
Abstract
Histone deacetylases (HDACs) and histone acetyl transferases (HATs) are two counteracting enzyme families whose enzymatic activity controls the acetylation state of protein lysine residues, notably those contained in the N-terminal extensions of the core histones. Acetylation of histones affects gene expression through its influence on chromatin conformation. In addition, several non-histone proteins are regulated in their stability or biological function by the acetylation state of specific lysine residues. HDACs intervene in a multitude of biological processes and are part of a multiprotein family in which each member has its specialized functions. In addition, HDAC activity is tightly controlled through targeted recruitment, protein-protein interactions and post-translational modifications. Control of cell cycle progression, cell survival and differentiation are among the most important roles of these enzymes. Since these processes are affected by malignant transformation, HDAC inhibitors were developed as antineoplastic drugs and are showing encouraging efficacy in cancer patients.
Similar content being viewed by others
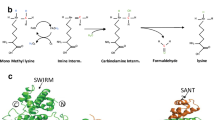

Introduction
Core histones have long N-terminal extensions that have been known for decades to undergo extensive post-translational modifications such as acetylation, methylation and phosphorylation, as well as ubiquitination, sumoylation and ADP-ribosylation 1, 2, 3, 4, 5. Interestingly, Vincent Alfrey et al. 6 proposed more than 40 years ago that modifications such as acetylation could have functional roles by modulating transcription efficiency, but it took about 30 years for the scientific community to follow-up on these observations and to establish a fundamental role for chromatin and its post-translational modification states in the epigenetic regulation of many processes impinging on DNA, such as transcription. The present review will focus on the histone deacetylase (HDAC) family of chromatin-modifying enzymes. Aberrant regulation of gene expression is at the basis of many human diseases and notably of many forms of cancer. Yet, attempts to pharmacologically target transcription complexes have been frustrated by the difficulty to disrupt protein-protein interactions with small molecules. The discovery that inhibition of chromatin-modifying enzymes allows to modulate transcription in eukaryotic cells paved the way for the development of novel pharmacologic agents. Recently, the HDAC inhibitor (HDACi) vorinostat was approved by the US FDA for the treatment of the cutaneous manifestations in patients with advanced, refractory cutaneous T-cell lymphoma demonstrating the therapeutic potential of HDAC inhibition. Our understanding of the biology of individual HDAC family members is rapidly increasing, showing that there is a considerable division of labor between the different HDAC subtypes that often have unique functions and that are promising targets for therapeutic intervention. The aim of this review is to highlight the diversity within the HDAC family, the mechanisms by which HDAC activity is regulated and the emerging insights into the structural biology of deacetylases. This knowledge will be fundamental for fully exploiting the therapeutic applications of HDAC inhibition. The last part of the review will therefore specifically focus on our ability to influence HDAC activity in vivo with small molecule inhibitors and the exciting possibility to modulate transcription (as well as non-transcriptional processes) with HDACi's to treat human disease.
The HDAC family
Acetylation occurs at the e-amino groups of lysine residues present within the N-terminal extensions of the core histones. In all documented cases, the acetylation status of target lysines is tightly controlled by the balance of two counteracting enzymatic activities: those of histone acetyl transferases (HATs) and HDACs 7, 8. Histone deacetylation represses transcription by different mechanisms. On the one hand, this process increases the charge density on the N-termini of the core histones thereby strengthening histone tail-DNA interactions and blocking access of the transcriptional machinery to the DNA template. In addition, histone modifications are specifically recognized by chromatin-interacting proteins. Bromodomains are found in several chromatin-associated proteins, such as some HAT family members, and are used for the recognition of acetylated lysine residues 9. Deacetylation of a given lysine residue may allow its further modification by histone methyl-transferases 10. Methylated lysine residues can in turn be recognized by proteins harboring chromodomains, Tudor domains or WD40 repeats 10, 11. HP1 is a chromodomain protein that specifically recognizes trimethylated lysine 9 of histone H3, thereby favoring the assembly of heterochromatin 12, 13, 14. Further heterochromatinization occurs by recruitment of DNA methyl transferases that will specifically methylate cytosine in the 5 position 15, 16. Together with these post-translational modifications, there is a progressive decrease in the accessibility of DNA to the transcription machinery and hence an increasing transcriptional silencing (Figure 1).

Chromatin modifications that induce progressively increasing transcriptional silencing. Note that modification by acetylation is fully reversible. Only recently enzymes that are able to remove methyl groups from histones (histone demethylases) have been identified. The occurrence of DNA demethylation in adult organisms is still debated.
In addition to histones, an increasing number of non-histone proteins are shown to undergo lysine acetylation: many of these proteins are transcription factors, but heat-shock proteins and structural proteins were also shown to be acetylated. Generally, acetylation has profound influences either on metabolic stability or on the biological function of the modified protein, and can be regarded akin to phosphorylation in terms of being a widely used post-translational mechanism of controlling protein function.
HATs utilize acetyl-CoA as a cofactor in the acetylation reaction. About 30 different HATs were identified that are grouped into five different families 17. In the eukaryotic cell, HATs may reside either in the nucleus or in the cytosol. HDACs catalyze the inverse reaction by removing the acetyl group from the acetyl lysine residue. Higher organisms have evolved a considerable complexity in the HDAC family that was divided into three classes according to phylogenetic analyses and sequence homologies with the yeast proteins Rpd3, Hos1 and Hos2 (class I), HDA1 and Hos3 (class II) and the sirtuins (class III) 18, 19, 20. Class I and class II proteins are evolutionarily related and share a common enzymatic mechanism, the Zn-catalyzed hydrolysis of the acetyl-lysine amide bond. Higher eukaryotes also express an additional Zn-dependent HDAC (HDAC11 in mammals) that is phylogenetically different from both class I and class II enzymes and is therefore regarded as a separate class (class IV). Class III proteins are evolutionarily unrelated to class I, II or IV and catalyze the transfer of the acetyl group onto the sugar moiety of NAD 20. This latter reaction is strictly dependent on NAD as a cofactor, and class III HDACs are thought to link transcriptional regulation to energy metabolism that regulates NAD levels. In addition, class III enzymes have been suggested to also play a role in life span regulation by caloric restriction. This review will not deal in detail with class III enzymes but primarily focus on the Zn-dependent HDACs.
Yeast and Drosophila HDACs: division of labor
Genome-wide roles of Saccharomyces cerevisiae HDACs were studied by microarray profiling of yeast HDAC knockout strains 21. In those studies a division of labor among the different classes emerged, with HDA1 primarily regulating genes involved in carbon metabolism, Rpd3 controlling primarily cell cycle genes and Sir2 emerging as a player in the regulation of amino-acid biosynthesis. Transcriptional profiles of Sin3 mutants showed striking similarity to Rpd3 mutants, in line with the notion that transcriptional repression by Sin3 involves the recruitment of Rpd3. This same paradigm was also found by others in higher organisms (see below).
The use of acetylation-site-specific antibodies for chromatin immunoprecipitation (ChIP) in yeast HDAC KO strains to probe intergenic region microarrays (ChIP on CHIP) has generated a very detailed genome-wide HDAC activity map of S. cerevisiae 22. The functional division of labor highlighted by gene expression profiling was further substantiated by these studies. Thus, HDA1 and Rpd3 deacetylate distinct intergenic regions, with HDA1 being preferentially recruited to subtelomeric (HAST) regions containing a high proportion of genes induced by nutritional stress. From these studies, clues to substrate specificity also emerged, providing evidence for the preference of Rpd3 for H4K12ac, H4K5ac and H4K18ac over H4K16ac. Interestingly, the yeast HOS2 deacetylase was shown to be preferentially recruited to highly transcribed genes indicating that, in contrast to other HDACs, HOS2 is required for gene activations 23. More recently, ChIP on CHIP analysis was performed by using a novel crosslinking technique in conjunction with anti-Rpd3 antibodies, leading to an Rpd3 binding map in S. cerevisiae 24. In one of these studies, a similar distribution of Rpd3 and Sin3 was noted 25, consistent with the gene expression profiling data.
We used RNAi in combination with microarray analysis in Drosophila S2 cells to define the role of each of the five individual Zn-dependent HDACs known in this organism 26. Unexpectedly, deregulated transcription was observed only upon silencing of Drosophila class I HDACs 1 and 3, but not upon RNAi of other family members. This suggests that many HDACs may either have primarily non-transcriptional roles or participate in transcription regulation only as a result of certain specific environmental stimuli. Indeed, many class II enzymes seem to follow the latter paradigm (see below), which also appears to be evolutionarily conserved, taking into consideration the preferred recruitment of HDA1 into HAST regions of the yeast genome. The major “housekeeping” deacetylase in Drosophila seems to be HDAC1. This protein is involved in the regulation of very diverse sets of genes, most notably those involved in cell proliferation and mitochondrial energy metabolism. As observed in the yeast system, in Drosophila too there is a noticeable functional overlap between HDAC1 and the transcriptional co-repressor Sin3 26, 27, suggesting that most of its repressor function is exerted through deacetylase recruitment, a feature that has been evolutionarily conserved.
No systematic “omic” approaches to unveil individual roles of HDACs have been reported so far in mammalian cells, possibly because of the greater complexity of the family that has 18 known members in these organisms. Figure 2 shows a comparison between Zn-dependent HDACs in Drosophila and in mammals. Still, many aspects of the biology of individual subtypes have emerged, highlighting how each subtype has very specialized and mostly non-redundant functions.

Zn-dependent histone deacetylases in Drosophila and mammals and their domain structure. Catalytic domains are shown in blue. Note that HDAC6 and DHDAC2 have two catalytic domains, whereas the second deacetylase domain (brown) is dysfunctional in HDAC10. C-terminal extensions are found in HDACs 1, 2 and 3 (class I) and in their Drosophila homologs (light brown and yellow). Also the N-terminal extensions found in class IIa HDACS (red) are conserved in DHDAC4, whereas the C-terminal extensions found in HDACs 4, 5 and 7 are not.
Mammalian deacetylases
Among mammalian class I deacetylases (subtypes 1, 2, 3 and 8), HDACs 1 and 2 are most closely related (82% sequence identity) and found in the ubiquitously expressed mSin3A, NURD/Mi2/NRD and CoREST corepressor complexes 28. HDAC1 KO mice die during embryonic development and their ES cells show decreased growth and enhanced expression of the cyclin-dependent kinase inhibitors p21 and p27 29. Compensatory upregulation of the expression of other class I HDACs in HDAC1 KO cells is apparently insufficient to counterbalance the loss of this subtype, suggesting both its functional uniqueness and the existence of regulatory cross-talk 30. Both HDACs 1 and 2 (in addition to HDAC3) seem to be involved in the regulation of key cell cycle genes such as p21 (31, 32, 33 and MP et al., manuscript in preparation). Still, HDAC2 also has some specialized functions, e.g. in counterbalancing proapoptotic signals that ensue from aberrant Wnt pathway activation in colon cancer 34. HDACs 1 and 2 are not exclusively HDACs but may deacetylate non-histone substrates as well 35.
HDAC3 associates to and is activated by SMRT and NCoR co-repressors that play an important role in the regulation of gene expression by nuclear hormone receptors 28, which, in their unliganded state, recruit these corepressors and utilize the deacetylase activity to silence transcription. Studies on the retinoic acid receptor suggest that a specific lysine (H4K5) is preferentially deacetylated by HDAC3 recruited to the unliganded receptor 36. Intriguingly, the SMRT/N-CoR corepressors are themselves capable of interacting with histones via an SANT domain and this interaction is strengthened upon deacetylation of H4K5, suggesting the existence of a positive feedback loop in the silencing mechanism. HDAC3 was found to be upregulated in CD34+ CD133+ early hematopoietic progenitors 37 and to play a transcription-independent role in mitosis 38, possibly pointing to functions in cell cycle progression and stem cell self-renewal. In contrast to HDACs 1 and 2 that are exclusively nuclear, HDAC3 may also be found in the cytoplasm and even associated with the plasma membrane, where it can be phosphorylated by Src 39. Also HDAC3 is not an exclusive HDAC but may also deacetylate non-histone substrates, such as the RelA subunit of NF-κB, thereby affecting its stability and DNA-binding properties 40, 41. The last member of class I, HDAC8, was recently found to be expressed in smooth muscle where it is required for muscle contractility 42. This protein has also been linked to cancer since it is recruited by the leukemic INV 16 protein 43; it regulates telomerase activity 44 and siRNAs targeting HDAC8 were shown to have antitumor effects in cell culture 45.
The class IIa HDACs (subtypes 4, 5, 7 and 9) are characterized by tissue-specific expression and stimulus-dependent nucleo-cytoplasmic shuttling 19. They are target of several kinases, and some phosphorylated forms are confined to the cytosol by interaction with 14-3-3 proteins (see below). In the nucleus they associate with transcription factors, notably of the MEF and Runx families, and control differentiation and cellular hypertrophy in muscle and cartilage tissues 46, 47. HDAC4 KO mice have a pronounced chondrocyte hypertrophy and die of aberrant ossification 46. HDAC9 KO mice show cardiac hypertrophy 47 that is further exacerbated in the HDAC9+HDAC5 double KO animals 48. HDAC7 has a specific role in the clonal expansion of T cells by suppressing Nur77-dependent apoptosis 49 and in vascular integrity through suppression of MMP10 50. Class IIb subtypes 6 and 10 have a duplication of their catalytic domains, but the second catalytic domain is thought to be dysfunctional in HDAC10. HDAC6 is the only deacetylase known to act on tubulin 51, 52. Tubulin deacetylation is required for disposal of misfolded proteins in aggresomes 53. HDAC6 also deacetylates Hsp90, pointing to a broader role of this subtype in protein folding 54, 55, 56. Finally, very little is known about HDAC11, which cannot be clearly assigned to either class I or class II HDACs based on sequence motifs 57.
Cellular mechanisms controlling HDAC activity
Mammalian HDACs play global roles in the regulation of gene transcription, cell growth, survival and proliferation, and their aberrant expression or activity lead to cancer development. As expected for vital cellular regulators, the activities of HDACs are tightly controlled through a multitude of mechanisms, among which the recruitment into different co-repressor complexes and the modulation of deacetylase activity by protein-protein interactions or by post-translational modifications are particularly relevant ones.
With the exception of HDAC8, functional HDACs are never found as single monomeric polypeptides, rather they accumulate in high molecular weight multi-protein complexes in which different HDAC subtypes are often associated with specific co-regulators as well as with other chromatin-modifying enzymes 28.
HDAC biological activity can be separated into two distinct, not always interdependent areas, enzymatic activity (the ability to deacetylate histone or other non-histone protein substrates) and functional activity (the ability to regulate transcription and other biological processes). There are two orders of considerations that are worth mentioning to clarify the relevance of this distinction. First, not in all circumstances HDACs' biological functions are strictly dependent on their enzymatic activity. There is ample evidence that class IIa HDACs exert transcriptional repression, thanks to their ability to directly interact and inactivate specific target transcription factors (see 19 and 58 and references therein). In addition, they can recruit a number of distinct corepressors and/or protein-modifying enzymes, which in turn directly switch target transcription factors to their inactive form. Most of these multiple protein-protein interactions occur via class IIa HDACs' N-terminal regulatory domain, while the activity or even the physical presence of the C-terminal catalytic domain is often not strictly required. The existence of the MEF2-interacting transcription repressor (MITR; a naturally occurring HDAC9 splice variant) is paradigmatic, which retains transcriptional repressive functions despite lacking the C-terminal catalytic domain. Furthermore, the enzymatic activity of HDAC4, 5 and 7 was shown to be dependent on the association with the HDAC3/SMRT/N-CoR complex, thus suggesting that class IIa HDACs are not active deacetylases, but operate by recruiting preexisting enzymatically active HDAC3 protein complexes 19. A second important consideration is that, with the exception of mammalian HDAC8, most purified recombinant HDACs are enzymatically inactive 59. Any protein that associates with HDACs, therefore, has the potential to exert an enzymatic co-activating function. The deacetylase activity of HDAC3 strictly requires the interaction with its transcriptional corepressor protein partners SMRT or N-CoR, which contact and activate HDAC3 via a conserved deacetylase-activating domain (DAD) 60, 61. Known HDAC1/2 complexes contain transcriptional corepressors mSin3A 62, MTA (metastasis-associated protein in NuRD complex) 63 and CoREST (corepressor of REST) 64. MTA-2 is an essential component of an enzymatically active recombinant HDAC1/HDAC2 protein complex, while in CoREST the SANT domain is necessary for recruiting HDAC1 activity. SDS3 is a key component of the Sin3 corepressor complex that increases the enzymatic activity of HDAC1 in yeast and mammalian cells 65, 66. Enzymatic activation by specific HDAC-cofactor interactions could therefore represent a general mechanism for spatially restraining HDAC activities to the specific promoter sites where corepressor complexes are targeted by sequence-specific DNA-binding proteins.
While the recruitment into different corepressor complexes and the modulation of deacetylase activity by protein-protein interactions mainly contribute to the functional diversity of different HDAC subtypes, post-translational modifications, such as phosphorylation and sumoylation, add additional complexity to the regulatory networks controlling these enzymes. All mammalian HDACs contain potential phosphorylation sites and the majority of them have been found to be phosphorylated in vitro and in vivo 59. In the case of class IIa HDACs, phosphorylation regulates their subcellular localization and therefore their biological activities 19, 59. The N-terminal regions of these HDACs contain a set of conserved serine residues that control their subcellular localization and confer signal responsiveness to downstream target genes 67, 68. Phosphorylation of these residues creates binding sites for the 14-3-3 chaperone protein, which escorts phospho-HDACs from the nucleus to the cytoplasm, with consequent activation of HDAC target genes. Protein kinase D and various Ca2+/calmodulin-dependent kinases transmit signals triggered by different extracellular stimuli to class IIa HDACs via their regulatory phosphorylation sites in a variety of cell types 69, 70, 71, 72, 73, 74, 75, 76.
CK2 is a key modulator of class I HDAC activity
With the exception of HDAC8, shown to be phosphorylated and thus inactivated by protein kinase A (PKA) 77, casein kinase 2 (CK2) emerged as a key enzyme for all other class I HDAC isotypes 78, 79, 80, 81, 82. CK2 is a multifunctional protein kinase, ubiquitously distributed in both the cytoplasm and the nucleus of eukaryotic cells and most prominently involved in the regulation of cell growth, survival and proliferation 83, 84, 85, and transcription-related chromatin remodeling 86, 87, 88. Its activity has been consistently found elevated in many human cancers and experimental tumors, and its deregulated expression was shown to contribute to tumorigenesis and impart oncogenic potential 85. In this regard, it is relevant that class I HDACs play similar global roles in homeostasis, signal transduction, cell cycle control and cancer development 89, 90. HDAC1 and HDAC2 C-terminal regions contain at least two serine residues at equivalent positions (S421/S422 and S423/S424, respectively) that are phosphorylated by CK2 78, 80. Based on site-directed mutagenesis studies, HDAC3 S424 was more recently found to be phosphorylated in cells and to be a CK2 phosphoacceptor site in vitro 82. Mass spectrometry analysis 91 reinforced this evidence directly identifying S424 as a unique HDAC3 phosphoacceptor site in vivo. This residue resides in a canonical CK2 consensus sequence and is highly conserved among HDAC3 homologs from different species. However, the C-terminal sequence of HDAC3 diverges significantly from the corresponding regions of HDAC1 and HDAC2, and by sequence alignment comparisons S424 has no equivalent in the other HDAC class I members. These studies raise a number of yet to be reconciled contradictory conclusions, mainly concerning the regulatory effects exerted by phosphorylation on the enzymatic activity, transcriptional repression potential and functional protein-protein interactions 78, 79, 80, 81, 91, 92. Interestingly, HDAC1 and HDAC2 phosphoserine mutations, shown to be deleterious for the enzymatic activity, also disrupt interactions with endogenous associated proteins 78, 79, 80 including MTA2 and CoREST, essential members of HDAC1/2 enzymatically proficient complexes 63, 64. Phosphorylation might therefore increase the affinity of HDACs for key interacting proteins, which in turn enhance their enzymatic activity. Phospho-serine mutagenesis approaches, however, include the caveat that the mutations themselves could alter the HDACs conformation inducing an enzymatically less active form and/or decreasing protein-binding affinity. Published evidence has shown that HDAC1 and HDAC2 immunocomplexes contain functional CK2 79, 81. More recently, we have identified active CK2 in the HDAC3 immunocomplex capable of efficiently phosphorylating both β-casein and the associated HDAC3 and of autophosphorylating its own regulatory β subunit 91. In the same work, phosphorylation of HDAC3 did not affect either the deacetylase activity of the protein or its ability to stably interact with N-CoR. Our findings are therefore not in agreement with recently published evidence showing a three- to four-fold reduction of HDAC3 enzymatic activity upon mutation of S424 82. Under our experimental conditions, N-CoR and CK2 co-elute with HDAC3 in sub-stoichiometric amounts and only a fraction of ectopically expressed HDAC3 is phosphorylated on S424. Collectively, these findings support a model in which phosphorylation and association with N-CoR could represent two distinct and mutually exclusive mechanisms regulating HDAC3 enzymatic activity, the latter being dominant under our experimental conditions. Concurrent interactions with both N-CoR and CK2 could be incompatible so that only the cofactor-free HDAC3 fraction would be a potential phosphorylation substrate. Alternatively, since S424 is located at the extreme HDAC3 C-terminus previously found to critically contribute to the interaction with SMRT/N-CoR DAD 60, 93, the stable association with the cofactor might interfere with the accessibility of the CK2 phosphoacceptor site to the kinase. Future studies aimed to map the HDAC3 region responsible for CK2 interaction would help clarifying these points. In a number of circumstances, HDAC3 is capable of establishing direct protein-protein interactions and displays biological activity independent of its recruitment in N-CoR/SMRT complexes, suggesting alternative mechanism(s) through which it can exert its transcriptional co-repression function in normal and tumor cells 94, 95, 96, 97, 98, 99, 100, 101. It would be tempting to speculate that HDAC3 phosphorylation might assume a critical role in activating the enzyme in N-CoR/SMRT-independent contexts.
Non-catalytic functions of class I HDACs
In addition to the pleiotropic biological roles accomplished through their deacetylase activities, in some circumstances both class I and class II HDACs have been recently shown to regulate key cellular processes by interacting with and targeting different enzymes to their specific substrates. For instance, HDAC1 and HDAC6 can influence the phosphorylation-dephosphorylation state of CREB and Akt, respectively, by forming reversible HDAC-PP1 complexes, thus regulating the delivery of PP1 to these substrates 102, 103. In this respect, the interaction of class I HDACs with CK2 could lead to two different regulatory mechanisms, one related with direct phosphorylation of the HDAC within the complex, and the other relying on delivering the protein kinase to specific targets. Interestingly, two of the better-characterized HDAC3-interacting proteins, i.e. SMRT and NF-κB RelA/p65 subunit, are also CK2 substrates 104, 105. Little is known about how CK2 activity is regulated in vivo. The association with key regulators such as HDACs could represent one mechanism for restraining CK2 activity in different cellular compartments and on different protein targets.
The role of phosphatases in the regulation of HDAC activity
While most studies have focused on HDAC phosphorylation, an increasing number of published data suggest the involvement of protein phosphatases being equally relevant 82, 102, 103, 106. Although PP1 was shown to associate specifically with HDAC1, 6 and 10, it is not known whether these HDAC isotypes are direct substrates of this phosphatase. In the case of HDAC3, however, a distinct serine/threonine protein phosphatase, PP4, has been recently identified that functionally interacts with and dephosphorylates HDAC3, thus negatively modulating its deacetylase activity 82. Interestingly, the HDAC3 N-terminal region involved in PP4 interaction was also shown to be essential for the association with the SMRT DAD 60, thus reinforcing the hypothesis that HDAC3 can be subjected to two independent regulatory mechanisms, one mediated by its activating cofactor and the other by phosphorylation/dephosphorylation.
Sumoylation
In addition to phosphorylation, another post-translational modification that has been shown to regulate HDAC activity and function is the conjugation of small ubiquitin-related modifier (SUMO-1) 59. Two independent reports 107, 108 identified HDAC1 as a substrate for SUMO-1 modification in vitro and in vivo at K444 and K476. However, as for HDACs phosphorylation studies, these studies also lead to opposite conclusions regarding the functional relevance of sumoylation on HDAC1 activity. Also some class II HDAC members (HDAC4, HDAC6 and MITR) have been found to be modified by SUMO-1 109. In particular, HDAC4 sumoylation was shown to positively modulate its repressive and deacetylase activities and to be linked to nuclear import regulatory mechanisms. It is worth noting that the reduced repressor activity of sumoylation-deficient HDAC1 and HDAC4 was found to be independent of their ability to associate with known binding proteins including mSin3A and N-CoR. However, several recent studies have determined that sumoylation of some transcriptional regulators, such as p300, Dnmt3a and ELK-1, regulates their association with HDACs and, consequently, their capacity to repress transcription 110, 111. More recently, a number of independent publications have shown that class IIa HDACs, in particular HDAC4, can stimulate MEF2 sumoylation, and thus MEF2-dependent transcriptional repression 113, 114, 115.
Knowledge gained from understanding the many different aspects of HDAC regulation is expected not only to broaden our comprehension of fundamental cellular events but also to be translated into the design of a new generation of mechanism-based therapeutic agents urgently needed for the treatment of major human diseases, such as cancer.
Structure and function of HDACs
The understanding of the mechanism of action and the rational design of HDACi's are greatly facilitated by the availability of structural information on the molecular architecture of HDAC enzymes. The first structural and mechanistic information on HDACs was derived from the crystal structure of an HDAC-like protein (HDLP) with unknown function, from the hyperthermophilic bacterium Aquifex aeolicus 116. The structure revealed a compact domain belonging to the α/β fold with a central eight-stranded parallel β-sheet flanked on each side by four α-helices. Seven additional α-helices are grouped close to the C-terminal end of the β-sheet. Portion of these helices together with loops connecting the secondary structure elements form, along one face of the structure, a narrow apolar channel with a depth of 11 Å at the bottom of which the polar catalytic core containing one zinc ion is found. In the X-ray structures of the HDACi's, Trichostatin A (TSA) and vorinostat (suberoylanilide hydroxamic acid, SAHA) in complex with HDLP 116, the aromatic end of the inhibitors (cap moiety) points away from the protein, while the long carbon chain (spacer) fits into the narrow hydrophobic pocket and ends with the hydroxamic acid establishing polar interactions with protein residues and the zinc at the bottom (zinc-binding, enzyme-inhibiting group). For 5 years, the HDLP crystal structure was the only model available for structure-activity relationship studies of HDACi's. In 2004, two independent groups published X-ray structures of inhibitor-bound human HDAC8 (approximately 30% sequence identity with HDLP; 117, 45). The structure of HDAC8 forms a single compact α/β domain composed of a central eight-stranded parallel β-sheet and eleven α-helices (Figure 3A), similar to HDLP 116 and to the dimanganese enzyme arginase 118. Striking differences between HDAC8 and HDLP are observed for most of the loops emerging from the core of the protein, for the distal helices and for a long extended loop, which connects the last two helices at the C-terminus. This region in HDAC8 is completely different from HDLP, including the loss of two helices (Figure 3A). Overall, these variations give rise to a more accessible active site in HDAC8 with a slightly deeper and larger pocket (Figure 3A).

Crystal structure of inhibitor-bound HDAC8 and comparison with inhibitor-bound HDAC-like enzymes. (A) Left, Ribbon diagram of human HDAC8 monomer in complex with the hydroxamic acid inhibitor, N-hydroxy-4-{methyl [(5-pyridin-2-yl-2-thienyl) sulfonyl] amino} benzamide, compound 1, pdb code 1w22. The protein is shown as a yellow ribbon and the inhibitor is drawn in stick representation (oxygen, nitrogen, sulfur and carbon are colored red, blue, yellow and green, respectively). Zn2+ and K+ ions are indicated and drawn as violet spheres. The HDACi pharmacophore consists of a metal binding moiety at the zinc site, a linker moiety in the channel and a surface recognition moiety on the rim of the channel. Center, Superposition of human HDAC8 (yellow) and HDLP (magenta) in complex with TSA, pdb code 1c3r. Compound 1 and TSA are drawn in stick representation and colored as in (A) left. In the HDLP structure, only the Zn2+ ion is present and indicated as a violet sphere. Right, Superposition of human HDAC8 (yellow) and FB188 HDAH (cyan) in complex with SAHA (116), pdb code 1zz1. Compound 1 and SAHA are drawn in stick representation and colored as in (A) left. In the FB188 HDAH structure, both Zn2+ and K+ ions are present and indicated as violet spheres. (B) Molecular surface representation at the entrance of the active site channel for HDAC8 (yellow), HDLP (magenta) and FB188 HDAH (cyan). Compound 1, TSA and SAHA are drawn in stick representation and colored as in (A). (C) Expanded view showing HDAC8 active site with the bound inhibitor, compound 1. Residues relevant for catalysis and involved in inhibitor binding are indicated and drawn in line representation. The inhibitor is drawn in stick representation. The Zn2+ ion is indicated as a violet sphere. Polar interactions are shown as dashed yellow lines. Red, oxygen; blue, nitrogen; yellow, sulphur; and green, carbon.
Recently, the structure of the bacterial FB188 HDAH (HDAC-like amidohydrolase from Bordetella/Alcaligenes strain FB188) was also determined 119. This protein has about 30% sequence identity with the two catalytic domains of HDAC6, and 20% and 21% sequence identity with HDLP and HDAC8, respectively. The overall structure of FB188 HDAH reveals an α/β domain similar to HDLP and HDAC8 (Figure 3A). It consists of a seven-stranded parallel β-sheet flanked on one side by three α-helices and on the other side by four α-helices with additional seven α-helices shielding the top of the β-sheet. The differences with HDLP and HDAC8 are predominantly restricted to the loops surrounding the entrance of the active site, especially the long N-terminal loop connecting α-helices 1 and 3 and the C-terminal loop regions (Figure 3A). The structural variability in the loop regions at the entrance of the active site channel results in different molecular surfaces for HDLP, HDAC8 and HDAH (Figure 3B). It is reasonable to assume that this structural variability will be found across the entire family of HDACs and points to a critical role of these regions in conferring different substrate specificities and different protein-protein interactions.
Like HDLP, one zinc ion is bound penta-coordinated to Asp, His, Asp and the hydroxamate moiety of several inhibitors 117, 45 (Figure 3C). This zinc ion binds to a site corresponding to the higher affinity catalytic Mn2+site of arginase: Zn2+ ligands Asp168/Asp178, His170/His180 and Asp258/Asp267 of HDLP/HDAC8 correspond to Mn2+(B) ligands Asp124, His126 and Asp234 of arginase 118. In addition, also the number and the spacing between the zinc ligands are typical of zinc enzymes where this metal is catalytically active: three protein side chains bind to the catalytic zinc ion, the first two ligands are separated by a short spacer of two amino acids and these ligands are separated from the third ligand by a long spacer of 87 amino acids 120. An exact conservation of the catalytic center is found also in HDAH 119, where the zinc ion is penta-coordinated by Asp180, His182 and Asp268 and the hydroxamate moiety of the inhibitor.
The structure of HDAC8 revealed another important difference with HDLP, which is the presence of two potassium-binding sites (Figure 3A, 44): site 1 located near (7.0 Å) the zinc-binding site and directly connected to it by coordinating Asp178 and His180 (both involved in zinc chelation); site 2 located 15 Å away from site 1. The two potassium ions seem to anchor helix α7 and its loops to the central β-sheet, and CD spectroscopy data suggest that these ions are required for the structural stability of the enzyme, while sodium ions that have the same external configuration but a smaller ionic radius, decrease HDAC8 stability 45, 118. Purified HDAC8 in the presence of NaCl has sodium instead of potassium in their structures. The two potassium-binding sites are also found in the HDAH crystal structures 120, and the high degree of conservation of ligands in the entire HDAC family suggests that bound potassium is a feature of all HDAC proteins 45.
The development of HDACi's as anticancer agents
Imbalance of histone and non-histone protein acetylation is common in human cancer. HDACs are overexpressed, aberrantly recruited by oncoproteins or mutated in malignant cells, and several mutations affecting HATs were discovered in tumors 121. TSA, a natural product, was among the first HDACi's to be discovered 122. This molecule shows pronounced anticancer activity and has generated much interest in the discovery of novel HDACi chemotypes. Only a minor fraction of genes is deregulated by HDACi and among these are genes involved in cell cycle progression and apoptosis 123, 124. In addition to transcriptional effects, HDACi also affect progression through mitosis by altering chromatin acetylation, and may alter expression of HSP90 client proteins, among which there are many players in oncogenesis, by affecting HSP90 acetylation. Detailed descriptions on the role of HDACs in cancer and the mechanisms by which HDACi's interfere with tumor cell growth and survival can be found in reviews elsewhere 121, 125, 126. Major hurdles in the discovery of new HDACi have been recently solved. First of all, several groups have been able to express and purify functional HDACs from human, insect and bacterial cells. The technique is mostly based on tagging the COOH terminus of the target HDAC with a short amino-acidic sequence like FLAG, Myc or His[6x] 127, 128, 129, 130, 131, 132 that allows a straightforward purification from transfected/transformed cells. Usually these affinity purified proteins are over 90% pure and often allow co-purification of many of the cofactors described in the HDAC complexes, indicating that these protein complexes are representative of the natural ones. Several groups have made use of stable clones and large-scale transient transfection mostly in HeLa and HEK293 cells, which allows purification of large amounts of recombinant HDACs. Availability of highly purified HDAC preparations is an important achievement that allows for a classic biochemical approach to the discovery of subtype-specific HDACi.
The capability to produce recombinant, tagged versions of all known HDACs needs to integrate with relevant cell-based functional assays. Up to now, very few cell-based assays have been described. Among these, a more limited subset was suitable for high throughput screening; in all these cases, HDACi upregulated the expression of a reporter gene, usually luciferase, under the control of portions of the natural p21WAF1/Cip1 and 5-lipoxygenase gene promoters or of a minimal SV40 promoter containing multiple SMAD-binding motifs. Despite these efforts, all cell-based assays described require several manipulation steps, and as is the case with an end point assay, do not permit continuous monitoring of the enzymatic reaction. To solve the problem, we have taken advantage of the β-lactamase (Bla) reporter gene, and selected a stable HeLa cell clone expressing the Bla reporter under the control of a p21WAF1/Cip1 minimal promoter (−183 through +25 bp) (Pallaoro et al., manuscript in preparation). A relevant advantage of the Bla reporter system versus all other commercial reporter genes is its superior sensitivity 133. Assay sensitivity derives from the combination of two features, an enzymatic reaction and its FRET detection system. The combined features allow detection of the activity of as little as 50 Bla molecules per cell compared to the 105 of GFP or 104 of luciferase molecules. Clearly, such sensitivity is very demanding in terms of fine tuning of reporter gene expression and requires no or extremely low basal expression levels and high inducibility. By combining the indicated minimal promoter lacking the p53 response elements and the HeLa cell line (which has a p53 null background) with the sensitivity features described earlier, we assured robustness, a straightforward readout format and a low probability of identifying false positives that would activate p21 via HDAC-inhibition unrelated mechanisms. A combination of ChIP, siRNA and HDAC overexpression approaches showed that this assay selectively measures the inhibition of class I HDACs in cells.
Classes of HDACi's and their anticancer properties
To date, there are five distinct classes of HDACi as illustrated in Figure 4: short-chain fatty acids like butyric acid (BA), hydroxamic acids such as vorinostat, electrophilic ketones, aminobenzamides exemplified by MS-275, and natural cyclic peptides like romidepsin and apicidin. Various HDACi from several of these chemical classes have already advanced into clinical trials, and recently Zolinza (vorinostat) was approved by the US FDA for the treatment of the cutaneous manifestations in patients with advanced, refractory cutaneous T-cell lymphoma following two or more prior systemic therapies.

Representative structures of HDACi's with anticancer activity.
Among the first HDACi to be identified were the short chain fatty acids like BA and valproic acid 134, 135, 136, 137. These agents are the least potent of the various classes of HDACi with IC50s in the millimolar range. Indeed in 1975, BA was shown to induce differentiation in Friend Leukemia cells, and then 3 years later to cause a dramatic and reversible increase in histone acetylation 136. These findings together with growth arrest seen in many cell lines caused a child with acute myelogenous leukemia to be treated with 500 mpk i.v. infusion of BA daily, resulting in reduction and then disappearance of the myeloblasts. However, more extensive clinical trials failed to reproduce these effects, possibly due to the short half-life (t1/2 = 6 min) and low Cmax (50 μM) achievable. In an effort to obtain higher plasma levels, several prodrugs of BA have been developed. One such prodrug, Pivanex (AN9), is highly lipophilic and is able to cross cell membranes more efficiently and has been shown to affect the growth, differentiation and apoptosis of various cancer cell lines faster and at lower concentrations than BA 137.
TSA, a fungistatic antibiotic, was the first of a series of hydroxamic acid HDACi to be identified and has become one of the main research tools for probing the function of histone acetylation 122, 138. Like BA, interest in this natural product arose due to the demonstration that it was able to induce Friend leukemia cell differentiation, as well as G1 and G2/M cell cycle arrest. In 1990, TSA was identified to be a potent HDACi, giving rise to a marked accumulation of acetylated histones in vivo and strongly inhibiting HDAC, with a Ki of 3.4 nM 122. More recently, an X-ray crystal structure of this compound bound to HDAC8 has revealed that the hydroxamic acid group tightly coordinates the active site zinc ion 117. Poor pharmacokinetics preclude any in vivo work with this HDACi; nevertheless, these findings stimulated the development of structurally related HDACi.
Zolinza, formerly known as SAHA, or generically known as vorinostat, is the most clinically advanced HDACi 139, 140, 141 and was approved by the FDA for the treatment of cutaneous manifestations of advanced, refractory cutaneous T-cell lymphoma. In a phase IIb study, ∼30% of the patients responded to the daily dose of 400 mg orally 139. Vorinostat was identified in the mid-1990s as an agent capable of inducing differentiation of murine erythroleukemia cells and subsequently has been shown to induce differentiation, cell growth arrest and/or apoptosis in a wide variety of cell lines at low micromolar concentrations 140, 142. Vorinostat is able to inhibit both class I and II HDACs at nanomolar concentrations, and like TSA has been shown to bind at the active site of the enzyme. Vorinostat has been demonstrated to have antiproliferative effects against a range of cultured transformed cells and these effects in part are due to altered gene expression where vorinostat causes both increased and decreased expression of about 2-10% of expressed genes. In vivo vorinostat inhibits tumor growth with little toxicity in a wide range of animal models of solid tumors (including breast, prostate, lung and gastric cancers) and hematological malignancies. Clinical studies have been initiated against diverse tumor types, and meaningful clinical responses have been observed in patients with T-cell and B-cell lymphomas, acute myeloid leukemia, mesothelioma, and laryngeal or thyroid cancers. Stable disease has also been achieved in patients with renal-cell carcinoma, urothelial cancer or Hodgkin's lymphoma. Vorinostat has also been shown to have synergistic or additive effects with a wide range of chemotherapeutics in preclinical studies, and clinical combination studies are underway. Additional examples of hydroxamic acid HDACi include LAQ824 and LBH589, PXD-101, and CRA-24781 143, 144, 145, 146, 147, 148.
In an attempt to move away from the hydroxamic acid zinc-binding group, which causes these HDACi to display short half-lives, a series of electrophilic ketones have been developed 149, 150, 151. Electrophilic ketones had previously been shown to be readily hydrated and be able to inhibit zinc-dependent enzymes. Various activated ketones were tested including trifluoromethyl ketones such as 1 in Figure 4, which was shown to be a modest HDACi (IC50 = 310 nM), with weak antiproliferative activity (IC50 = 4.2 μM) against the mouse HT1080 tumor cell line. The corresponding methyl ketone was inactive, demonstrating the requirement for the electrophilic carbonyl group. Unfortunately, these compounds are readily reduced to the inactive alcohol, displaying poor PK parameters, short t1/2 and low exposure upon i.v. dosing. A related series of heterocyclic ketones suffered a similar fate; and despite oxazoles 2 (Figure 4) being a potent HDACi (IC50 = 30 nM), only modest cellular effects were observed. Related α-ketoamides displayed the same liability of rapid reduction, yet efficacy in vivo was achieved with 3 (Figure 4), a potent non-hydroxamic acid HDACi (IC50 = 9 nM) showing antiproliferative activity on HT1080 cells. Indeed, in a mouse HT1080 xenograft model 3 gave rise to tumor growth inhibition when dosed every other day at 30 or 100 mpk i.p., although some toxicity was seen at the highest dose.
A fourth class of HDACi is the aminobenzamides exemplified by CI-994, MS-275 and MGCD0103 152, 153, 154, 155, 156. CI-994 was the first member of this class to be identified and arose from the serendipitous observation that dinaline, a potential anticonvulsant and the des-acetyl derivative of CI-994, caused bone marrow suppression. Investigation into the potential antitumor activity revealed efficacy in vivo in preclinical models 152, 153. It was identified that dinaline is rapidly acetylated in vivo and CI-994 is the active agent. More recently, the mechanism of action has been determined to be due to HDAC inhibition. The most advanced aminobenzamide currently in clinical trials is MS-275, which is in development for melanoma 154, 155. Recently disclosed clinical data showed that this compound administered orally at either 3 mg biweekly or 7 mg weekly was well tolerated and 7/28 patients had stable disease for 8 to more than 48 weeks. The infrequent dosing regime is due to this compound's extended half-life in man, t1/2 = 45-100 h. Preclinically MS-275 was shown to inhibit HDACs with IC50 = 2.0 μM and was demonstrated to cause hyperacetylation of histone H4, induce p21WAF1/CIP and cause G1 cell cycle arrest. Antiproliferative activities ranging from 42 nM to 4.7 μM were seen against a wide panel of cancer cell lines, and in vivo studies with this agent in several xenograft models showed that tumor growth inhibition was observed in seven out of eight models at non-toxic doses when administered orally. The exact manner by which this and related compounds inhibit HDACs is not understood, although the 2′-amino group on the benzanilide has been shown to be crucial for activity, as its removal or isomerization to the related 3′-amino position resulted in loss of HDACi activity. A related aminobenzamide, MGCD0103 156, is undergoing phase I/II trials in solid tumors and hematological cancers. This compound is a selective inhibitor of HDACs 1, 2, 3 and 11 with IC50s = 0.1, 0.2, 2 and 2 μM respectively, and in contrast to hydroxamic acid HDACi, it does not inhibit the other isoforms.
Romidepsin (also known as FK228 or depsipeptide) is a natural product produced by Chromobacterium violaceum 157, 158. The compound was demonstrated to show potent antitumor activity both in vitro and in vivo although several years passed before the mechanism of action of this agent was elucidated. Although romidepsin apparently contains no functional group able to interact with the active site zinc ion, the compound was shown to have appreciable HDACi activity, and have effects on cell cycle, chromatin structure and cause accumulation of acetylated histones. Further work revealed that it is a natural pro-drug, activated by cellular reduction of the disulfide bond to the corresponding thiols 158. Molecular modeling suggests that these groups are capable of acting as zinc-binding groups. In the presence of the reductant DTT, the potency of romidepsin increases 36-fold to 1 nM; furthermore, alkylation of these thiol groups almost abolishes the HDAC inhibitory activity. Romidepsin has been shown to be a selective class I HDACi, with IC50s of 36, 47, 510 and 14 000 nM respectively on HDACs 1, 2, 4 and 6. Clinical studies with this agent are under way and interim phase II data in patients with advanced CTCL has been reported. Following a 4-h i.v. infusion on days 1, 8 and 15 of a 28-day cycle, the compound was reported to be well tolerated with manageable toxicities, including nausea, vomiting and fatigue 158.
A final class of HDACi is a series of cyclic peptides exemplified among others by Trapoxin A, H-C toxin, chlamydocin and apicidin 159, 160, 161, 162, 163. All these structures contain a proline or pipecolinic acid residue and a non-proteinogenic long aliphatic amino acid (L-Aoe or L-Aoda) that may act as a substrate analog. Most of these natural products contain the epoxide group that was believed to be essential for biological activity and this was supported by the fact that trapoxin was shown to potently and irreversibly inhibit HDACs163. More recently, apicidin was isolated and demonstrated to have potent, broad spectrum antiprotozoal activity and was shown to be a potent HDACi 162. It displays antiproliferation activity against a wide panel of cancer cell lines. Apicidin is unique in that it lacks the epoxide whose presence is integral for biological activity in closely related cyclic tetrapeptide.
We have used apicidin as a starting point for a drug discovery program and were to evolve a series of potent, low molecular weight, non-hydroxamic acid HDACi's, exemplified by 4 (Figure 4), which is selective for HDACs 1, 2, 3 and 6, and displays only weak or no activity on HDACs 4, 5, 7 and 8 164. Furthermore, 4 showed submicromolar activity against several human tumor cell lines.
In a search for selective deacetylase inhibitors, Schreiber designed a library of 7 392 members capable of inhibiting deacetylase activity and used a multi-dimensional, high throughput, cell-based assay to screen for induction of α-tubulin or histone acetylation165. Follow-up of the hits enabled a selective, reversible inhibitor of α-tubulin deacetylation to be identified – Tubacin. Tubacin was shown to induce a three-fold increase in α-tubulin acetylation at the 10 μM concentration, with EC50 = 2.5 μM. At these concentrations no alterations in gene expression were seen nor did it affect cell cycle progression, demonstrating the selectivity of this compound. Further studies identified that HDAC 6 was the target of Tubacin and that this isoform plays a role in microtubule stability and cell mobility. Subsequent deconvolution of the library also identified a selective inducer of histone acetylation, Histacin 166.
Summary and perspectives
The possibility of using lysine acetylation in a highly diversified manner in different biological processes seems to be the evolutionary driver behind the complexity of the HDAC family in mammals. Not only has lysine acetylation emerged as a common post-translational mechanism that has many different roles that go beyond chromatin conformation but also distributing the task of controlling lysine acetylation levels on different players has largely expanded the possibility to fine tune this reaction and to incorporate it in many different physiological contexts. In line with this notion, the tight regulation of deacetylase activity at different levels and through different mechanisms is a common theme for probably all HDAC family members. Interfering with the expression or function of individual HDACs was shown to elicit unique biological responses. Since HDACs are involved in many pathological processes, their inhibition could provide clinical benefits. This has been extensively proven for cancer but there is hope that the present compounds may prove useful in several other human diseases. Even though many aspects of the functions of individual HDAC subtypes in mammals were elucidated, more comprehensive in vivo data are needed to extrapolate potential therapeutic benefits. Conditional knockout animals or catalytically-dead knock-ins would be tremendously important in this assessment. The medicinal chemistry efforts have shown that it is possible to extensively modulate the subtype specificity of HDACi. Highly selective compounds may turn out to be formidable tools for improving our understanding of the biology of this fascinating class of enzymes and they may also open new avenues for selective targeting of pathology-associated HDAC subtypes in a therapeutic setting.
References
Jenuwein T, Allis CD . Translating the histone code. Science 2001; 293:1074–1080.
Lachner M, Jenuwein T . The many faces of histone lysine methylation. Curr Opin Cell Biol 2002; 14:286–298.
Nowak SJ, Corces VG . Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet 2004; 20:214–220.
Jason LJ, Moore SC, Lewis JD, Lindsey G, Ausio J . Histone ubiquitination: a tagging tail unfolds? Bioessays 2002; 24:166–174.
Nathan D, Sterner DE, Berger SL . Histone modifications: Now summoning sumoylation. Proc Natl Acad Sci USA 2003; 100:13118–13120.
Allfrey VG, Faulkner, R, Mirsky AE . Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Acad Sci USA 1964; 51: 786–794.
Roth SY, Denu JM, Allis DC . Histone acetyltransfereases. Annu Rev Biochem 2001; 70:81–120.
Marks PA, Miller T, Richon VM . Histone Deacetylases. Curr Opin. Pharmacol 2003; 3:344–351.
Yang XJ . Lysine acetylation and the bromodomain: a new partnership for signaling. Bioessays 2004; 26:1076–1087.
Martin C, Zhang Y . The diverse function of histone lysine methylation. Nat Rev Mol Cell Biol 2005; 6:838–849.
Jones DO, Cowell IG, Singh PB . Mammalian chromodomain proteins: their role in genome organization and expression. Bioessays 2000; 22:124–137.
Lachner M, O'Carrol D, Rea S, et al. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001; 410:116–120.
Bannister AJ, Zegeman P, Patridge JF, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001; 401:120–124.
Nakayama JI, Rice JC, Strahl BD, et al. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 2001; 292:110–113.
Robertson KD . DNA methylation and human disease. Nat Rev Genet 2005; 6:597–610.
Lande-Diner L, Cedar H . Silence of the genes: mechanisms of long term repression. Nat Rev Genet 2005; 8:648–654.
Carrozza MJ, Utley RT, Workman JL, et al. The diverse functions of histone acetyltransferase complexes. Trends Genet 2003; 19:321–329.
de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB . Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003; 370:737–749.
Verdin E, Dequiedt F, Kasler HG . Class II histone deacetylases: versatile regulators. Trends Genet 2003; 19:286–293.
Blander G, Guarente L . The Sir2 family of protein deacetylases. Annu Rev Biochem 2004; 73:417–435.
Bernstein BE, Tong JK, Schreiber SL . Genomewide studies of histone deacetylase function in yeast. Proc Natl Acad Sci USA 2000; 97:13708–13713.
Robyr D, Suka Y, Xenarios I, et al. Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell 2002; 109:437–446.
Wang A, Kurdistani SK, Grunstein M . Requirement of Hos2 histone deacetylase for gene activity in yeast. Science 2002; 298:1412–1414.
Kurdistani SK, Robyr D, Tayazoie S, et al. Genome-wide binding map of the histone deacetylase Rpd3 in yeast. Nat Genet 2002; 31:248–254.
Robert F, Pokholok DK, Hannett NM, et al. Global position and recruitment of HATs and HDACs in the yeast genome. Mol Cell 2003; 16:199–209.
Foglietti C, Filocamo G, Cundari E, et al. Dissecting the biological functions of histone deacetylases by RNA interference and transcriptional profiling. J Biol Chem 2006; 281:17968–17976.
Pile LA, Spellman PT, Katzenberger RJ, et al. The SIN3 deacetylase complex represses genes encoding mitochondrial proteins: implications for the regulation of energy metabolism. J Biol Chem 2003; 278:37840–37848.
Yang XJ, Seto E . Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr Opin Genet Dev 2003; 13:143–153.
Lagger G, O'Carroll D, Rembold M, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J 2002; 21:2672–2681.
Zupkovitz G, Tischler J, Posch M, et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol Cell Biol 2006; 26:7913–7928.
Gui CY, Ngo L, Xu WS, et al. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci USA 2004; 101:1241–1246.
Lagger G, Doetzlhofer A, Schuettengruber B, et al. The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol Cell Biol 2003; 23:2669–2679.
Wilson AJ, Byun DS, Popoya N, et al. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem 2006; 281:13548–13558.
Zhu P, Martin E, Mengwasser J, et al. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004; 5:455–463.
Glozak MA, Sengupta N, Zhang X, et al. Acetylation and deacetylation of non-histone proteins. Gene 2005; 363:15–23.
Hartman HB, Yu J, Alenghat T, et al. The histone-binding code of nuclear receptor co-repressors matches the substrate specificity of histone deacetylase 3. EMBO Rep 2005; 6:445–451.
He X, Gonzalez V, Tsang A, Thompson J, Tsang TC, Harris DT . Differential gene expression profiling of CD34+ CD133+ umbilical cord blood hematopoietic stem progenitor cells. Stem Cells Dev 2005; 14:188–198.
Li Y, Kao GD, Garcia BA, et al. A novel histone deacetylase pathway regulates mitosis by modulating Aurora B kinase activity. Genes Dev 2006; 20:2566–2579.
Longworth MS, Laimins LA . Histone deacetylase 3 localizes to the plasma membrane and is a substrate of Src. Oncogene 2006; 25:4495–4500.
Chen LF, Fischle W, Verdin E, et al. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001; 293:1653–1657.
Kieman R, Bres V, Ng RW, et al. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem 2003; 278:2758–2766.
Waltregny D, Glenisson W, Tran SL, et al. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J 2005; 19:966–968.
Durst KL, Lutterbach B, Kummalue T, et al. The inv(16) fusion protein associates with corepressors via a smooth muscle myosin heavy-chain domain. Mol Cell Biol 2003; 23:607–619.
Lee H, Sengupta N, Villagra A, et al. Histone deacetylase 8 safeguards the human ever-shorter telomeres 1B (hEST1B) protein from ubiquitin-mediated degradation. Mol Cell Biol 2006; 26:5259–5269.
Vannini A, Volpari C, Filocamo G, et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci USA 2004; 101:15064–15069.
Zhang CL, McKinsey TA, Chang S, et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 2002; 110:479–488.
Vega RB, Matsuda K, Oh J, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 2004; 119:555–566.
Chang S, McKinsey TA, Zhang CL, et al. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol 2004; 24:8467–8476.
Dequiedt F, Kasler H, Fischle W, et al. HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity 2003; 18:687–698.
Chang S, Young BD, Li S, et al. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 2006; 126:321–334.
Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature 2002; 417:455–458.
Matsuyama A, Shimazu T, Sumida Y, et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J 2002; 21:6820–6831.
Kawaguchi Y, Kovacs JJ, McLaurin A, et al. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003; 115:727–738.
Yu X, Guo ZS, Marcu MG, et al. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst 2002; 94:504–513.
Kovacs JJ, Murphy PJ, Gaillard S, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 2005; 18:601–607.
Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 2005; 280:26729–26734.
Gao L, Cueto MA, Asselbergs F, et al. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem 2002; 277:25748–25755.
Yang XJ, Gregoire S . Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol Cell Biol 2005; 25:2873–2884.
Sengupta N, Seto E . Regulation of histone deacetylase activities. J Cell Biochem 2004; 93:57–67.
Guenther MG, Barak O, Lazar MA . The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol 2001; 21:6091–6101.
Zhang J, Kalkum M, Chait BT, et al. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol Cell 2002; 9:611–623.
Hassig CA, Fleischer TC, Billin, AN, et al. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell 1997; 89:341–347.
Zhang Y, Ng HH, Erdjument-Bromage H, et al. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev 1999; 13:1924–1935.
You A, Tong JK, Grozinger CM, et al. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc Natl Acad Sci USA 2001; 98:1454–1458.
Alland L, David G, Shen-Li H, et al. Identification of mammalian Sds3 as an integral component of the Sin3/histone deacetylase corepressor complex. Mol Cell Biol 2002; 22:2743–2750.
Lechner T, Carrozza MJ, Yu Y, et al. Sds3 (suppressor of defective silencing 3) is an integral component of the yeast Sin3 Rpd3 histone deacetylase complex and is required for histone deacetylase activity. J Biol Chem 2000; 275:40961–40966.
McKinsey TA, Zhang CL, Lu J, et al. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 2000; 408:106–111.
McKinsey TA, Zhang CL, Olson EN, et al. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol 2001; 21:6312–6321.
McKinsey TA, Zhang CL, Olson EN . Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14–3-3 to histone deacetylase 5. Proc Natl Acad Sci USA 2000; 97:14400–14405.
Vega RB, Harrison BC, Meadows E, et al. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol 2004; 24:8374–8385.
Dequiedt F, Van Lint J, Lecomte E, et al. Phosphorylation of histone deacetylase 7 by protein kinase D mediates T cell receptor-induced Nur77 expression and apoptosis. J Exp Med 2005; 201:793–804.
Harrison BC, Kim MS, van Rooij E, et al. Regulation of cardiac stress signaling by protein kinase d1. Mol Cell Biol 2006; 26:3875–3888.
Davis FJ, Gupta M, Camoretti-Mercado B, et al. Calcium/calmodulin-dependent protein kinase activates serum response factor transcription activity by its dissociation from histone deacetylase, HDAC4. Implications in cardiac muscle gene regulation during hypertrophy. J Biol Chem 2003; 278:20047–20058.
Zhao X, Ito A, Kane CD, et al. The modular nature of histone deacetylase HDAC4 confers phosphorylation-dependent intracellular trafficking. J Biol Chem 2001; 276:35042–35048.
Liu Y, Randall WR, Schneider MF, et al. Activity-dependent and -independent nuclear fluxes of HDAC4 mediated by different kinases in adult skeletal muscle. J Cell Biol 2005; 168:887–897.
Bolger TA, Yao TP . Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J Neurosci 2005; 25:9544–9553.
Lee H, Rezai-Zadeh N, Seto E, et al. Negative regulation of histone deacetylase 8 activity by cyclic AMP-dependent protein kinase A. Mol Cell Biol 2004; 24:765–773.
Pflum MK, Tong JK, Lane WS, et al. Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J Biol Chem 2001; 276:47733–47741.
Cai R, Kwon P, Yan-Neale Y, et al. Mammalian histone deacetylase 1 protein is posttranslationally modified by phosphorylation. Biochem Biophys Res Commun 2001; 283:445–453.
Tsai SC, Seto E . Regulation of histone deacetylase 2 by protein kinase CK2. J Biol Chem 2002; 277:31826–31833.
Sun JM, Chen HY, Moniwa M, et al. The transcriptional repressor Sp3 is associated with CK2-phosphorylated histone deacetylase 2. J Biol Chem 2002; 277:35783–35786.
Zhang X, Ozawa Y, Lee H, et al. Histone deacetylase 3 (HDAC3) activity is regulated by interaction with protein serine/threonine phosphatase 4. Genes Dev 2005; 19:827–839.
Meggio F, Pinna LA . One-thousand-and-one substrates of protein kinase CK2? FASEB J 2003; 17:349–368.
Uhle S, Medalia O, Waldron R, et al. Protein kinase CK2 and protein kinase D are associated with the COP9 signalosome. EMBO J 2003; 22:1302–1312.
Unger GM, Davis AT, Slaton JW, et al. Protein kinase CK2 as regulator of cell survival: implications for cancer therapy. Curr Cancer Drug Targets 2004; 4:77–84.
Guo C, Davis AT, Yu S, et al. Role of protein kinase CK2 in phosphorylation nucleosomal proteins in relation to transcriptional activity. Mol Cell Biochem 1999; 191:135–142.
Guo C, Davis AT, Ahmed K . Dynamics of protein kinase CK2 association with nucleosomes in relation to transcriptional activity. J Biol Chem 1998; 273:13675–13680.
Barz T, Ackermann K, Dubois G, et al. Genome-wide expression screens indicate a global role for protein kinase CK2 in chromatin remodeling. J Cell Sci 2003; 116:1563–1577.
Hess-Stumpp H . Histone deacetylase inhibitors and cancer: from cell biology to the clinic. Eur J Cell Biol 2005; 84:109–121.
Momparler RL . Cancer epigenetics. Oncogene 2003; 22:6479–6483.
Dello Russo C, Talamo F, Paolini C, et al. Histone deacetylase 3 (HDAC3) is phosphorylated on a unique serine residue by casein kinase 2 that is physically associated with HDAC3 in mammalian cells, submitted
Galasinski SC, Resing KA, Goodrich JA, et al. Phosphatase inhibition leads to histone deacetylases 1 and 2 phosphorylation and disruption of corepressor interactions. J Biol Chem 2002; 277:19618–19626.
Yang WM, Tsai SC, Wen YD, et al. Functional domains of histone deacetylase-3. J Biol Chem 2002; 277:9447–9454.
Fajas L, Egler V, Reiter R, et al. The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev Cell 2002; 3:903–910.
Kim SC, Kim YS, Jetten AM . Kruppel-like zinc finger protein Gli-similar 2 (Glis2) represses transcription through interaction with C-terminal binding protein 1 (CtBP1). Nucleic Acids Res 2005; 33:6805–6815.
Li D, Yea S, Li S, et al. Kruppel-like factor-6 promotes preadipocyte differentiation through histone deacetylase 3-dependent repression of DLK1. J Biol Chem 2005; 280:26941–26952.
Ozawa Y, Towatari M, Tsuzuki S, et al. Histone deacetylase 3 associates with and represses the transcription factor GATA-2. Blood 2001; 98:2116–2123.
Yuan ZL, Guan YJ, Chatterjee D, et al. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005; 307:269–273.
Franco PJ, Farooqui M, Seto E, et al. The orphan nuclear receptor TR2 interacts directly with both class I and class II histone deacetylases. Mol Endocrinol 2001; 15:1318–1328.
Somech R, Shaklai S, Geller O, et al. The nuclear-envelope protein and transcriptional repressor LAP2beta interacts with HDAC3 at the nuclear periphery, and induces histone H4 deacetylation. J Cell Sci 2005; 118:4017–4025.
Li G, Franco PJ, Wei LN . Identification of histone deacetylase-3 domains that interact with the orphan nuclear receptor TR2. Biochem Biophys Res Commun 2003; 310:384–390.
Canettieri G, Morantte I, Guzman E, et al. Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat Struct Biol 2003; 10:175–181.
Chen CS, Weng SC, Tseng PH, et al. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J Biol Chem 2005; 280:38879–38887.
Zhou Y, Gross W, Hong SH, et al. The SMRT corepressor is a target of phosphorylation by protein kinase CK2 (casein kinase II). Mol Cell Biochem 2001; 220:1–13.
Wang D, Westerheide SD, Hanson JL, et al. Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem 2000; 275:32592–32597.
Brush MH, Guardiola A, Connor JH, et al. Deactylase inhibitors disrupt cellular complexes containing protein phosphatases and deacetylases. J Biol Chem 2004; 279:7685–7691.
David G, Neptune MA, DePinho RA SUMO-1 modification of histone deacetylase 1 (HDAC1) modulates its biological activities. J Biol Chem 2002; 277:23658–23663.
Colombo R, Boggio R, Seiser C, et al. The adenovirus protein Gam1 interferes with sumoylation of histone deacetylase 1. EMBO Rep 2002; 3:1062–1068.
Kirsh O, Seeler JS, Pichler A, et al. The SUMO E3 ligase RanBP2 promotes modification of the HDAC4 deacetylase. EMBO J 2002; 21:2682–2691.
Girdwood D, Bumpass D, Vaughan OA, et al. P300 transcriptional repression is mediated by SUMO modification. Mol Cell 2003; 11:1043–1054.
Ling Y, Sankpal UT, Robertson K, et al. Modification of de novo DNA methyltransferase 3a (Dnmt3a) by SUMO-1 modulates its interaction with histone deacetylases (HDACs) and its capacity to repress transcription. Nucleic Acids Res 2004; 32:598–610.
Yang SH, Sharrocks AD . SUMO promotes HDAC-mediated transcriptional repression. Mol Cell 2004; 13:611–617.
Zhao X, Sternsdorf T, Bolger TA, et al. Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol Cell Biol 2005; 25:8456–8464.
Gregoire S, Tremblay AM, Xiao L, et al. Control of MEF2 transcriptional activity by coordinated phosphorylation and sumoylation. J Biol Chem 2006; 281:4423–4433.
Gregoire S, Yang XJ . Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol Cell Biol 2005; 25:2273–2287.
Finnin MS, Donigian JR, Cohen A, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999; 401:188–193.
Somoza JR, Skene RJ, Katz BA, et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004; 12:1325–1334.
Christianson DW . Arginase: structure, mechanism, and physiological role in male and female sexual arousal. Acc Chem Res 2005; 38:191–201
Nielsen TK, Hildmann C, Dickmanns A, et al. Crystal structure of a bacterial class 2 histone deacetylase homologue. J Mol Biol 2005; 354:107–120.
Vallee BL, Auld DS . Active-site zinc ligands and activated H2O of zinc enzymes. Proc Natl Acad Sci USA 1990; 87:220–224.
Minucci S, Pelicci PG . Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 2006; 6:38–51.
Yoshida M, Kijima M, Akita M, et al. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem 1990; 265:17174–17179.
Van Lint C, Emiliani S, Verdin E . The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr 1996; 5:245–253.
Mitsiades CS, Mitsiades NS, McMullan CJ, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci USA 2004; 101:540–545.
Rosato RR, Grant S . Histone deacetylase inhibitors: insights into mechanisms of lethality. Expert Opin Ther Targets 2005; 9:809–824.
Xu W, Ngo L, Perez G, et al. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc Natl Acad Sci USA 2006; 103:15540–15545.
Riester D, Wegener D, Hildmann C, et al. Members of the histone deacetylase superfamily differ in substrate specificity towards small synthetic substrates. Biochem Biophys Res Commun 2004; 324:1116–1123.
Schultz BE, Misialek S, Wu J, et al. Kinetics and comparative reactivity of human class I and class IIb histone deacetylases. Biochemistry 2004; 43:11083–11091.
Li J, Staver MJ, Curtin ML, et al. Expression and functional characterization of recombinant human HDAC1 and HDAC3. Life Sci 2004; 74:2693–2705.
Fischer DD, Cai R, Bhatia U, et al. Isolation and characterization of a novel class II histone deacetylase, HDAC10. J Biol Chem 2002; 277:6656–6666.
Hu E, Chen Z, Fredrickson T, et al. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J Biol Chem 2000; 275:15254–15264.
Sun JM, Spencer VA, Chen HY, et al. Measurement of histone acetyltransferase and histone deacetylase activities and kinetics of histone acetylation. Methods 2003; 31:12–23.
Zlokarnik G, Negulescu PA, Knapp TE, et al. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science 1998; 279:84–88.
Leder A, Leder P . Butyric acid, a potent inducer of erythroid differentiation in cultured erythroleukemic cells. Cell 1975; 5:319–322.
Riggs MG, Whittaker RG, Neumann JR, Ingram VM . n-Butyrate causes histone modification in HeLa and Friend erythroleukemia cells. Nature 1977; 268:462–464.
Novogrodsky A, Dvir A, Ravid A, et al. Effect of polar organic compounds on leukemic cells. Cancer 1983; 51:9–14.
Rephaeli A, Zhuk R, Nudelman A . Prodrugs of butyric acid from bench to bedside: synthetic design, mechanisms of action, and clinical applications. Drug Dev Res 2000; 50:379–391.
Tsuji N, Kobayashi M, Nagashima K, et al. A new antifungal antibiotic, trichostatin. J Antibiotics 1976; 29:1–6.
Olsen E, Kim YH, Kuzel T, et al. Vorinostat (suberoylanilide hydroxamic acid, SAHA) is clinically active in advanced cutaneous T-cell lymphoma (CTCL): results of a phase IIb trial. J Clin Oncol; 2006 ASCO Annual Meeting Proceedings Part I 24(18S):abstr 7500.
Richon VM, Webb Y, Merger R, et al. Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc Natl Acad Sci USA 1996; 93:5705–5708.
Kelly WK, O'Connor OA, Krug L, et al. Phase I study of the oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), in patients with advanced cancer J Clin Oncol 2005; 23:3923–3931.
Kelly WK, Marks PA . Drug insight: HDACi-development of the new targeted anticancer agent SAHA. Nat Clin Pract Oncol 2005; 2:150–157.
Remiszewski SW, Sambucetti LC, Bair KW, et al. N-hydroxy-3-phenyl-2-propenamides as novel inhibitors of human HDAC with in vivo antitumor activity: discovery of NVP-LAQ824. J Med Chem 2003; 46:4609–4624.
Ottmann OG, Deangelo DJ, Stone RM . A phase I, pharmacokinetic (PK) and pharmacodynamic (PD) study of a novel HDACi LAQ824 in patients with hematologic malignancies. J Clin Oncol; 2004 ASCO Annual Meeting Proceedings (Post-Meet Ed) 22(14S):abstr 3024.
Prince HM, et al. Oral LBH589: a novel deacetylase inhibitor demonstrates clinical efficacy I patients with CTCL. Annual Meeting of the American Association of Canadian Research, Washington, DC. 2006:abstr 1146.
Plumb JA ; Finn PW, Williams RJ, et al. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel HDACi PXD101. Mol Canc Ther 2003; 2:721–728.
Steele N . A phase 1 pharmacokinetic (PK) and pharmacodynamic (PD) study of the HDACi PXD101 in patients (pts) with advanced solid tumours. Annual Meeting of the American Society of the Clinical Oncology, Orlando, FL. 2005:abstr 3035.
Buggy JJ, Cao ZA, Bass KE, et al. CRA-024781: a novel synthetic inhibitor of HDAC enzymes with antitumor activity in vitro and in vivo. Mol Cancer Ther 2006; 5:1309–1317.
Frey RR, Wada CK, Garland RB, et al. Trifluoromethyl ketones as inhibitors of HDAC. Bioorg Med Chem Lett 2002; 12:3443–3447.
Wada CK, Frey RR, Ji Z, et al. α-Keto amides as inhibitors of HDAC. Bioorg Med Chem Lett 2003; 13:3331–3335.
Vasudevan A, Ji Z, Frey RR, et al. Heterocyclic ketones as inhibitors of HDAC. Bioorg Med Chem Lett 2003; 13:3909–3913.
Kraker AJ, Mizzen CA, Hartl BG, et al. Modulation of histone acetylation by [4-(acetylamino)-N-(2-amino-phenyl) benzamide] in HCT-8 colon carcinoma. Mol Can Ther 2003; 2:401–408.
Prakash S, Foster BJ, Meyer M, et al. Chronic oral administration of CI-994: a phase 1 study. Invest New Drugs 2001; 19:1–11.
Saito A, Yamashita T, Mariko Y, et al. A synthetic inhibitor of HDAC, MS-27–275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci USA 1999; 96:4592–4597.
Hauschild A, Trefzer U, Garbe C, et al. A phase II multicenter study on the histone deacetylase (HDAC) inhibitor MS-275, comparing two dosage schedules in metastatic melanoma. J Clin Oncol; 2006 ASCO Annual Meeting Proceedings Part I 24(18S):abstr 8044.
Vaisburg A . Discovery and development of MGCD0103 – an orally active HDAC inhibitor in human clinical trials. XIXth International Symposium on Medical Chemistry, Istanbul, Aug 29-Sep 2, 2006.
Furumai R, Matsuyama A, Kobashi N, et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I HDACs. Cancer Res 2002; 62:4916–4921.
Whittaker S, McCulloch W, Robak T, et al. International multicentre phase II study of the HDACi depsipeptide in CTCL – interim report. J Clin Oncol; 2006 ASCO Annual Meeting Proceedings Part I 24(18S):abstr 3063.
Itazaki H, Nagashima K, Sugita K, et al. Isolation and structural elucidation of new cyclotetrapeptides, trapoxins A and B, having detransformation activities as antitumor agents. J Antibiotics 1990; 43:1524–1532.
Pringle RB . Chemical constitution of the host-specific toxin of Helminthosporium carbonum. Plant Phys 1970; 46:45–49
Closse A, Huguenin R . Isolation and structure elucidation of chlamydocin. Helv Chim Acta 1974; 57:533–545
Darkin-Rattray SJ, Gurnett AM, Myers RW, et al. Apicidin: A novel antiprotozoal agent that inhibits parasite HDAC. Proc Natl Acad Sci USA 1996; 93:13143
Kijima M, Yoshida M, Sugita K, et al. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian HDAC. J Biol Chem 1993; 268:22429–22435.
Jones P, Altamura S, Chakravarty PK, et al. A series of novel, potent, and selective histone deacetylase inhibitors. Bioorg Med Chem Lett 2006; 16:5948–5952.
Haggarty SJ, Koeller KM, Wong JC, et al. Domain-selective small-molecule inhibitor of HDAC6-mediated tubulin deacetylation. Proc Natl Acad Sci USA 2003; 100:4389–4394.
Haggarty SJ, Koeller KM, Wong JC, et al. Multidimensional chemical genetic analysis of diversity-oriented synthesis-derived deacetylase inhibitors using cell-based assays. Chem Biol 2003; 10:383–396.
Acknowledgements
This work was in part supported by a grant from the Ministero per l'Università e la Ricerca Scientifica (MIUR)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gallinari, P., Marco, S., Jones, P. et al. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res 17, 195–211 (2007). https://doi.org/10.1038/sj.cr.7310149
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7310149
Keywords
This article is cited by
-
Epigenetic regulation of the nuclear genome associated with mitochondrial dysfunction in Leber’s hereditary optic neuropathy (LHON)
Human Genome Variation (2024)
-
Progress in discovery and development of natural inhibitors of histone deacetylases (HDACs) as anti-cancer agents
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Butyrate limits human natural killer cell effector function
Scientific Reports (2023)
-
Advances in the Mechanistic Study of the Control of Oxidative Stress Injury by Modulating HDAC6 Activity
Cell Biochemistry and Biophysics (2023)
-
Indole-based LpxC (UDP-3-O-(R-3-hydroxyacyl)-N-acetylglucosaminedeacetylase) inhibitors for Salmonella typhi: rational drug discovery through in silico screening
3 Biotech (2023)
