diode-array detector (HP 1100 series). The amounts of BP metabolites were quantified by means of an internal standard, INF.
Statistical analysis
Statistical analyses were performed with SigmaStat ® software. Differences between mean values were determined by a Student's t -test or a one-way ANOVA followed by a Tukey–Kramer post-test. Statistically significant differences were set at P ≤ 0.05 and at P ≤ 0.001 for highly significant data.
Results
Transport of BP3S in the presence of inhibitors of BCRP and MRP2
To test whether the ABC transport protein BCRP is involved in the transport of BP3S in intestinal Caco-2 cells, we investigated its transport in the presence of the selective BCRP inhibitor Ko 143 (5 μM) ( 23 ) and the inhibitor reserpine (25 μM) ( 23 ). To further confirm our previous findings that MRP2 is not involved in the transport of BP conjugates, we also included the chemical inhibition of MRP2 by the specific MRP2 inhibitor S 3025 (50 μM) ( 15 ). Our results clearly indicate that BCRP is involved in the transport of BP3S.
As shown in Figure 1A , co-incubation of Caco-2 cells with the substrate BP-3-OH and the inhibitor Ko 143 almost completely blocked the apically directed transport of BP3S to 36% (187.41 ± 6.22 pmol/cm 2 ) of the control value (524.93 ± 37.8 pmol/cm 2 ) after incubation for 3 h. This effect declined thereafter (70% of control after 6 h and 73% after 9 h). In the absence of inhibitors BP3S was found to be transported mainly to the apical compartment, resulting in an apically directed net transport. Inhibition of the apical ABC transporter BCRP was so efficient that the cells excreted even more BP3S to the basolateral compartment of the Transwell™ plate, at least after the shortest incubation time (3 h). This reversed the excretion to a basolaterally directed net transport of 14% (compared with control) to the basal chamber. The total amount of the apically and basolaterally excreted BP3S was the same with both treatments and the control (data not shown). This indicates that the inhibitors Ko 143 and reserpine did not interfere with the sulfation of the substrate BP-3-OH.
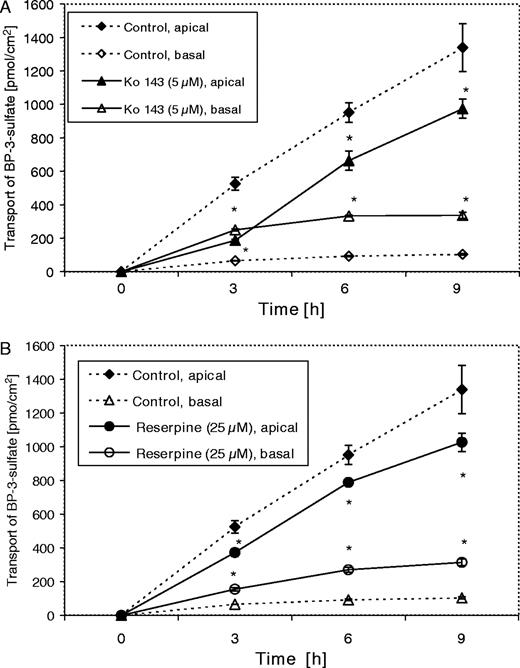
Transport of BP3S (in pmol/cm 2 ) in the presence of Ko 143 (5 μM) ( A ) and reserpine (25 μM) ( B ). Caco-2 cells were cultured on Transwell™ plates for 18 days and pre-incubated with Ko 143 (5 μM), reserpine (25 μM) or vehicle (DMSO, 0.1%) for 1 h, and subsequently co-incubated with 3-hydroxy-BP (5 μM) and Ko 143 (5 μM), reserpine (25 μM), or vehicle (DMSO, 0.1%) for 3, 6 and 9 h. All treatments were carried out on both chambers of the Transwell™ plates. Medium samples were collected from both chambers of the Transwell™ plates and analyzed for BP3S. Data are expressed as means ± SD of four individual monolayers. This experiment has been repeated with similar results. An asterisk (*) indicates statistically significant differences compared with control ( P < 0.05; one-way ANOVA, Tukey Kramer post-test).
Co-incubation with 25 μM of reserpine had a lower inhibitory effect on the apically directed transport of BP3S than with Ko 143. Reserpine reduced the amount of apically transported BP3S to 71% of the control value after 3 h ( Figure 1B ). This observation is consistent with the reported higher affinity (lower EC 90 value) of Ko 143 (∼25 nM) to BCRP compared with that of reserpine, which is 5 μM ( 23 ). Due to the lower inhibitory effect of reserpine, there was no reversal of the net transport. As described for cells treated with Ko 143, inhibition of the apically directed net transport declined after 6 and 9 h of incubation time, most likely due to endogenous metabolism of the inhibitor.
There was no difference in the transport of BP3S in the presence of the specific MRP2 inhibitor S 3025 from that in the control cells, which indicates that MRP2 is not involved in the transport of BP3S. S 3025 had no effect on the total amount of BP3S transported by Caco-2 cells (data not shown).
Transport of BP3G in the presence of Ko 143
The most potent inhibitor of BCRP, Ko 143 (5 μM), was chosen for the investigation of the inhibitory effect on the transport of a glucuronic acid conjugate of BP, BP3G. For these experiments we used TC7 cells, a subclone of Caco-2 cells with higher UGT activity than that of the parental cell line ( 24 ). The TC7 cells (both control and treated cells) were also pre-treated with oltipraz for 48 h to induce UGT1A6 ( 18 ).
As shown in Figure 2A , the amount of apically excreted BP3G was diminished in cells treated with Ko 143 to 24% (48.57 ± 2.24 pmol/cm 2 ) of the control value (203.88 ± 25.33 pmol/cm 2 ) after 3 h incubation. This effect was subsequently weakened, with 37% (118.38 ± 6.68 pmol/cm 2 ) of the control level present after 6 h and 67% (179 ± 9.09 pmol/cm 2 ) after 9 h incubation. It should be noted that the total amount of BP3G excreted by untreated cells also declined after 9 h of incubation, most likely due to the metabolic activity of endogenous glucuronidases, which deconjugate the metabolite that is formed. Comparison of the effect of Ko 143 on the transport of both phase-2 metabolites (BP3S and BP3G) clearly shows that the reduction in the transport of BP3S to the apical compartment was greater than that of BP3G [to 67.08 ± 7.09 (17%) of the control after 3 h, to 171.04 ± 22.63 (20%) after 6 h and to 273.63 ± 10.57 pmol/cm 2 (24%) after 9 h; see Figure 2B ].
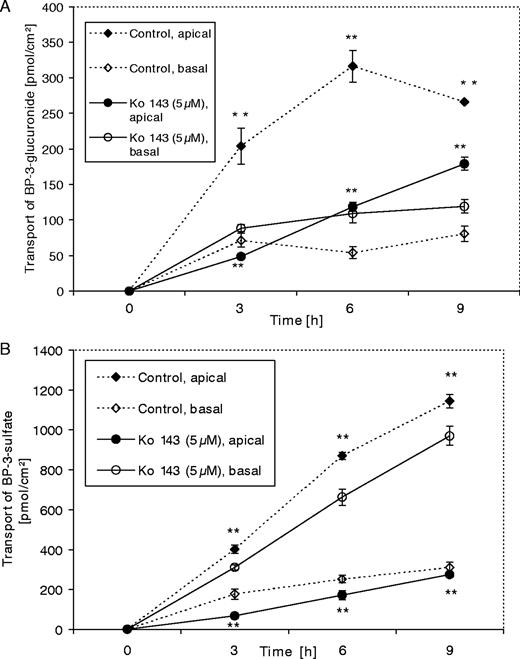
Transport of BP3G ( A ) and BP3S ( B ) in the presence of Ko 143 (5 μM) led to a reversal of the net transport to the basal compartment for all incubation times. TC7 cells were cultured on Transwell™ plates for 14 days. The cells were treated with 50 μM of oltipraz for 48 h to induce UGT1A6. The transport experiment was started by pre-treatment of the cells with Ko 143 (5 μM) for 1 h, followed by co-incubation with 3-hydroxy-BP (5 μM) and Ko 143 (5 μM) for 3, 6 and 9 h. Treatments were performed on both sides of the Transwell™ plates, and medium samples were collected (from both chambers) and analyzed for BP3G and BP3S. Data are expressed as means ± SD of four individual monolayers. This experiment has been repeated with similar results. ** P < 0.001 (Student's t -test).
Interestingly, the apically directed transport of BP3S was inhibited by Ko 143 in TC7 cells more effectively than in Caco-2 cells; this is consistent with the higher BCRP protein expression in Caco-2 cells than in its subclone TC7 (Figure 6B). The inhibition of BCRP in TC7 cells resulted in a basolateral net transport at all times (3, 6 and 9 h; data not shown). Overall, our results show that Ko 143 had a lower inhibitory effect on the transport of BP3G to the apical region than on that of BP3S.
Transport of BP3S by HCT 116 NRI cells
The BCRP-overexpressing cell line HCT 116 NRI was used as an additional tool to test whether BP3S is a substrate for BCRP. High overexpression of BCRP in HCT 116 NRI cells was confirmed by western blotting ( Figure 3 ). The net transport of endogenously formed BP3S to the apical compartment of the Transwell™ plate was significantly higher for HCT NRI cells after 3 h of incubation (23.31 ± 1.03 pmol/cm 2 ) than for HCT116 WT cells (12.71 ± 2.30 pmol/cm 2 , P < 0.05; Student's t -test; data not shown), but this net transport declined after 6 and 9 h of incubation.
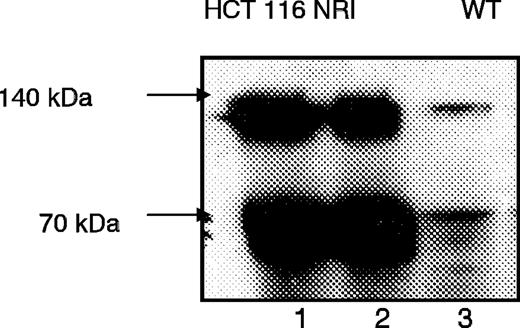
Western blot analysis of membrane-enriched cell fractions of drug-selected (NB-506) HCT 116 NRI cells (lanes 1 and 2) and HCT116 WT cells (lane 3) cells clearly indicates the overexpression of BCRP in HCT 116 NRI cells. Cells were cultured for 5 days, and 80 μg of the membranes so obtained were subjected to western blot analysis as described in the ‘Materials and methods’ section. Reduced BCRP protein (monomer) migrated at 70 kDa, and the dimeric form at 140 kDa ( 1 ) (indicated by arrows). A representative blot of three is shown.
Transport of BP3S by TC7 cells treated with AhR agonists
As shown in Figure 4 , TC7 cells treated with all three test compounds for 3 days (oltipraz, 50 μM; ICZ, 2.5 μM; B[ k ]F, 5 μM) transported significantly more BP3S to the apical chamber of the Transwell™ plate after 3 and 6 h of incubation. After 9 h, BP3S levels were elevated only slightly (112% of the control in cells treated with oltipraz) or not at all (101% in cells treated with ICZ).
![Transport of BP3S to the apical compartment of the Transwell™ plates. TC7 cells were grown on Transwell™ plates for 7 days and treated on both chambers of the Transwell™ plate for 3 days either with ICZ (2.5 μM), B[ k ]F (5 μM), oltipraz (50 μM) or vehicle (DMSO, 0.1%). To block the activity of CYP1A1 and CYP1B1, cells were pre-incubated with 150 μM of α-NF for 1 h and then co-incubated with the substrate 3-hydroxy-BP (5 μM) and α-NF for 3, 6 and 9 h (both sides of the Transwell™ plate). Medium samples were analyzed for BP3S. Data are expressed as percent of control (from values of three individual monolayers). This experiment has been repeated with similar results. An asterisk (*) indicates statistically significant differences compared with control ( P < 0.05; one-way ANOVA, Tukey Kramer post-test). ** P < 0.001.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/carcin/26/10/10.1093_carcin_bgi139/2/m_bgi139f4.jpeg?Expires=1716349319&Signature=o49IraEE2W-413eXRrP3VSd-LPGNWcLbzCOrKdZ31fA1pySyCQ8UPyobdBXG9HtCqs5E~XSk29kIYPQbtQqCIGRTLQRqiVTdI3pU1h0KEN9nKTj1coUDjbGM0VIELWZ2VIUAgJ8gNk3zB4FNswYdKm7XiBDksre1kY1E6u0RQvuVKbim5gM4wtgFfIqnFzFbcmFbZFsZJ97HHv46ms-RJzVawTE-n61bj-3UVwVnqo7Oos1qayct8FdpYTmjMQiUwD5qa8-Mn3MGOPLHCllmvcPf~CB0hOGGmnhp4RWBSoQbGfrwjXyBYq3-AGOcg3swzLGGxK3mEZFgd6yKjylt2w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Transport of BP3S to the apical compartment of the Transwell™ plates. TC7 cells were grown on Transwell™ plates for 7 days and treated on both chambers of the Transwell™ plate for 3 days either with ICZ (2.5 μM), B[ k ]F (5 μM), oltipraz (50 μM) or vehicle (DMSO, 0.1%). To block the activity of CYP1A1 and CYP1B1, cells were pre-incubated with 150 μM of α-NF for 1 h and then co-incubated with the substrate 3-hydroxy-BP (5 μM) and α-NF for 3, 6 and 9 h (both sides of the Transwell™ plate). Medium samples were analyzed for BP3S. Data are expressed as percent of control (from values of three individual monolayers). This experiment has been repeated with similar results. An asterisk (*) indicates statistically significant differences compared with control ( P < 0.05; one-way ANOVA, Tukey Kramer post-test). ** P < 0.001.
Cells treated with ICZ showed the greatest increase in apically transported BP3S after 3 h (185% of control), followed by those treated with oltipraz (182% of control) and B[ k ]F (171% of control, Figure 4 ). Interestingly, B[ k ]F treatment led to the greatest elevation in the amount of BP3S transported to the apical chamber of the Transwell™ plate after 6 h (162% of control), whereas the increase was lower in the other treatment groups (111% of control with oltipraz; 138% of control with ICZ).
The design of this experiment required the inhibition of CYP1A1 and CYP1B1 by α-NF (see ‘Materials and methods’ section), which are co-induced with BCRP due to treatment with the above-mentioned AhR agonists.
A preliminary experiment showed that, without this inhibition, the substrate BP-3-OH would undergo further hydroxylation thereby decreasing the substrate concentration available for phase-2 enzymes. The presence of α-NF had no effect on the total amount of BP3S formed by Caco-2 cells (data not shown).
Effect of oltipraz, ICZ and B[k]F on BCRP mRNA expression
Fully differentiated Caco-2 cells were treated for 24 h with different AhR agonists such as TCDD, B[ k ]F, ICZ, oltipraz and BP. Measured by real-time quantitative PCR and shown in Figure 5 , all AhR agonists were able to induce mRNA expression of BCRP. The stronger the AhR agonist, the stronger the induction of BCRP. TCDD seems to be the strongest inducer, followed by ICZ, B[ k ]F and BP, and oltipraz is the weakest. Furthermore, the induction of all investigated compounds was concentration dependent (data not shown).
![Effects of TCDD, B[ k ]F, BP, ICZ or oltipraz on mRNA expression of BCRP. Real time PCR was used to determine the effects of treatment of Caco-2 cells with with 0.1% DMSO (control), BP (10 μM), oltipraz (50 μM), ICZ (2.5 μM), B[ k ]F (5 μM), or TCDD (50 nM) for 24 h on the levels of BCRP in Caco-2 cells relative to the housekeeping gene β-actin. Experiments were carried out at least in triplicate. A two-tail, paired Student’s t -test was performed, and P values of the mRNA levels of the treated samples were calculated in reference to the solvent control ( *P < 0.05).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/carcin/26/10/10.1093_carcin_bgi139/2/m_bgi139f5.jpeg?Expires=1716349319&Signature=xPyWI7BmJzVVAu3boswHwwNpe3us7FGQMHECe6afMUP2kZXiMKkEjPTCrRLhdbhOmtUKyXFnpx1HUvopccwXqLWV~Cw7qONQd9VFsd7tcQIlZ5viPLPFML9VfzfM48dGWqiv-IXa-aMN-l472eaH~ASTP6VDs8RPGKQkXQeMAwcWqhQmwVk7ghbOGrhH3YJgbJq~t-c~az1dUDwcAbX5DaYR6rrqF64Hk0fCy35KpTMO-g-bUFi1bz0QhvKuyVqz~ClYsBKZjj34udo2sFX9q8f5qt5pKGUF54hRsEn9c6xf4HbQLG0rqu2PU19OSRSDHFklxwgRbyjP9xgO-l8F1w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effects of TCDD, B[ k ]F, BP, ICZ or oltipraz on mRNA expression of BCRP. Real time PCR was used to determine the effects of treatment of Caco-2 cells with with 0.1% DMSO (control), BP (10 μM), oltipraz (50 μM), ICZ (2.5 μM), B[ k ]F (5 μM), or TCDD (50 nM) for 24 h on the levels of BCRP in Caco-2 cells relative to the housekeeping gene β-actin. Experiments were carried out at least in triplicate. A two-tail, paired Student’s t -test was performed, and P values of the mRNA levels of the treated samples were calculated in reference to the solvent control ( *P < 0.05).
Effect of AhR agonists on BCRP protein expression in Caco-2 and TC7 cells
The treatment of Caco-2 cells for 3 days with various AhR agonists (TCDD, 3-MC, BP, oltipraz, ICZ or B[ k ]F) was found to markedly enhance the expression of BCRP protein as detected by western blotting. The ability to induce BCRP protein expression apparently correlates with the AhR affinity of the treatment compounds. Treatment with TCDD, a prototypical AhR ligand with an AhR affinity in the picomolar range ( Kd = 7.1 × 10 −12 mol ( 25 ), resulted in the greatest increase in the expression of BCRP protein, followed by 3-MC and BP ( Figure 6A ).
![( A ) Western blot analysis of BCRP in Caco-2 cells after 3 days of treatment with vehicle (0.1% DMSO, lane 1), BP (10 μM, lane 2), 3-MC (5 μM, lane 3), TCDD (50 nM, lane 4). (The arrow indicates 75 kDa protein of the standard; M = marker.) Whole cell lysates were prepared, and 75 μg of protein were resolved by SDS–PAGE (10% acrylamide) and blotted as described in the ‘Materials and methods’ section. One representative of three similar experiments is shown. ( B ) Western blot analysis of BCRP in Caco-2 cells and its subclone TC7. TC7 cells were treated for 3 days with either vehicle (0.1% DMSO, lane 1), ICZ (5 μM, lane 2), B[ k ]F (5 μM, lane 3) or oltipraz (50 μM, lane 4). Caco-2 cells were treated for 3 days with either (0.1% DMSO, lane 5), ICZ (5 μM, lane 6), oltipraz (50 μM, lane 7) or B[ k ]F (5 μM, lane 8). Proteins (75 μg) from whole cell lysates were loaded onto an 8% acrylamide gel and separated by SDS–PAGE as described in the ‘Materials and methods’ section. One representative of three similar experiments is shown.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/carcin/26/10/10.1093_carcin_bgi139/2/m_bgi139f6.jpeg?Expires=1716349319&Signature=jvNzeHJ6YIBBxwxLNKNGjK-mNAcOGTJozKt3c1ih1BUpbqCvkJHWS14L3RhJZLIEqiydqHNeCRuOHC~OGUzA7WD6X-NDFKpgsRDnJozB-03lfE-wz8ZvB7n3H98r0I0OwcbeSZcjzlwk4eoOziUFs~aXLYu-bVwzPGm3Qy9fSoFkOoPjF-1A0env3mCxLrMbzzMNIIy7Vfu63lcc1UJweT73n8AJWTmMPqFUZHpM3pB1nIR~P~LZ6gGC39Jw5t~ob2ROKbVj~587LIS8oRmc2XEkrqTmOKEFnhurO50OSDQP2TgBPn8kjWqXFukQohxGT39KzKUBLa7RpGoUUhmxqg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
( A ) Western blot analysis of BCRP in Caco-2 cells after 3 days of treatment with vehicle (0.1% DMSO, lane 1), BP (10 μM, lane 2), 3-MC (5 μM, lane 3), TCDD (50 nM, lane 4). (The arrow indicates 75 kDa protein of the standard; M = marker.) Whole cell lysates were prepared, and 75 μg of protein were resolved by SDS–PAGE (10% acrylamide) and blotted as described in the ‘Materials and methods’ section. One representative of three similar experiments is shown. ( B ) Western blot analysis of BCRP in Caco-2 cells and its subclone TC7. TC7 cells were treated for 3 days with either vehicle (0.1% DMSO, lane 1), ICZ (5 μM, lane 2), B[ k ]F (5 μM, lane 3) or oltipraz (50 μM, lane 4). Caco-2 cells were treated for 3 days with either (0.1% DMSO, lane 5), ICZ (5 μM, lane 6), oltipraz (50 μM, lane 7) or B[ k ]F (5 μM, lane 8). Proteins (75 μg) from whole cell lysates were loaded onto an 8% acrylamide gel and separated by SDS–PAGE as described in the ‘Materials and methods’ section. One representative of three similar experiments is shown.
In the second set of experiments, the expression of BCRP in parental Caco-2 cells was compared with that in its subclone TC7 ( Figure 6B ). Western blot analysis of untreated cells showed a higher basal expression of BCRP in Caco-2 cells than in TC7 cells, while treatment with B[ k ]F, ICZ and oltipraz induced BCRP protein expression in both cell lines; here, B[ k ]F was the most potent inducer of BCRP protein, followed by ICZ and oltipraz.
Effect on BCRP mRNA expression in the presence of ICZ and the AhR antagonist PD 98059 or 3′-methoxy-4′-nitroflavone
Treatment of Caco-2 cells with the potent AhR agonist ICZ (2.5 μM) markedly increased the expression of BCRP mRNA after 8 and 24 h of incubation ( Figure 7A ). Cells co-incubated with ICZ and the AhR antagonist PD98059 (2′-amino-3′-methoxyflavone) showed a clearly reduced induction of the BCRP mRNA expression by ICZ. The effects of the same treatment on the expression of the established AhR target gene CYP1A1 are shown in Figure 7B . The induction of CYP1A1 mRNA expression induced by ICZ was also inhibited by co-treatment of the cells with ICZ and PD98059. Furthermore, cells incubated with ICZ and the well known AhR antagonist 3′-methoxy-4′-nitroflavone ( 26 ) showed also a clear inhibition of the induction of BCRP mRNA expression by ICZ ( Figure 7C ).
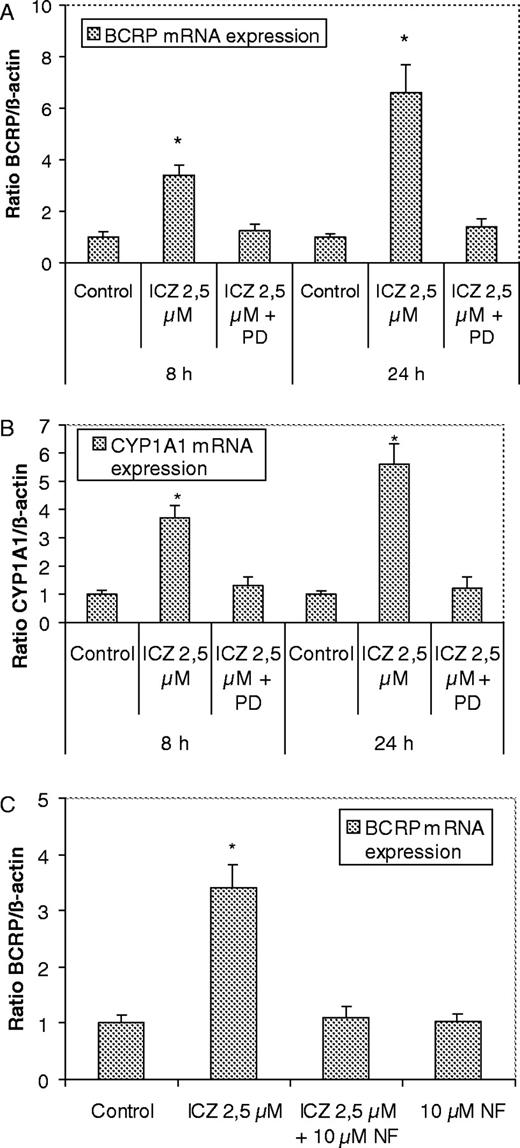
Effects of AhR antagonists on BCRP mRNA expression in Caco-2 cells. Effect of co-treatment of Caco-2 cells with ICZ (2.5 μM) and the AhR antagonist PD98059 (10 μM) on the expression of BCRP ( A ) and CYP1A1 mRNA ( B ). BCRP, CYP 1A1, and β-actin gene expression was measured by real-time PCR after treatment of Caco-2 cells for 8 h with vehicle (control, 0.2% DMSO), ICZ (2.5 μM), ICZ 2.5 μM + PD98059 (10 μM) and 24 h: vehicle (control, 0.2% DMSO), ICZ (2.5 μM), ICZ 2.5 μM + PD98059 (10 μM). ( C ) Effect of AhR antagonist 3′-methoxy-4′-nitroflavone (NF): BCRP and β-actin gene expression was measured by real-time quantitative PCR after treatment of Caco-2 cells for 8 h with vehicle (control, 0.2% DMSO), ICZ (2.5 μM), ICZ 2.5 μM + NF (10 μM), NF (10 μM). Experiments were carried out at least in triplicate. A two-tail, paired Student’s t -test was performed, and P values of the mRNA levels of the treated samples were calculated in reference to the solvent control ( *P < 0.05).
Effect of the AhR antagonist PD98059 on the induction of BCRP protein expression by ICZ
As shown in Figure 8 , treatment of Caco-2 cells with the potent AhR agonist ICZ (2.5 μM) markedly increased the BCRP protein expression. Cells co-incubated with ICZ and the AhR antagonist PD98059 showed a clear inhibition of the induction of the BCRP protein expression by ICZ, indicating involvement of AhR.

Western blot analysis of BCRP in Caco-2 cells after 3 days of treatment with vehicle (0.1% DMSO = C), ICZ (2.5 μM, lane 1), ICZ (2.5 μM) plus PD98059 (10 μM, lane 2), ICZ (2.5 μM) plus PD98059 (20 μM, lane 3), PD98059 (10 μM, lane 4). Whole cell lysates were prepared and 75 μg of protein were resolved by SDS–PAGE and blotted as described in the ‘Materials and methods’ section. Similar results were obtained in three independent experiments.
Discussion
Due to its rather broad substrate specificity, BCRP is not only able to transport large lipophilic compounds, which can be either negatively or positively charged, but also conjugates with glucuronic acid and sulfates such as estrone sulfate, dehydroepiandrosterone sulfate and 4-methylumbelliferone glucuronide ( 13 ). Further support for the important role of BCRP in the transport of sulfate conjugates is given by a recent in vivo study of Mizuno et al . ( 27 ) employing bcrp1 knock-out mice. These animals showed impaired renal excretion of a thiazolium sulfate conjugate due to the lack of bcrp1 expression. Our findings provide a new example of the transport of conjugates with sulfate and glucuronic acid by BCRP in Caco-2 cells and its subclone TC7: phase-2 metabolites of the carcinogen BP, BP3S and BP3G. To the best of our knowledge, this is the first demonstration of the involvement of BCRP in the transport of sulfate and glucuronide conjugates of a toxicologically relevant PAH in human intestinal cells.
In addition to BCRP, two further members of the ABC superfamily of drug transporters, namely P-gp and MRP2, are known to be expressed on the apical domain of Caco-2 cells. Since overlapping substrate specificities for all three transporters have been described ( 11 ), the possible involvement of P-gp and MRP2 in the transport of BP3S and BP3G had to be taken into consideration. Published data on the P-gp-mediated transport of BP are somewhat contradictory ( 28 , 29 ). Our own previous studies indicate at least that P-gp is not involved in the transport of BP1S and BP3S ( 9 , 10 ).
Another possible candidate included in this study in addition to BCRP was MRP2, since sulfate and glucuronic acid conjugates of organic compounds are known to be substrates for MRP2 ( 11 ). Furthermore, the heterocyclic aromatic amine PhIP has, for instance, been shown to be a substrate for both BCRP ( 12 ) and MRP2 ( 30 ). As was shown in the present study, chemical inhibition of MRP2 by S3025, a synthetic chlorogenic acid derivative and effective MRP2 inhibitor ( 15 ), had no effect on the transport of BP3S in Caco-2 cells. Involvement of MRP2 in the transport of BP3S can therefore be ruled out.
The investigation of the transport of an endogenously formed compound in the intact cell is more complex than a translocation assay of an unaltered substrate. Because the compound which is transported first has to be formed, any disruption of this process may affect the results. However, none of the inhibitors employed here had any effect on phase-2 metabolism of the substrate 3-hydroxy-BP, as all treatments resulted in the same total amount of apically and basolaterally excreted BP3S or BP3G.
Interestingly, the apically directed transport of BP3S was inhibited by Ko 143 to a different extent in Caco-2 cells than in its subclone TC7. Whereas the amount of apically excreted BP3S was reduced in TC7 cells to 17% (corresponding to 83% inhibition) of the control level after 3 h of incubation, the amount of apically transported BP3S was reduced in Caco-2 cells to only 36% (corresponding to 64% inhibition) of the control level (after 3 h). This suggests different levels of expression of the BCRP protein in Caco-2 cells and in those of its subclone TC7, and this was confirmed by western blot analysis ( Figure 6B ).
It is significant that inhibition of BCRP with the most effective inhibitor Ko 143 was even able to reverse the direction of the net transport of BP3S and BP3G to the basolateral compartment of the Transwell™ plate. Reversal of the transport direction indicates the existence of another transport system on the basolateral domain of the Caco-2 cells, which mediates the export of both phase-2 metabolites of BP. Two members of the multidrug resistance-associated protein family, MRP1 and MRP3, are possible candidates since they are expressed in the basolateral region of polarized cells, and both transport proteins show overlapping substrate specificity with BCRP [e.g. estrone-3-sulfate ( 11 )].
Several regulatory mechanisms have been reported for the expression of ABC transport proteins. The regulation of MRP2 has been shown to involve several nuclear receptors ( 31 ), and the expression of P-gp can be stimulated by certain environmental stress factors such as heat shock, hypoxia and UV irradiation ( 32 – 34 ). Interestingly, there are some similarities between the regulation of P-gp and BCRP mediated by stress factors, since long exposure of human gastric carcinoma cells to increasing temperatures elevated the expression of BCRP ( 35 ); very recently, Bcrp was reported to be upregulated under hypoxic conditions in murine stem cells ( 36 ).
In the present study, we provide for the first time strong evidence that the regulation of the BCRP gene is most likely mediated via an Ah receptor-dependent pathway. Known AhR agonists such as TCDD, B[ k ]F, 3-MC, BP, ICZ and oltipraz elevated the levels of BCRP mRNA and protein expression. In addition to the induction of the BCRP protein expression by several AhR agonists, we showed here that treatment of TC7 cells with B[ k ]F, ICZ and oltipraz led to an increased apical transport of BP3S, indicating that the expressed protein is functionally active with respect to the transported substrates. The increase in the amount of apically transported BP3S seems to correspond to the potency of the AhR agonist used for the treatment. In addition to TCDD, ICZ is known to be one of the strongest AhR agonists, with an AhR binding affinity only ∼200 times lower ( Kd = 1.9 × 10 −10 M) than that of TCDD ( Kd = 7.1 × 10 −12 M) ( 25 , 37 ). Another strong AhR agonist is the planar and lipophilic PAH, B[ k ]F ( 38 ). Treatment with ICZ and B[ k ]F elicited the greatest increase in the amount of apically transported BP3S, and this was statistically significant for incubation times of 3 and 6 h, but not for 9 h. After 9 h incubation, there was no longer sufficient inhibition of CYP1A1 and CYP1B1 by α-NF due to its metabolic degradation (as detected by HPLC; not shown). Apparently, α-NF was metabolized more rapidly in cells treated with the more potent AhR agonists and with the CYP1A1 inducers ICZ and B[ k ]F. In contrast, treatment with oltipraz led to a statistically significant increase in the amount of apically excreted BP3S after 3 and 9 h, which implies a lower affinity of AhR for oltipraz than for B[ k ]F and ICZ.
Moreover, the BCRP overexpressing colonic cell line HCT116 NRI showed a higher net transport of BP3S than wild-type HCT 116 cells. Although a high overexpression of BCRP in the drug-selected cell line was confirmed by western blot analysis, there was only very little difference in the amount of transported BP3S between overexpressing clone and wild-type cells, which was observed only after an incubation of 3 h but not after 6 and 9 h. The absolute amount of BP3S formed from the substrate BP-3-OH was ∼10 times less than the amount of BP3S formed by TC7 cells, and this is most likely due to a lower level of expression of phenol-sulfotransferases (aside from to the fact that the concentation of substrate in this experiment was lower, 2.5 μM instead of 5 μM). Xia et al . ( 8 ) found that a lower TEER value in late passage Caco-2 cells correlated with a decreased polarity of the transport of estrone-3-sulfate. The higher polarity of the transport of BP3S observed in TC7 cells and Caco-2 cells compared with HCT116 cells is most likely a result of the lower TEER of cultured HCT 116 cells and may be an explanation for the discrepancy between the high expression of BCRP and the observed low transport of BP3S.
To confirm the possible involvement of the AhR in the transcriptional regulation of BCRP, we investigated the expression of BCRP (mRNA and protein levels) in the presence of the AhR antagonists such as PD98059, a substituted flavone ( 14 ) or the 3′-methoxy-4′-nitroflavone ( 26 ). The outcome of these experiments supports our hypothesis that the AhR is most likely involved in the transcriptional regulation of BCRP. It is clear that additional investigations are needed to further substantiate this finding.
Numerous naturally occurring AhR agonists are presently known (e.g. flavonoids, dietary indoles, dibenzoylmethane), which suggests the possible modulation of BCRP expression by the consumption of food containing these compounds. This can result in decreased bioavailability of potential carcinogens such as BP; we are, therefore, currently investigating the impact of food-derived AhR agonists on the expression of BCRP.
In summary, our results indicate the involvement of BCRP in the apically directed transport of BP3S and BP3G in Caco-2 and TC7 cells, and provide strong evidence that regulation of the BCRP gene is most likely AhR dependent. While progress is being made in the clarification of further physiological roles of BCRP, it is very likely that other control mechanisms will also be revealed that govern the expression of this half-transporter. We conclude that BCRP may play an important role in the restriction of systemic exposure of humans to ingested BP and related food-associated PAH by the active transport of their metabolically formed sulfate and glucuronide conjugates back to the gut lumen.
This study was supported by DFG grants (LA1177, SE552) EU-RIN-2002-00268, women promotion programme of the School of Veterinary Medicine Hannover for BE, and the BfR Berlin. We thank N.Brauer for technical help. We thank Professor A.H.Schinkel (The Netherlands Cancer Institute, Amsterdam, The Netherlands) for generously providing Ko 143; Dr H.Komatani (Merck Research Laboratories, Ibaraki, Japan) for the HCT116 NRI cells, Dr A.W.Herling for the S3025; and Professor J.Bergman (Karolinska Institute, Stockholm, Sweden) for kindly donating the ICZ and Professor J.Abel (University of Duesseldorf, Germany) for providing 3′-methoxy-4′-nitroflavone. We thank Dr Yan Lee for doing parts of the optimerisation of the QPCR.
Conflict of Interest Statement : None declared.
References
Kage,K., Tsukahara,S., Sugiyama,T., Asada,S., Ishikawa,E., Tsurou,T. and Sugimoto,Y. (
Maliepaard,M., Scheffer,G.L., Faneyte,I.F., van Gastelen,M.A., Pijnenborg,A.C., Schinkel,A.H., van De Vijver,M.J., Scheper,R.J. and Schellens,J.H. (
Van Herwaarden,A.E., Jonker,J.W., Wagenaar,E., Brinkhuis,R.F., Schellens,J.H.M., Beijnen,J.H. and Schinkel,A.H. (
Phillips,D.H. (
Kazerouni,N., Sinha,R., Hsu,C.H., Greenberg,A. and Rothman,N. (
Hattemer-Frey,H.A. and Travis,C.C. (
Robinson,J.R., Felton,J.S., Levitt,R.C., Thorgeirsson,S.S. and Nebert,D.W. (
Xia,C.Q., Liu,N., Yang,D., Miwa,G. and Gan,L.S. (
Buesen,R., Mock,M., Nau,H., Seidel,A., Jacob,J. and Lampen,A. (
Buesen,R., Mock,M., Seidel,A., Jacob,J. and Lampen,A. (
Haimeur,A., Conseil,G., Deeley,R.G. and Cole,S.P.C. (
Pavek,P., Merino,G., Wagenaar,E., Bolscher,E., Novotna,M., Jonker,J.V. and Schinkel,A.H. (
Suzuki,M., Suzuki,H., Sugimoto,Y. and Sugiyama,Y. (
Reiners,J.J.,Jr, Lee,J.-Y., Clift,R.E., Dudley,D.T. and Myrand,S.P. (
Herling,A.W., Schwab,D., Burger,H.J., Maas,J., Hammerl,R., Schmidt,D., Strohschein,S., Hemmerle,H., Schubert,G., Petry,S. and Kramer,W. (
Chantret,I., Rodolosse,A., Barbat,A., Dussaulx,E., Broth-Laroche,E., Zweibaum,A. and Rousset,M. (
Komatani,H., Kotani,H., Hara,Y., Nakagawa,R., Matsumoto,M., Arakawa,H. and Nishimura,S. (
Le Ferrec,E., Lagadic-Gossmann,D., Rauch,C., Bardiau,C., Maheo,K., Massiere,F., Le Vee,M., Guilluozo,A. and Morel,F. (
Gentilini,M., Duflo,B., Richard-Lenoble,D., Brucker,G., Danis,M., Niel,G. and Meunier,Y. (
Shimada,T., Yamazaki,H., Foroozesh,M., Hopkins,N.E., Alworth,W.L. and Guengerich,F.P. (
Smith,P.K., Krohn,R.I., Hermanson,G.T., Mallia,A.K., Gartner,F.H., Provenzano,M.D., Fujimoto,E.K., Goeke,N.M., Olson,B.J. and Klenk,D.C. (
Lampen,A., Ebert,B., Stumkat,L., Jacob,J. and Seidel,A. (
Allen,J.D., van Loevezijn,A., Lakhai,J.M., van der Valk,M., van Tellingen,O., Reid,G., Schellens,J.H.M., Koomen,G.-J. and Schinkel,A.H. (
Muenzel,P.A., Bookjans,G., Mehner,G., Lehmköster,T. and Bock,K.W. (
Safe,S.H. (
Lu,Y.F., Santostefano,M., Cunningham,B.D., Threadgill,M.D. and Safe,S. (
Mizuno,N., Suzuki,M., Kusuhara,H., Suzuki,H., Takeuchi,K., Niwa,T., Jonker,J.W. and Sugiyama,Y. (
Schuetz,E.G., Yasuda,K., Arimori,K. and Schuetz,J.D. (
Yeh,G.C., Lopaczynska,J., Poore,C.M. and Phang,J.M. (
Dietrich,C.G., de Waart,D.R., Ottenhoff,R., Bootsma,A.H., van Gennip,A.H. and Oude Elferink,R.P.J. (
Kast,H.R., Goodwin,B., Tarr,P.T., Jones,S.A., Anisfeld,A.M., Stoltz,C.M., Tontonoz,P., Kliewer,S., Willson,T.M. and Edwards,P.A. (
Kioka,N., Yamano,Y., Komano,T. and Ueda,K. (
Scotto,K.W. (
Hu,Z., Jin,S. and Scotto,K.W. (
Stein,U., Lage,H., Jordan,A., Walther,W., Bates,S.E., Litman,T., Hohenberger,P. and Dietel,M. (
Krishnamurthy,P., Ross,D.D., Nakanishi,T., Bailey-Dell,K., Zhou,S., Mercer,K.E., Sarkadi,B., Sorrentino,B.A.P. and Schuetz,J.D. (
Bjeldanes,L.F., Kim,J.-Y., Grose,K.R., Bartholomew,J.C. and Bradfield,C.A. (
Jacob,J., Grimmer,G. and Schmoldt,A. (
Christians,U., Jacobsen,W., Benet,L.Z. and Lampen,A. (
Author notes
1Institute for Food Toxicology, University of Veterinary Medicine Hannover, Foundation, Bischofsholern Damm 15/115, 30173 Hannover, Germany, 2Biochemical Institute for Environmental Carcinogens, Prof. Dr Gernot Grimmer Foundation, Lurup 4, D-22927 Grosshansdorf, Germany and 3Federal Institute for Risk Assessment (BfR), Thielallee 88-92, D-14195 Berlin, Germany

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}