
Abstract
Biliary cancers (BC) are rare, chemoresistant and are associated with a poor prognosis. Targeting the Akt pathway is of significance in BC. We hypothesized that the allosteric inhibitor MK-2206 will be active in BC. This was a multi-institutional phase II study of MK-2206 given to patients with advanced, refractory BC. The primary end point was overall response rate. We also characterized pharmacokinetic profiles of MK-2206 in these patients and explored its potential correlation with clinical outcomes. Eight patients were enrolled prior to early termination of the trial. All patients had received prior systemic therapy. The best response observed was stable disease, exceeding 12 weeks in two patients. Toxicities were mild and tolerable. MK-2206 exhibited a pharmacokinetic profile with an apparent slow absorption followed by biphasic elimination in these patients with BC. No significant association was observed between the pharmacokinetic properties of MK-2206 and clinical outcomes. MK-2206 as a single-agent in BC is tolerable with pharmacokinetic properties similar to patients with other solid tumors. No clinical activity was observed in this limited population. Further development of Akt inhibitors may need to focus on combinations with other molecular targeted agents, conventional cytotoxic chemotherapy and prospective patient selection.
Similar content being viewed by others

Introduction
Biliary cancers (BC) are rare, chemoresistant and are associated with a poor prognosis. The tumor arises from the ductal epithelium of the biliary tree within the liver (intrahepatic), extrahepatic ducts (extrahepatic) or gallbladder1. The mechanisms of cholangiocarcinogenesis are complex and involve multiple molecular signaling pathways and inflammatory cytokines that contribute to tumor growth, chemoresistance and cachexia in biliary cancer2,3. The current standard regimen for untreated advanced biliary cancer is the combination of cytotoxic chemotherapy with gemcitabine and cisplatin, but the disease is nearly always fatal, with a median survival that remains less than one year4. In addition, trials for second-line therapy in refractory biliary cancer have been disappointing, highlighting the urgent need to develop new and effective therapies5,6,7.
The PI3k/Akt pathway is downstream of the common growth factor receptor tyrosine kinases (RTKs), including EGFR, HER2 and IGFR and is a likely driver of tumor progression in most carcinomas8,9,10. Akt, also known as protein kinase B, is activated in a substantial proportion of human solid tumors (breast, endometrial, ovarian, prostate, pancreatic, gastric and non-small cell lung cancer). Upregulation of Akt can be caused by direct amplification and mutation of Akt or by overexpression of TKR, PI3K and RAS, and/or by inactivation of the tumor suppressor, PTEN11,12,13. Because of its key function in cell survival, Akt plays a pivotal role in rendering tumor cells insensitive or resistant to chemotherapy or targeted agents, making it an increasing area of interest in development of targeted therapies.
Pre-clinical data has shown activated Akt overexpression in biliary cancers and has demonstrated growth inhibition with Akt dephosphorylation11,14. Treatment of cholangiocarcinoma cell lines with PI3K inhibitor (LY294002) or the MEK 1/2 (UO126) attenuated the effect of CXCL12-induced cholangiocarcinoma cell invasion. These findings indicate that signaling pathways (MEK 1/2 and Akt) are essential for CXCL12-induced cholangiocarcinoma proliferation and cell invasion, implying a potential role for inhibition of Akt and or MEK signaling cascades in the treatment of biliary cancers.
MK-2206 is an oral selective allosteric inhibitor of Akt that targets all three isoforms of human Akt (Akt-1, Akt-2 and Akt-3) with 50% inhibitory concentration (IC50) values of 8, 12 and 65 nM, respectively. In a phase I study of solid tumors, MK-2206 demonstrated evidence of target modulation and anti-proliferative activity as a single agent and in combination with other agents15. Previous studies have shown that oxidation and/or glucuronidation are the primary elimination pathways of MK-2206. While oxidation is primarily mediated by CYP3A4, it is unknown which UGT enzyme isoforms are responsible for glucuronidation of MK-2206. Interestingly, MK-2206 is not a significant inhibitor or inducer of major CYP enzymes (IC50 > 35 μM for CYP3A4, 2C9 and 2D6 inhibition and has insignificant effect on CYP3A mRNA and activity at 0.1 to 10 μM)16, therefore, it is anticipated that MK-2206 does not perpetuate significant drug-drug interactions at the clinical dose levels. This premise is supported by a recent phase 1 study demonstrating that the combination of MK-2206 with standard chemotherapy agents, such as carboplatin/paclitaxel, docetaxel, or erlotinib, does not significantly influence the pharmacokinetic properties and potency of MK-2206 in solid tumors16.
Considering these findings, we hypothesized that MK-2206 would be active in patients with advanced, refractory BC as a single-agent or in combination with other standard cytotoxic agents. We also hypothesized that beneficial clinical effects of MK-2206 would correlate with the presence of activation of the PI3K/Akt pathway. We conducted and report here a phase II study of single-agent MK-2206 in BC to evaluate its efficacy and tolerability at a dose of 200 mg given weekly.
Patients and Methods
The protocol of this study were reviewed and approved by the Ohio State University, Georgetown University and Case Western University Institutional Review Board. The methods were carried out in accordance with the approved guidelines and regulations. Eligible patients were required to have histologically confirmed biliary tract carcinoma that was surgically unresectable. All patients provided written informed consent before the initiation of the study. All patients were required to have either fresh or paraffin-embedded tissue from tumor blocks less than a year prior to enrolling onto the study. Patients had to have measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST)17, one prior therapy for metastatic disease and no prior exposure to Akt inhibitors. Patients with prior cryotherapy, radiofrequency ablation, ethanol injection, transarterial chemoembolization (TACE) or photodynamic therapy were included provided that greater than 6 weeks had elapsed and indicator lesion(s) were outside the area of prior treatment. Patients who received prior radiation therapy with or without the use of fluoropyrimidine as a radiosensitizer in the adjuvant setting were allowed if greater than 12 weeks elapsed since therapy. Additional criteria included age ≥18 years, life expectancy ≥12 weeks, Eastern Cooperative Oncology Group performance status ≤2 and the ability to take and absorb oral medications. Patients were required to have normal organ function, including total bilirubin ≤1.5 times the upper limit of normal and AST/ALT ≤2.5 times the upper limit of normal. Selected exclusion criteria included prior treatment with Akt inhibitors; brain metastases; prior recipient of other investigational agents; diabetes mellitus; history of cardiac disorders including arrhythmias or QTc >450 milliseconds for males and >470 milliseconds for females; pregnant women; and HIV infection.
Study Design
This was a National Cancer Institute (NCI)/Cancer Therapy Evaluation Program (CTEP)-sponsored phase II, open label, multicenter trial led by The Ohio State University with the participation of Georgetown University and Case Western University (ClinicalTrials.gov NCT01425879, date of registration 08/27/2011). MK-2206 was provided by NCI/CTEP. The primary objective of this study was to determine the overall response rate (complete response and partial response) as defined by RECIST17. Tumor tissue samples were required from all patients prior to enrollment. Secondary objectives included overall survival (OS), progression-free survival (PFS) and the presence of any mutations in the pI3k/AKT pathway and the evaluation of toxicities related to MK-2206.
MK-2206 Administration and Dose Modifications
The starting dose and schedule for MK-2206 was 200 mg given weekly in 28-day cycles without interruption. Treatment was administered on an outpatient basis. Two levels of dose reductions were planned (135 mg weekly and 90 mg weekly) with patients taken off the study for additional dose reductions.
Assessment of Response and Toxicity
Radiologic assessment was done by computed tomography every 8 weeks and responses were measured according to RECIST 1.117. Toxicities were defined by the NCI-Common Terminology Criteria of Adverse Events version 4.0.
Correlative Studies
Pharmacokinetics
Plasma samples were collected from all patients at Week 1 of Cycle 1 immediately prior to dose administration and at 30 min, 1, 2, 3, 4, 6, 8, 24, 48 and 96 hours after dose administration. The 24, 48 and 96-hour samples were collected on the mornings of days 2, 3 and 5 within 4 hours of the target time (e.g. 24 ± 4, 48 ± 4 and 96 ± 4 hours). Plasma samples were also collected immediately prior to the oral intake of MK-2206 tablet(s) on days 8, 15 and 29 (i.e. Cycle 2, Day 1) so that trough pre-dose levels of MK-2206 could be determined in the following studies. The plasma levels of MK-2206 were evaluated using quantitative liquid chromatography/tandem mass spectrometry (LC-MS/MS) at Merck & Co. Non-compartmental PK parameters were determined for MK-2206 using Phoenix WinNonlin (v6.3, Pharsight, Mountain View, CA).
Pharmacogenetics
Formalin-fixed/paraffin-embedded (FFPE) or fresh frozen tissue samples were obtained from all patients prior to enrollment. A biopsy was only required if there was insufficient material for analysis or if it was greater than 1 year from the time of biopsy to enrollment. Genomic DNA was extracted from frozen or FFPE tissue (20 micron slides) using FFPE DNA Purification Kit (Qiagen) according to the manufacturer’s instructions. For each DNA sample, the following four SNPs were analyzed using qPCR-based or PCR/RFLP-based assays as previously described: CYP3A4 *22 (C>T, rs35599367), CYP3A5*3 (A>G, rs776746), ABCB1 C1236T (rs1128503), ABCB1 G2677T/A (rs2032582) and ABCB1 C3435T (rs1045642)18,19,20,21. These pharmacogenetic endpoints were correlated to the pharmacokinetic parameters and response status. FFPE tissue samples for genomic DNA extraction were available for six of the eight patients on study.
Although patients were required to have tissue samples available prior to enrollment on the study, genomic DNA extraction was not feasible for two of the eight patients on study due to insufficient quantities of tissue. Due to the early termination of the trial from a loss in the funding mechanism for the study, additional correlative studies to assess pathway activation and tumor genetics were not conducted.
Statistical Methods
This study was designed using Simon’s two-stage approach; the true overall response rate was set at 5% and 20% under null and alternative hypotheses, respectively. With type-I and type-II error rates both at 10%, a total of 35 patients were needed. If 1 or more responses were seen from the first 13 patients, the study would proceed to the second stage. If four or more responses were observed in the whole cohort, the agent would be considered promising. Pharmacokinetics and pharmacogenetic correlative components were primarily descriptive and exploratory, with findings from each of these to guide future clinical use of MK-2206 by potentially identifying individual patient characteristics that may influence outcomes from therapy.
Results
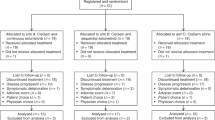
Eight patients were enrolled between September 2012 and December 2013 prior to early termination of the trial. Patients were followed until disease progression or death. The trial was ended by the sponsor prior to the interim analysis due to a loss in funding support from Merck & Co., Inc. The probability that the trial will stop early for futility at the end of stage I (interim analysis) was computed by estimating the conditional probability that there will be no response out of the first 13 patients given that the first 8 patients had no response. If the true overall response rate is 5%, the probability of stopping early for futility at the end of stage 1 is 77.4% when no response was observed in the first eight patients. If the true overall response rate is 20%, the probability of stopping early for futility at the end of stage 1 is 32.8% when no response was observed in the first 8 patients.
Patient characteristics are listed in Table 1. Of the 8 patients enrolled, 8 were evaluable for response and toxicity. Of the patients enrolled into the trial, six (75%) patients had intrahepatic cholangiocarcinoma while 2 (25%) patients had extrahepatic cholangiocarcinoma. Seven patients (88%) had metastatic disease and all the patients enrolled into the trial were exposed to prior chemotherapy.
Treatment Toxicity
The most common toxicities (Table 2) included lymphopenia (75%), rash (63%), fatigue (50%), fever (50%), vomiting (50%) and diarrhea (50%). Toxicities were mostly grade 1 or 2 and were reversible. Only one patient suffered a grade 4 toxicity (hyponatremia). This was felt to be unrelated to MK-2206. All toxicities were manageable and reversible. All 3 patients who experienced grade 3 rash required dose reductions.
Treatment Efficacy
A median of 2 cycles was administered per patient (range, 1 to 2). No patients had an objective response. The best response for all patients accrued in the study was stable disease (SD) lasting more than 12 weeks that was observed in two patients (Table 3). Median PFS was 1.7 months and median OS was 3.5 months.
Pharmacokinetics
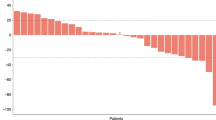
After oral administration of the first dose of MK-2206 (200 mg weekly), plasma samples from eight patients were collected and the concentrations of MK-2206 were determined. Plasma concentration-time curves of these patients are shown in Fig. 1. The median time to peak observed plasma concentrations (Tmax) was 6 hrs (range 4 to 8 hrs). After reaching maximum observed concentration (Cmax), MK-2206 plasma concentrations declined in a biphasic manner with a median terminal elimination half-life of 58 hrs (range 34 to 88 hrs). Table 4 summarizes the PK parameters of MK-2206 from non-compartmental analyses after the first dose. Of note, the Cmax and AUC for subject 1004 is significantly lower compared to the other seven subjects (1004 has approximately 6% to 25% the Cmax and AUC compared to others), suggesting a significantly higher clearance or lower absorption in this patient relative to others. The mechanisms underlying the unique PK properties of Subject 1004 remain unclear. While several potential explanations exist for the difference, no evidence of sampling errors could be identified, nor were co-medications deemed to be relevant for drug-drug interactions. One notable characteristic of subject 1004 is that this patient is of East Asian decent, while the other 7 patients were Caucasian. Differences in metabolism between East Asians and Caucasians are common, especially in the activity of several phase I enzymes including CYP2D6 and the CYP2C subfamily22.

MK-2206 plasma concentration profiles following the oral administration of the first dose for each patient.
Pharmacokinetic profile for MK-2206 is for each subject after receiving their first dose of MK-2206.
We also measured the trough pre-dose levels of MK-2206 in these patients at days 8 and 15. The average plasma concentrations of MK-2206 in these patients at days 8 and 15 were 14.7 +/− 10.6 ng/mL and 14.4 +/− 6.9 ng/ML, respectively. This data suggests there is no significant accumulation of MK-2206 in patients with the current regimen.
Overall, the PK properties of MK-2206 in these patients (excluding subject 1004) were comparable to those previously reported (Supplementary Table A)23. Additionally, the 48-hr plasma concentrations of MK-2206 in 6 of these 8 patients were higher than 22.7 ng/mL (i.e. 56.8 nM), for which this or higher plasma concentrations were reported to be effective in patients with breast cancer24. Plasma concentrations measured at 48 hours (hrs) for the other two patients were 18.5 ng/mL and 5.06 ng/mL (subject 1004). The relevance of this threshold plasma concentration could not be evaluated given the small number of patients treated on this trial.
Pharmacogenetics
While not a significant inhibitor or inducer of major CYP enzyme activity, MK-2206 is converted to oxidative metabolites in human microsomes via CYP3A4 and is a substrate of P-glycoprotein. Hence, we determined genotypes of selected SNPs in CYP3A4, CYP3A5 and ABCB1, including rs35599367 (CYP3A4), rs776746 (CYP3A5), rs1128503, rs2032582 and rs1045642 (ABCB1). The results are listed in Table 5. While all six tested patients were homozygous (GG) for CYP3A5 rs776746, Subject 1005 was heterozygous for CYP3A4 rs35599367 (CT). Moreover, Subjects 1001 and 1003 were heterozygous for all three ABCB1 SNPs (rs1128503, rs2032582 and rs1045642), whereas the genotypes of these three SNPs in Subject 1002 were homozygous (TT). However, there is no apparent association between the PK properties and the genotypes of these five SNPs in this small patient population.
Discussion
Biliary cancer remains a challenging cancer with a universally poor prognosis. The rationale for Akt inhibition was based on the demonstration of a potential role for Akt/PI3k signaling pathways in the carcinogenesis of biliary cancer. Findings from previous pre-clinical studies have shown Akt and MAPK overexpression in biliary cancer cell lines, suggesting Akt inhibition as a potential role for treatment25.
While the study was inconclusive due to its premature termination, our preliminary findings suggest that the oral Akt inhibitor, MK-2206, did not demonstrate meaningful clinical activity as a single-agent in patients with refractory biliary cancer at the doses administered. While no patients had an objective response, two patients experienced prolonged SD (>12 weeks) prior to early closure of the trial and before the interim analysis. Although none of the patients experienced an objective response, the premature termination of the trial did not allow us to fully assess the true efficacy and potential benefit of MK-2206 in biliary cancer. PFS could be a more desirable endpoint for studies incorporating single agent biologics. However, in the absence of randomization, measuring objective response rate may be more meaningful, especially given recent data with other targeted agents in advanced biliary cancers demonstrating objective responses in this disease population26,27. Taking into account the statistical design of the study, the probability of observing at least one response in the next five patients in the first stage of the study, which would allow the study to proceed to the second-stage was 22.6% to 67.2% when the true response rate ranges between 5% and 20%.
The plasma pharmacokinetics of weekly MK-2206 at a 200 mg dose in patients with biliary cancer is similar to that reported in patients with other solid tumors, including breast cancer and malignant glioma15,23,24. For example, the average Cmax value achieved after the first dose in biliary cancer patients (excluding subject 1004) was 114 (+/− 41.2 ng/ mL), whereas the Cmax in a group of children with refractory malignancies was 171 (+/− 89.3 ng/mL). Moreover, there is no substantial difference in BSA-adjusted CL/F between biliary cancer patients (16.5 +/− 7.2 L/hr/m2) and children with refractory malignancies (18.0 +/− 18.8 L/hr/m2). The limited pharmacogenetic data available for CYP3A4, CYP3A5 and ABCB1 did not reveal any major factors associated with drug disposition. In summary, the plasma exposures in these biliary cancer patients were similar to those reported in other disease populations where more clinical activity was observed. Therefore, the lack of observed efficacy achieved in these 8 patients was not due to differences in whole-body disposition of MK-2206. A high incidence of grade 3 rash (38%) was observed in patients who received MK-2206. Despite plasma pharmacokinetics being similar in these patients to previous reports, it is unclear if plasma drug levels are representative of tumor site concentrations. Drug exposure at the tumor site could be significantly lower due to poor access to biliary tumors or by metabolic/catabolic influences acting locally within the hepato-biliary region surrounding the tumor. Further understanding of this will be important for further development of MK-2206 combination therapy in this disease population.
The absence of response may be related to the lack of an improved selection of patients. There is a noticeable absence of potential biomarkers for MK-2206 that could have helped with this process. It is possible that a select subset of patients with phosphorylated Akt (pAkt) overexpression may benefit from MK-2206 and previous studies demonstrated a substantial proportion of patients with advanced biliary cancer exhibit pAkt overexpression26. The clinical efficacy of Akt inhibition in advanced biliary cancers with pAkt overexpression may merit furhter investigation in future studies. Additionally, the lack of response to MK-2206 may be due to co-activation of Akt and MAPK signaling pathways within the tumors. Previous studies indicated that the co-activation and cross talk between the Akt/PI3k and MAPK pathways constitute a potential mechanism of resistance to Akt monotherapy28,29,30. High levels of MEK can lead to resistance to Akt inhibition, thus, even with adequate Akt inhibition, cells with MEK or ERK overexpression may continue to influence several of these downstream proteins and thus prevent apoptosis and promote cellular proliferation26. In Akt resistant cells, MEK inhibition restores susceptibility to Akt inhibition, suggesting negative modulation of the MAPK pathway may be a key to overcoming resistance to Akt inhibitors29. With previous studies showing promising single-agent activity with MEK inhibition in biliary cancer26, future studies combining Akt and MEK inhibitors may be warranted in patients with biliary cancers. Lastly, Akt overexpression has been associated with resistance of biliary cancer cells to chemotherapy and radiotherapy, suggesting a potential role for Akt inhibitors in combination with cytotoxic treatments16. Furthermore, in vitro studies have demonstrated a decreased sensitivity to allosteric Akt inhibitors in cell lines with somatic mutations in Akt, suggesting that appropriate screening and selection of patients without Akt mutations may have an improved clinical benefit from MK-220631.
In conclusion, MK-2206 as a single-agent in biliary cancer was generally tolerable, although no objective clinical activity was observed in this small sample of patients. This is the first report of the use of Akt-targeted therapies in biliary cancers. Monotherapy targeting Akt inhibition may have clinical relevance for a subset of patients with biliary cancer, however further studies assessing the utility of MK-2206 in combination with other molecular targeted agents or in conjunction with conventional cytotoxic chemotherapy are warranted. Correlative investigation confirming the potential benefit in responders and identifying new positive predictors of clinical response are needed to better understand the mechanisms of activity and to select patients that will derive benefit from treatment.
Additional Information
How to cite this article: Ahn, D. H. et al. Results of an abbreviated phase-II study with the Akt Inhibitor MK-2206 in Patients with Advanced Biliary Cancer. Sci. Rep. 5, 12122; doi: 10.1038/srep12122 (2015).
References
Khan, S. A., Thomas, H. C., Davidson, B. R. & Taylor-Robinson, S. D. Cholangiocarcinoma. Lancet 366, 1303–1314 (2005).
Goydos, J. S., Brumfield, A. M., Frezza, E., Booth, A., Lotze, M. T. & Carty, S. E. Marked elevation of serum interleukin-6 in patients with cholangiocarcinoma: validation of utility as a clinical marker. Ann Surg 227, 398–404 (1998).
Park, J., Tadlock, L., Gores, G. J. & Patel, T. Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology 30, 1128–1133 (1999).
Valle, J. et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 362, 1273–1281 (2010).
Kruth, J. et al. Efficacy and safety of capecitabine in combination with docetaxel and mitomycin C in patients with pre-treated pancreatic, gallbladder and bile duct carcinoma. J Cancer Res Clin Oncol 136, 1845–1851 (2010).
Lee, S. et al. Second-line treatment with a combination of continuous 5-fluorouracil, doxorubicin and mitomycin-C (conti-FAM) in gemcitabine-pretreated pancreatic and biliary tract cancer. Am J Clin Oncol 32, 348–352 (2009).
Oh, S. Y. et al. Phase II study of second line gemcitabine single chemotherapy for biliary tract cancer patients with 5-fluorouracil refractoriness. Invest New Drugs 29, 1066–1072 (2011).
Altomare, D. A. & Testa, J. R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 24, 7455–7464 (2005).
Hennessy, B. T., Smith, D. L., Ram, P. T., Lu, Y. & Mills, G. B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 4, 988–1004 (2005).
Steelman, L. S. et al. Akt as a therapeutic target in cancer. Expert Opin Ther Targets 12, 1139–1165 (2008).
Deshpande, V. et al. Mutational profiling reveals PIK3CA mutations in gallbladder carcinoma. BMC Cancer 11, 60 (2011).
Nagata, Y. et al. PTEN activation contributes to tumor inhibition by trastuzumab and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6, 117–127 (2004).
Oki, E. et al. Akt phosphorylation associates with LOH of PTEN and leads to chemoresistance for gastric cancer. Int J Cancer 117, 376–380 (2005).
Leal, P. et al. AKT/mTOR substrate P70S6K is frequently phosphorylated in gallbladder cancer tissue and cell lines. Onco Targets Ther 6, 1373–1384 (2013).
Yap, T. A. et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol 29, 4688–4695 (2011).
Molife, L. R. et al. Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J Hematol Oncol 7, 1 (2014).
Therasse, P. et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92, 205–216 (2000).
Dong, L., Mao, M., Luo, R., Tong, Y. & Yu, D. Common ABCB1 polymorphisms associated with susceptibility to infantile spasms in the Chinese Han population. Genet Mol Res 10, 2569–2577 (2011).
Wang, D., Guo, Y., Wrighton, S. A., Cooke, G. E. & Sadee, W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J 11, 274–286 (2011).
Min, D. I., Ellingrod, V. L., Marsh, S. & McLeod, H. CYP3A5 polymorphism and the ethnic differences in cyclosporine pharmacokinetics in healthy subjects. Ther Drug Monit 26, 524–528 (2004).
Owen, A., Goldring, C., Morgan, P., Chadwick, D., Park, B. K. & Pirmohamed, M. Relationship between the C3435T and G2677T(A) polymorphisms in the ABCB1 gene and P-glycoprotein expression in human liver. Br J Clin Pharmacol 59, 365–370 (2005).
Kim, K., Johnson, J. A. & Derendorf, H. Differences in drug pharmacokinetics between East Asians and Caucasians and the role of genetic polymorphisms. J Clin Pharmacol 44, 1083–1105 (2004).
Fouladi, M. et al. A phase I trial of MK-2206 in children with refractory malignancies: a Children’s Oncology Group study. Pediatr Blood Cancer 61, 1246–1251 (2014).
Hudis, C. et al. A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res 15, R110 (2013).
Javle, M. M. et al. Akt expression may predict favorable prognosis in cholangiocarcinoma. J Gastroenterol Hepatol 21, 1744–1751 (2006).
Bekaii-Saab, T. et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol 29, 2357–2363 (2011).
Bendell, J. et al. A phase I dose-escalation study of MEK inhibitor MEK162 (ARRY-438162) in patients with advanced solid tumors. Mol Cancer Ther 10 (11 Suppl): Abstract nr B243 (2011).
Leelawat, K., Udomchaiprasertkul, W., Narong, S. & Leelawat, S. Induction of MKP-1 prevents the cytotoxic effects of PI3K inhibition in hilar cholangiocarcinoma cells. J Cancer Res Clin Oncol 136, 1537–1544 (2010).
Meng, J. et al. High level of AKT activity is associated with resistance to MEK inhibitor AZD6244 (ARRY-142886). Cancer Biol Ther 8, 2073–2080 (2009).
Meng, J. et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLoS One 5, e14124 (2010).
Parikh, C. et al. Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc Natl Acad Sci USA 109, 19368–19373 (2012).
Acknowledgements
This study was supported in part by NCI N01 HHSN261201100070C to MAV.
Author information
Authors and Affiliations
Contributions
D.A. reviewed the data, wrote the manuscript and prepared tables 1, 2, 3. J.L. performed the pharmacokinetic and pharmacogenomic experiments, assisted in writing the manuscript and prepared tables 4, 5 and supplementary table A. L.W. performed all the statistical calculations and manuscript preparation. A.D. assisted in study design and manuscript preparation. J.M. assisted in study design, patient accrual and manuscript preparation. L.S. assisted in study design and manuscript preparation. M.P. assisted in supervising all pharmacokinetic and pharmacogenomic experiments and assisted in manuscript preparation. M.V. assisted in study design and manuscript preparation. T.B. assisted in the study concept and design and manuscript preparation and review.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ahn, D., Li, J., Wei, L. et al. Results of an abbreviated phase-II study with the Akt Inhibitor MK-2206 in Patients with Advanced Biliary Cancer. Sci Rep 5, 12122 (2015). https://doi.org/10.1038/srep12122
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep12122
This article is cited by
-
AKT1-CREB stimulation of PDGFRα expression is pivotal for PTEN deficient tumor development
Cell Death & Disease (2021)
-
Targeting cancer via ribosome biogenesis: the cachexia perspective
Cellular and Molecular Life Sciences (2021)
-
SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals
Nature Communications (2021)
-
Overview of current targeted therapy in gallbladder cancer
Signal Transduction and Targeted Therapy (2020)
-
AKT1 restricts the invasive capacity of head and neck carcinoma cells harboring a constitutively active PI3 kinase activity
BMC Cancer (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
