
Abstract
Deregulated expression of genes encoding members of the S100 family of calcium-binding proteins has been associated with the malignant progression of multiple tumour types. Using a pharmacological expression reactivation approach, we screened 16 S100 genes for evidence of epigenetic regulation in medulloblastoma, the most common malignant brain tumour of childhood. Four family members (S100A2, S100A4, S100A6 and S100A10) demonstrated evidence of upregulated expression in multiple medulloblastoma cell lines, following treatment with the DNA methyltransferase inhibitor, 5′-aza-2′-deoxycytidine. Subsequent analysis revealed methylation of critical CpG sites located within these four genes in an extended cell line panel. Assessment of these genes in the non-neoplastic cerebellum (from which medulloblastomas develop) revealed strong somatic methylation affecting S100A2 and S100A4, whereas S100A6 and S100A10 were unmethylated. Assessed against these normal tissue-specific methylation states, S100A6 and S100A10 demonstrated tumour-specific hypermethylation in medulloblastoma primary tumours (5 out of 40 and 4 out of 35, respectively, both 12%) and cell lines (both 7 out of 9, 78%), which was associated with their transcriptional silencing. Moreover, S100A6 hypermethylation was significantly associated with the aggressive large cell/anaplastic morphophenotype (P=0.026). In contrast, pro-metastatic S100A4 displayed evidence of hypomethylation relative to the normal cerebellum in a significant proportion primary tumours (7 out of 41, 17%) and cell lines (3 out of 9, 33%), which was associated with its elevated expression. In summary, these data characterise complex patterns of somatic methylation affecting S100 genes in the normal cerebellum and demonstrate their disruption causing epigenetic deregulation of multiple S100 family members in medulloblastoma development. Epigenetic events affecting S100 genes have potential clinical utility and merit further investigation as molecular biomarkers for this disease.
Similar content being viewed by others


Main
Epigenetic alterations play an important role in the development of medulloblastoma, an embryonal tumour of the cerebellum, which accounts for ∼20% of childhood brain tumours (Giangaspero et al, 2000; Lindsey et al, 2005). The S100 protein family is a large family of EF hand calcium-binding proteins of approximately 20 members (Donato, 2003; Marenholz et al, 2004), involved in the regulation of a variety of cellular processes including cell growth and cell cycle regulation, differentiation, transcription and motility. Sixteen members of the gene family (S100A1–S100A16) map within a 1.65 Mb region at chromosome 1q21.3 (Figure 1A) (Marenholz et al, 2004).

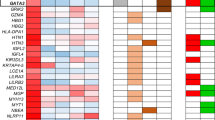
Methylation-dependent regulation of S100 genes in medulloblastoma. (A) The S100 gene cluster at 1q21.3, showing the position (vertical lines) and direction of transcription (>, <) of each gene. Physical positions are based on NCBI Assembly 36 of the human genome (see http://www.ncbi.nlm.nih.gov/). Genes not belonging to the S100 family have been omitted for clarity. (B) Methylation-dependent changes in expression of 16 S100 genes in three medulloblastoma cell lines (MEB-MED8A, D283Med, D425Med), following treatment with the DNA methyltransferase inhibitor, 5-aza-CdR (5 μ M, 72 h), determined by analysis using the Affymetrix U133A Human expression array (sequence identities of probes indicated were verified independently). Fold-changes in expression detected by each probe are shown, based on signal intensities assessed by MAS5 software (Affymetrix). Four genes (marked**) displayed evidence of positive methylation-dependent regulation in ⩾2 cell lines; S100A2, S100A4, S100A6 and S100A10.
Members of the S100 gene family show divergent patterns of cell and tissue-specific expression, and the expression of specific family members is disrupted in a range of diseases including cancer (Heizmann et al, 2002; Marenholz et al, 2004; Heizmann, 2005). Overexpression of several of the S100 proteins is thought to be involved in tumour progression, such as S100A4, which is upregulated in many cancers including medulloblastoma and promotes angiogenesis and metastasis (Hernan et al, 2003; Emberley et al, 2004; Helfman et al, 2005; Garrett et al, 2006) and S100P which is associated with metastasis in breast and pancreatic cancer (Arumugam et al, 2005; Wang et al, 2006). Other S100 proteins are downregulated in tumours and have putative tumour suppressor roles, including S100A2 and S100A6 in prostate cancer (Rehman et al, 2005).
Expression of several members of the S100 family, including S100A2, S100A4, S100A6 and S100P, is known to be regulated epigenetically, by methylation of key CpG sites within the genes or their promoters (Wicki et al, 1997; Rosty et al, 2002; Sato et al, 2004; Rehman et al, 2005). This regulation has been postulated to be important in controlling the cell type-specific expression of S100 genes, as methylation associated with transcriptional silencing of these genes in normal somatic tissues has been found to occur in a tissue-specific manner (Lesniak et al, 2000; Rosty et al, 2002; Sato et al, 2004). Accordingly, the normal regulation of individual S100 family members can be disrupted during tumourigenesis, by aberrant gene-specific methylation events, which have been demonstrated in diverse cancer types including lung, prostate, pancreatic and colon cancer (Wicki et al, 1997; Nakamura and Takenaga, 1998; Lesniak et al, 2000; Feng et al, 2001; Rosty et al, 2002; Rehman et al, 2004, 2005; Sato et al, 2004). However, the epigenetic status of S100 genes in medulloblastomas and the normal cerebellum has not been previously investigated.
In this study, we examined 16 members of the S100 gene family for evidence of methylation-dependent epigenetic regulation in medulloblastoma, using a pharmacological expression reactivation approach, involving microarray analysis of gene expression changes induced by the treatment of medulloblastoma cell lines with inhibitors of DNA methylation. Four gene family members, S100A2, S100A4, S100A6 and S100A10, displayed consistent and confirmed evidence of methylation-dependent upregulation following treatment. The methylation status of these genes was therefore investigated in detail in medulloblastoma primary tumours, cell lines and the normal cerebellum, to identify and characterise any epigenetic events affecting them, and to assess any potential biological roles and/or clinicopathological significance in medulloblastoma pathogenesis.
Materials and methods
Cell lines
Nine independently derived medulloblastoma cell lines (DAOY, D283 Med, MHH-MED-1, MEB-MED8A, D341 Med, D384 Med, D425 Med, D556 Med and UW228-3) were studied. All cells were grown under recommended culture conditions provided by suppliers (see acknowledgements) and cell line identity was confirmed before use by karyotyping (data not shown). Cell line DNA was extracted using the Qiagen DNeasy kit (Qiagen, Crawley, UK).
Primary tumours and normal control samples
A cohort of 41 primary medulloblastomas was analysed including representatives of all the major histopathological subtypes (25 classic, six large cell/anaplastic and 10 nodular/desmoplastic)(Giangaspero et al, 2000). Patients (15 females and 26 males) ranged in age from 1.3 to 19 years (seven infants <3 years, 30 children 3–16 years and four adults >16 years). Six cerebellar samples were analysed consisting of post-mortem material from three fetuses (18- 19- and 22-weeks gestational age), one infant (newborn) and two adults (60 and 67 years) who had died of non-neoplastic conditions. DNA was extracted from frozen tissues using standard methods and from formalin-fixed, paraffin-embedded tissue using a Nucleon hard tissue kit (Amersham Biosciences, Little Chalfont, UK). Local Ethical Committee and Institutional Review Board approval was obtained for the collection, storage and biological study of all material.
5′-Aza-2′-deoxycytidine (5-aza CdR) treatment
Four cell lines (DAOY, D283 Med, D425 Med and MEB-MED8A) were grown in the presence or absence of the demethylating agent 5-aza CdR (5 μ M) (Sigma-Aldrich, Poole, UK) for 3 days. Medium was renewed daily.
Microarray analysis
Total RNA was extracted from cell lines D283 Med, D425 Med and MEB-MED8A, which had been grown in parallel cultures with or without 5-aza CdR treatment. These cell lines were selected for analysis as they exhibit characteristic chromosomal defects representative of primary medulloblastomas (Langdon et al, 2006). RNA was extracted from 107 cells using Trizol reagent (Invitrogen, Paisley, UK) according to manufacturer's instructions. Microarray expression analysis was performed at the Newcastle University microarray facility; RNA was converted into biotin-labelled cRNA and hybridised to the Human U133A array according to manufacturer's protocols (Affymetrix, Santa Clara, CA, USA). MAS5 software (Affymetrix) was used for data processing, normalisation and calculation of signal intensities. Signal intensities were compared between treatment conditions using Microsoft Excel software (Microsoft, Reading, UK).
Analysis of S100 gene methylation status
Bisulphite treatment of DNA was carried out using a CpG genome DNA modification kit (Serologicals, Livingston, UK) according to the manufacturer's instructions. The promoter and exon1 regions of S100A2 and S100A6 and a region within the first intron of S100A4 were amplified following bisulphite treatment using previously published primers and conditions (Rosty et al, 2002; Rehman et al, 2005). The promoter region of S100A10 (Huang et al, 2003) was analysed using primers designed by ‘MethPrimer’ (Li and Dahiya, 2002) to amplify a region of the CpG island 400–652 bp relative to the transcriptional start site. Primer sequences were S100A10F 5′-ATTATTTGTTGGATGATTTTGTAGG-3′ and S100A10R 5′-ACACAAAAAAATAAATCCCCTATTC-3′ (designed to amplify the antisense strand following bisulphite conversion). Thirty nanograms of bisulphite treated DNA was used per reaction. PCR products were amplified using standard conditions with an annealing temperature of 60°C. PCR products were directly sequenced with a CEQ DTCS kit (Beckman Coulter, High Wycombe, UK). Sequenced products were analysed on a CEQ 2000XL DNA analysis system (Beckman Coulter), and the methylation status at each CpG residue determined by assessment of the relative peak intensities.
Combined bisulphite and restriction analysis (COBRA (Xiong and Laird, 1997) of S100A4 was carried out by overnight digestion of the 142 bp PCR product at 37°C with the HypCH4IV restriction endonuclease (New England Biolabs, Hitchin, UK), which has the recognition sequence 5′-ACGT-3′ and which cuts twice within the product if fully methylated before bisulphite conversion to give 100, 26 and 16 bp fragments. Digested PCR products were separated on a 4% Nusieve 3 : 1 agarose gel in 1 × TBE (0.09 M Tris-Borate, 0.002 M EDTA pH 9) and stained with ethidium bromide, before visualisation by transillumination under ultraviolet (UV) light. The unmethylated control for COBRA and bisulphite sequence analysis consisted of a pool of 20 newborn cord blood DNAs, the methylated control was universal methylated DNA (Serologicals Corporation, Livingston, UK). Representative examples of PCR products showing different digestion patterns were sequenced as described above, to determine relative peak intensities.
Reverse transcription PCR (RT–PCR)
RNA was extracted from 107 subconfluent cells using Trizol reagent (Invitrogen, Paisley, UK) according to manufacturer's instructions. One microgram of total RNA was used to synthesise cDNA using a reverse transcription system (Promega, Southampton, UK). Equivalent amounts of this cDNA were used for PCR amplification of the respective S100 gene, RASSF1A and ACTB transcripts. RT–PCRs for S100A2, S100A4 and S100A6 were carried out using previously published primers and conditions (Rosty et al, 2002; Rehman et al, 2005). RT primers for S100A10 were designed using transcript information obtained from the Ensembl genome browser (www.ensembl.org; Gene ID, ENSG00000197747). Primer sequences were S100A10rtF (in exon 2) 5′-TTCACAAATTCGCTGGGGATAA-3′ and S100A10rtR (in exon 3) 5′-AACTGCTCATTTCTGCCTACTTCT, which amplified a 264 bp product. PCR products were amplified using standard conditions with an annealing temperature of 54°C. RT–PCR of RASSF1A was used as a positive control and was carried out according to procedures published previously (Lusher et al, 2002). RT–PCR of ACTB (encoding β-actin, a housekeeping gene) was used as a control for RNA concentration and integrity and was carried out using previously published primers and conditions (Horikoshi et al, 1992). PCR products were electrophoresed on a 2% agarose gel in 1 × TBE and stained with ethidium bromide, before visualisation by transillumination under UV light.
Results
Re-expression of S100 family members following demethylating treatment of medulloblastoma cell lines: identification of candidate epigenetically regulated genes
To identify S100 genes showing evidence of methylation-dependent transcriptional regulation in medulloblastoma, three medulloblastoma cell lines (D425Med, D283Med and MEB-MED8A) were cultured in the presence or absence of the demethylating agent, 5-aza CdR. Resultant expression changes were assessed using the Affymetrix Human U133A array. The U133A array contains probe sets which recognise 16 S100 genes (S100A1-S100A14, S100β and S100P), for which probe sequence identity and specificity could be verified using the BLAST sequence alignment tool (http://www.ncbi.nlm.nih.gov/BLAST/). Changes in probe signal intensity following 5-aza CdR treatment were calculated and genes showing an expression increase >2-fold were classed as being significantly upregulated, and thus showing evidence of methylation-dependent transcriptional regulation. Four independent S100 transcripts showed evidence of transcriptional upregulation in multiple cell lines (Figure 1B); S100A6 and S100A10 were upregulated in all three cell lines, while S100A2 and S100A4 were upregulated in two cell lines. In addition, S100A3, S100A11 and S100P were each upregulated in one cell line, and the remaining S100 genes were not significantly upregulated in any cell line.
The expression changes observed for S100A6, S100A10, S100A2 and S100A4 on array analysis were next validated in an independent series of experiments by RT–PCR (Figure 2A). The increases in expression observed by microarray for these genes following 5-aza CdR treatment were also clearly seen using RT–PCR methods, with good concordance between results obtained using the two techniques (Figure 2A). Reverse transcription–PCR analysis following demethylation treatment was also extended to a fourth medulloblastoma cell line, DAOY, which did not appear to show altered expression of any of the four S100 genes examined following 5-aza CdR treatment, despite clear evidence of re-expression of the RASSF1A control transcript.

Epigenetic inactivation of S100 genes by promoter hypermethylation in medulloblastoma cell lines. (A) Expression analysis of S100 genes in medulloblastoma cell lines. RT–PCR analysis of S100A2, S100A4, S100A6 and S100A10 is shown for cell lines grown in the presence (+) or absence (−) of the demethylating agent 5-aza CdR (5 μ M, 72 h). Probe signal intensities generated for each gene in independent microarray experiments (see Figure 1A) are also given for comparison. RASSF1A (positive methylation-regulated gene) and ACTB (housekeeping) controls are also shown. (B) Representative examples of bisulphite sequence analysis. Two electropherograms showing analysis of three CpG sites within the S100A10 promoter from a highly methylated cell line (D384) and an unmethylated cell line (DAOY). Individual CpG sites are underlined. (C) Representative examples of COBRA analysis of S100A4. PCR products from bisulphite-treated cell line DNA were digested with HpyCH4IV, which cuts the product twice when CpG sites are methylated, generating products of 100 bp (shown) and 26 and 16 bp (not shown). Unmethylated control (U) was normal blood DNA, methylated control (M) was universal methylated DNA (Serologicals Corporation, Livingston, UK). (D) Methylation status analysis of CpG residues within the promoter-associated CpG islands of S100A2, S100A6 and S100A10 and first intron of S100A4 in nine medulloblastoma cell lines, determined by bisulphite sequencing and estimation of relative peak heights. S100A4 PCR products representative of different digestion patterns were sequenced and used to assign methylation levels to samples showing equivalent digestion patterns. Filled circles, >75% methylation; half-filled circles, 25–75% methylation; empty circles, <25% methylation; NA, not analysed.
The methylation status of S100A2, S100A4, S100A6 and S100A10 in medulloblastoma cell lines is associated with methylation-dependent transcriptional silencing
S100 genes with confirmed evidence of methylation-dependent upregulation in ⩾2 medulloblastoma cell lines were selected for direct analysis of their DNA methylation status. The methylation status of CpG sites within the promoter/exon 1 regions of S100A6 and S100A2, which have previously been identified as being involved in their epigenetic regulation (Rehman et al, 2005), was established in a panel of nine medulloblastoma cell lines by bisulphite sequencing. Both genes were frequently methylated (in 7 out of 9 and 8 out of 9 lines, respectively). S100A6 displayed extensive methylation in D283 Med, D425 Med, MEB-MED8A, MHH-MED-1, D341 Med, D384 Med and D556 Med; the methylation of S100A6 in D283 Med, D425 Med, MEB-MED8A is in accordance with its observed methylation-dependent transcriptional silencing and re-activation by 5-aza CdR in these cell lines. In contrast, DAOY and UW228-3 were completely unmethylated, correlating with the higher level of expression and the lack of expression change following demethylating treatment, seen in DAOY (Figure 2A and D). S100A2 showed more variable methylation patterns, with methylation ranging from partial methylation at one site (DAOY) to complete methylation at all sites (D283 Med) (Figure 2D). However, the observed methylation patterns again correlated well with methylation-dependent transcriptional silencing (Figure 2A and D), except in the case of D425 Med, which is densely methylated and transcriptionally silenced, but does not undergo significant re-expression following 5 aza CdR treatment, suggesting that methylation-independent mechanisms are involved in its silencing.
Methylation analysis of S100A10 has not been performed previously; however, the promoter and associated CpG island have been characterised. (Huang et al, 2003) Primers were designed using MethPrimer (Li and Dahiya, 2002) to assess the methylation status of a region of the S100A10 promoter-associated CpG island (400–652 bp relative to the transcriptional start) site by bisulphite sequencing (see Figure 2B and D). This region was methylated at multiple CpG residues in 7 out of 9 cell lines, including all three cell lines, which showed evidence of transcriptional silencing and methylation-dependent upregulation of S100A10 following treatment with 5-aza CdR (D283 Med, D425 Med, MEB-MED8A), but was not methylated in DAOY, which showed higher endogenous expression levels and no clear change in expression following demethylating treatment (Figure 2A), indicating that methylation of this region is involved in the transcriptional silencing and epigenetic regulation of S100A10.
S100A4 does not contain a promoter-associated CpG island, but has been shown to be epigenetically regulated by methylation of critical intragenic CpG sites (Rosty et al, 2002). We therefore assessed the methylation status of two CpG sites within its first intron, whose methylation has been previously correlated with expression (Rosty et al, 2002). Methylation was assessed by COBRA (Figure 1C), with representative examples (n=3) of each digestion pattern confirmed by bisulphite sequencing. These two CpG sites were fully methylated in 6 out of 9 cell lines, and their methylation status correlated well with the expression patterns observed; reduced expression and 5-aza CdR-associated upregulation was seen in methylated cell lines (D283 Med, D425 Med), but not in unmethylated cell lines (MEB-MED8A, DAOY) (Figure 1; Figure 2A and D). These data were reinforced by extension of RT–PCR expression analysis of S100A4 to two further cell lines (MHH-MED1, D556 Med), which produced the anticipated expression patterns; unmethylated MHH-MED1 showed higher endogenous expression and no evidence of methylation-dependent regulation, whereas the methylated D556 Med cell line displayed transcriptional silencing and methylation-dependent upregulation following 5-aza CdR treatment (data not shown).
Methylation status of S100 genes in the normal cerebellum
Gene-specific methylation can be a feature of normal somatic tissues (Strathdee et al, 2004). It is therefore crucial to establish the methylation status of candidate genes in normal non-neoplastic tissues before any role for aberrant methylation patterns in tumourigenesis can be assessed. Six normal cerebellar samples, comprising fetal, infant and adult examples were therefore assessed for the methylation status of all four S100 genes under investigation (Figure 3). For S100A4, S100A6 and S100A10, consistent methylation patterns were observed across all samples. S100A6 and S100A10 were somatically unmethylated, with no evidence of methylation of either promoter-associated CpG island observed, with the exception of one sample which showed minimal methylation (<25%) at 2 out of 13 sites in the S100A10 CpG island. In contrast, S100A4 was somatically methylated, showing evidence of high-level methylation at both CpG residues assessed in all normal cerebellar samples. S100A2 also showed evidence of methylation in the normal cerebellum, although the methylation patterns detected were variable between samples, both in terms of the number of methylated CpG residues and the extent of methylation observed at these residues.

Methylation status of S100 genes in primary medulloblastomas and the normal cerebellum. Analysis of methylation at CpG residues within the promoter-associated CpG islands of S100A2, S100A6 and S100A10 and the first intron of S100A4, is shown in normal cerebella and selected medulloblastoma primary tumours, determined by bisulphite sequencing and estimation of relative peak heights (as described in Figure 2). Filled circles, >75% methylation; half-filled circles, 25–75% methylation; empty circles, <25% methylation.
Aberrant methylation patterns identify roles for S100 genes in medulloblastoma pathogenesis
S100 gene methylation patterns were next investigated in a series of primary medulloblastomas, and, together with patterns previously observed for medulloblastoma cell lines (Figure 2), were assessed against the normal methylation patterns observed for each gene in the cerebellum, for evidence of altered methylation states. In the majority of cases, methylation patterns observed in medulloblastomas corresponded to the methylation status of the normal cerebellum (Figure 3). For S100A6 and S100A10, the majority of medulloblastomas were unmethylated (35 out of 40 and 31 out of 35 of analysable samples, 87.5 and 88.5% respectively); however five tumours (12.5%) and seven cell lines (78%) showed evidence of hypermethylation of the S100A6 promoter relative to the normal cerebellum. S100A10 was hypermethylated in four tumours (11.4%) and seven cell lines (78%), relative to the normal cerebellum. Direct analysis of expression in tumours was not possible, as RNA was not available for the majority of the cohort; however, the hypermethylation of S100A10 observed in primary tumours is of a comparable level to that seen in the cell lines, consistent with its epigenetic transcriptional inactivation and candidate tumour suppressor role in disease pathogenesis (Figures 2A, D and 3). The hypermethylation observed for S100A6 was less pronounced in primary tumours (affecting a limited number of CpG residues) than cell lines and further work is needed to determine whether this level of methylation leads to transcriptional silencing.
In contrast, all medulloblastomas showed some evidence of methylation of the CpG regions assessed in S100A4 and S100A2. Whereas the normal cerebellum and the majority of primary medulloblastomas (34 out of 41) and medulloblastoma cell lines (6 out of 9) demonstrated complete methylation (>75%) of the S100A4 intronic CpG sites analysed, a subset of primary tumours (17%; 7 out of 41) and cell lines (33%; 3 out of 9) were undermethylated in comparison, displaying either partial methylation or no evidence of methylation at each site (Figures 2D and 3). Considered alongside the methylation-dependent transcriptional silencing observed for S100A4 in medulloblastoma cell lines (Figure 2A), these findings support (i) the somatic hypermethylation-associated transcriptional silencing of S100A4 in the normal cerebellum, and (ii) tumour-specific hypomethylation of S100A4 in a subset of medulloblastomas, consistent with elevated expression and a pro-tumourigenic role.
Patterns of methylation observed for S100A2 were complex; evidence of methylation was observed in all primary tumours tested; however, considerable variation was seen in both the number of CpG residues affected and the extent of methylation observed at individual CpG residues (Figure 3). A comparable incidence and variability of patterns of methylation were also detected in medulloblastoma cell lines and the normal cerebellum, and our data do not therefore provide any clear evidence to indicate the existence of aberrant tumour-specific patterns of S100A2 methylation in medulloblastomas (Figures 2D and 3). In view of the methylation-dependent transcriptional silencing associated with a range of S100A2 methylation states in medulloblastoma cell lines (Figure 2A and D), our data suggest that S100A2 methylation in the normal cerebellum is associated with its somatic epigenetic transcriptional silencing, but more detailed investigations are now required to explore any role for the epigenetic deregulation of S100A2 in medulloblastoma.
Clinicopathological significance of S100 methylation events in medulloblastomas
Tumour-specific methylation patterns detected for the S100A4, S100A6 and S100A10 genes were next assessed against basic clinical and pathological information available for all tumours (age, sex, histopathological subtype), to make a preliminary assessment of any clinicopathological significance in disease development. S100A6 methylation showed a statistically significant association with the large cell/anaplastic histopathological morpho-phenotype (50%; (3 out of 6) of large cell/anaplastic tumours vs 8% (2 out of 25) of classic and 0% (0 out of 9) of nodular/desmoplastic tumours; P=0.026 by Fisher's exact test). No further significant associations were found for any gene.
Discussion
This study demonstrates a role for the epigenetic deregulation of three members of the S100 gene family, S100A4, S100A6 and S100A10, in medulloblastoma tumourigenesis. Furthermore, our data provide evidence for the control of S100 gene expression in the normal cerebellum by gene-specific somatic methylation events, and highlight the importance of assessing epigenetic events in tumour development in the context of normal tissue-specific methylation patterns.
Epigenetic deregulation of gene expression plays an important role in the development of medulloblastoma, and studies of DNA methylation have identified a series of putative tumour suppressor genes for this disease; however, analysis has so far been restricted to a limited number of candidate genes (Lindsey et al, 2005). In this study, we therefore used a pharmacological expression reactivation approach, to allow a more global analysis of gene expression changes using expression microarrays. Using this analysis to assess members of the S100 gene family for evidence of methylation-dependent regulation, we have successfully identified four S100 family members which showed upregulation in multiple cell lines following demethylation treatment, and were subsequently shown to be associated with specific DNA methylation events. Our findings thus strongly support the value of this experimental approach for the identification of novel methylated genes relevant to both normal development and oncogenesis, and provide important precedent for the epigenetic deregulation of S100 gene expression in medulloblastoma. Moreover, the role of methylation events in the regulation of S100 gene expression in the normal and malignant cerebellum may yet be more extensive than recognised currently. Our investigations of S100 gene DNA methylation status were limited to four genes that displayed evidence of methylation-dependent regulation in multiple cell lines. Additional S100 genes either (i) showed evidence of methylation-dependent regulation in a more limited number of cell lines (<2; S100A3, S100A11, S100P and S100G), and were therefore not investigated further in the present study, or (ii) were not represented on the U133A expression microarray (e.g., S100A15), suggesting that more systematic investigations are now required to investigate any roles for DNA methylation events in the epigenetic regulation of further S100 family members in medulloblastoma.
Two of the genes identified by this approach, S100A6 and S100A10, were each hypermethylated in a proportion of both medulloblastoma cell lines and primary tumours (each in ∼12% of cases), but not in the normal cerebellum. The higher frequency of hypermethylation observed for both genes in cell lines than tumours may reflect either a limited representation of the clinical and pathological diversity of primary medulloblastomas by these cell lines (Langdon et al, 2006), or an involvement of culture-related de novo methylation events (Jones et al, 1990; Pantoja et al, 2005). Nonetheless, for both genes a significant frequency of tumour-specific methylation was observed in primary tumours, which was associated with transcriptional silencing in medulloblastoma cell lines, indicating epigenetic inactivation and candidate tumour suppressor roles for S100A6 and S100A10 in primary medulloblastoma development. S100A6 has been shown to be expressed in a cell type-specific manner in the brain, with expression present in subsets of neurons including granule cells of the cerebellum (Filipek et al, 1993). Consistent with this observation, the S100A6 promoter was unmethylated in the normal cerebellum in the present study. In other tumour types, S100A6 has also been reported to be hypermethylated in prostate cancer (Rehman et al, 2005). S100A10 is also expressed in the brain (Zimmer et al, 2005). To our knowledge, the present study is the first to demonstrate that S100A10 expression can be deregulated by methylation of its promoter-associated CpG island in tumourigenesis. The finding that S100A10 was hypermethylated in medulloblastoma cell lines and tumours was unexpected, as its overexpression has previously been associated with plasminogen-dependent cellular invasiveness in fibrosarcoma and colorectal cancer cells (Choi et al, 2003; Zhang et al, 2004). However, S100A10 can interact with a variety of proteins and is involved in diverse processes (reviewed in Santamaria-Kisiel et al, 2006), and studies to clarify its role in medulloblastoma tumourigenesis are now required.
In contrast, we have demonstrated consistent evidence of methylation of S100A2 and S100A4 in the normal cerebellum. The methylation patterns observed for both genes are comparable to those associated with transcriptional silencing in medulloblastoma cell lines, suggesting that expression of these genes is reduced or silenced epigenetically in the normal non-neoplastic cerebellum. There is growing evidence that a number of genes are epigenetically silenced by somatic methylation in normal tissues (reviewed in Strathdee et al, 2004). Previously we have shown evidence of partial methylation of CASP8, HIC1 and EDNRB (Lindsey et al, 2004) in non-neoplastic cerebella. As less than 50 genes have been analysed for methylation in medulloblastoma and the normal cerebellum (Lindsey et al, 2005); this raises the possibility that somatic methylation in the normal cerebellum may be a feature of a significant number of genes, although for some it may be restricted to specific cell types within the cerebellum. In addition to cell type-specific methylation (Lesniak et al, 2000; Rosty et al, 2002; Sato et al, 2004), members of the S100 gene family have been shown to undergo temporal methylation changes during development, for example S100β, becomes demethylated during astrocyte development in the mouse brain correlating with the time when its expression commences at E14 (Namihira et al, 2004). This raises the possibility that aberrant methylation in embryonal tumours such as medulloblastoma may reflect a failure of developmentally regulated methylation changes. The identification of normal tissue-specific methylation in the non-neoplastic cerebellum emphasizes the importance of including relevant somatic tissue controls in medulloblastoma methylation studies (Lindsey et al, 2004; Lindsey et al, 2005).
S100A4 showed complete methylation of the intronic CpG sites analysed in all normal cerebellar samples, consistent with its epigenetic transcriptional silencing (see above). S100A4 has been previously shown to be methylated in other normal tissues, including pancreatic tissue (Rosty et al, 2002). Three medulloblastoma cell lines were unmethylated for S100A4, and were associated with higher gene expression unaffected by demethylation treatment (in contrast to methylated lines, which were associated with methylation-dependent transcriptional silencing), suggesting that they had undergone selection for hypomethylation and overexpression of S100A4 during the tumourigenic process. No tumour samples were completely unmethylated at S100A4; however, 7 out of 41 (17%) of tumours displayed reduced levels of methylation compared to the normal cerebellum. S100A4 hypomethylation is also a feature of other cancers, including pancreatic and colon adenocarcinoma (Nakamura and Takenaga, 1998; Rosty et al, 2002). S100A4 promotes angiogenesis and extracellular matrix degradation through its upregulation of specific matrix metalloproteases (reviewed in Helfman et al, 2005) and high levels of S100A4 expression are associated with metastatic progression in a wide range of cancers including medulloblastoma (Hernan et al, 2003). Current findings are therefore consistent with a role for the oncogenic activation of S100A4 by hypomethylation-associated expression upregulation in medulloblastoma tumourigenesis.
S100A2 has been postulated in certain studies to play a suppressor role in tumour development, as it shows reduced expression in a range of cancer types, while other investigators have been unable to show any clear difference in methylation patterns between tumours and normal tissue (Rehman et al, 2005). S100A2 methylation was detected in all normal cerebella, cell line and tumour samples tested; however, significant variability in methylation patterns and levels were observed in all sample groups. While these methylation patterns were associated with transcriptional silencing in medulloblastoma cell lines, suggesting that S100A2 is epigenetically silenced by hypermethylation in the normal cerebellum, no clear evidence of aberrant tumour-specific epigenetic regulation in medulloblastoma development was found, and more detailed examination of extended tumour cohorts are now required to further discern any role in medulloblastoma.
The majority of the S100 genes, including S100A2, S100A4, S100A6 and S100A10 are organised in a cluster on chromosome 1q21.3 (Figure 1). Recent studies have provided precedent for epigenetic silencing across an entire chromosome band in colorectal cancer (Frigola et al, 2006); however, this does not appear to be the case for the S100 gene cluster in medulloblastoma. Microarray analysis of S100 gene expression changes following demethylation treatment of medulloblastoma cell lines (Figure 1) shows that there is no positional clustering of upregulated genes and the expression of many genes in the cluster remains unchanged. Furthermore, analysis of DNA methylation changes shows that some medulloblastoma cell lines (e.g., MHH-MED1, MEB-MED8A) show both hypermethylation (e.g., of S100A6) and hypomethylation (e.g., of S100A4) of genes in close physical proximity, suggesting that epigenetic remodelling appears to be localised within the S100 cluster, involving gene-specific hypermethylation and hypomethylation events. Furthermore, our findings indicate that S100 gene methylation patterns may have clinical significance and potential utility as medulloblastoma biomarkers. Based on an initial analysis in our limited cohort, methylation of S100A6 was significantly associated with the more aggressive large cell/anaplastic histopathological variant (Giangaspero et al, 2000; McManamy et al, 2003), and investigations in large uniformly treated clinical trials cohorts are now required to investigate these relationships further.
A proportion of the CpG methylation found in normal tissue contributes to the silencing of transposable elements and foreign DNA sequences and the cancer genome is known to be globally hypomethylated at these sites, which contributes towards its overall genomic instability (Ehrlich, 2002). However, the role of methylation in the control of expression of individual genes in normal tissues has not been studied in detail and the majority of studies of the methylation status of individual genes in tumours have focused on hypermethylation events leading to the epigenetic inactivation of putative tumour suppressor genes. This has led to the advocation of demethylating agents as anti-cancer therapeutics (reviewed in Strathdee and Brown, 2002). The growing evidence that a number of genes are epigenetically silenced by methylation in normal tissues (reviewed in Strathdee et al, 2004) and that some of these genes such as S100A4 may be tumour promoting, now forces a reconsideration of our understanding of how alterations in the epigenome contribute to cancer development, and the rationale and mechanisms underlying the therapeutic efficacy of such ‘epigenetic’ therapies.
The epigenetic regulation of the S100 gene family in medulloblastoma and the normal cerebellum illustrates the complex nature of the epigenome, with different family members being subject to both tissue and tumour-specific epigenetic control by DNA methylation, involving both hypomethylation and hypermethylation events which occur during tumourigenesis. Further work is now needed to (i) understand the role of S100 gene family deregulation in medulloblastoma development and (ii) determine the relative contribution of hypo and hypermethylation events in the development of this tumour and its implications for cancer therapeutics.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Arumugam T, Simeone DM, Van Golen K, Logsdon CD (2005) S100P promotes pancreatic cancer growth, survival, and invasion. Clin Cancer Res 11: 5356–5364
Choi KS, Fogg DK, Yoon CS, Waisman DM (2003) p11 regulates extracellular plasmin production and invasiveness of HT1080 fibrosarcoma cells. FASEB J 17: 235–246
Donato R (2003) Intracellular and extracellular roles of S100 proteins. Microsc Res Tech 60: 540–551
Ehrlich M (2002) DNA methylation in cancer: too much, but also too little. Oncogene 21: 5400–5413
Emberley ED, Murphy LC, Watson PH (2004) S100 proteins and their influence on pro-survival pathways in cancer. Biochem Cell Biol 82: 508–515
Feng G, Xu X, Youssef EM, Lotan R (2001) Diminished expression of S100A2, a putative tumor suppressor, at early stage of human lung carcinogenesis. Cancer Res 61: 7999–8004
Filipek A, Puzianowska M, Cieslak B, Kuznicki J (1993) Calcyclin--Ca(2+)-binding protein homologous to glial S-100 beta is present in neurones. Neuroreport 4: 383–386
Frigola J, Song J, Stirzaker C, Hinshelwood RA, Peinado MA, Clark SJ (2006) Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat Genet 38: 540–549
Garrett SC, Varney KM, Weber DJ, Bresnick AR (2006) S100A4, a mediator of metastasis. J Biol Chem 281: 677–680
Giangaspero F, Bigner S, Kleihues P, Pietsch T, Trojanowski JQ (2000) Medulloblastoma. In World Health Organization Classification of Tumours. Pathology and Genetics - Tumours of the Nervous System. Kleihues P, Cavenee W (ed) pp 129–137. Lyon: IARC
Heizmann CW, Fritz G, Schafer BW (2002) S100 proteins: structure, functions and pathology. Front Biosci 7: d1356–d1368
Heizmann CW (2005) The importance of calcium-binding proteins in childhood diseases. J Pediatr 147: 731–738
Helfman DM, Kim EJ, Lukanidin E, Grigorian M (2005) The metastasis associated protein S100A4: role in tumour progression and metastasis. Br J Cancer 92: 1955–1958
Hernan R, Fasheh R, Calabrese C, Frank AJ, Maclean KH, Allard D, Barraclough R, Gilbertson RJ (2003) ERBB2 up-regulates S100A4 and several other prometastatic genes in medulloblastoma. Cancer Res 63: 140–148
Horikoshi T, Danenberg KD, Stadlbauer TH, Volkenandt M, Shea LC, Aigner K, Gustavsson B, Leichman L, Frosing R, Ray M, Gibson NW, Spears CP, Danenberg PV (1992) Quantitation of thymidylate synthase, dihydrofolate reductase, and DT-diaphorase gene expression in human tumors using the polymerase chain reaction. Cancer Res 52: 108–116
Huang X, Pawliczak R, Yao XL, Madara P, Alsaaty S, Shelhamer JH, Cowan MJ (2003) Characterization of the human p11 promoter sequence. Gene 310: 133–142
Jones PA, Wolkowicz MJ, Rideout III WM, Gonzales FA, Marziasz CM, Coetzee GA, Tapscott SJ (1990) De novo methylation of the MyoD1 CpG island during the establishment of immortal cell lines. Proc Natl Acad Sci USA 87: 6117–6121
Langdon JA, Lamont JM, Scott DK, Dyer S, Prebble E, Bown N, Grundy RG, Ellison DW, Clifford SC (2006) Combined genome-wide allelotyping and copy number analysis identify frequent genetic losses without copy number reduction in medulloblastoma. Genes Chromosomes Cancer 45: 47–60
Lesniak W, Swart GW, Bloemers HP, Kuznicki J (2000) Regulation of cell specific expression of calcyclin (S100A6) in nerve cells and other tissues. Acta Neurobiol Exp (Wars) 60: 569–575
Li LC, Dahiya R (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics 18: 1427–1431
Lindsey JC, Anderton JA, Lusher ME, Clifford SC (2005) Epigenetic events in medulloblastoma development. Neurosurg Focus 19: E10
Lindsey JC, Lusher ME, Anderton JA, Bailey S, Gilbertson RJ, Pearson AD, Ellison DW, Clifford SC (2004) Identification of tumour-specific epigenetic events in medulloblastoma development by hypermethylation profiling. Carcinogenesis 25: 661–668
Lusher ME, Lindsey JC, Latif F, Pearson AD, Ellison DW, Clifford SC (2002) Biallelic epigenetic inactivation of the RASSF1A tumor suppressor gene in medulloblastoma development. Cancer Res 62: 5906–5911
Marenholz I, Heizmann CW, Fritz G (2004) S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem Biophys Res Commun 322: 1111–1122
McManamy CS, Lamont JM, Taylor RE, Cole M, Pearson AD, Clifford SC, Ellison DW (2003) Morphophenotypic variation predicts clinical behavior in childhood non-desmoplastic medulloblastomas. J Neuropathol Exp Neurol 62: 627–632
Nakamura N, Takenaga K (1998) Hypomethylation of the metastasis-associated S100A4 gene correlates with gene activation in human colon adenocarcinoma cell lines. Clin Exp Metastasis 16: 471–479
Namihira M, Nakashima K, Taga T (2004) Developmental stage dependent regulation of DNA methylation and chromatin modification in a immature astrocyte specific gene promoter. FEBS Lett 572: 184–188
Pantoja C, de Los Rios L, Matheu A, Antequera F, Serrano M (2005) Inactivation of imprinted genes induced by cellular stress and tumorigenesis. Cancer Res 65: 26–33
Rehman I, Cross SS, Azzouzi AR, Catto JW, Deloulme JC, Larre S, Champigneuille J, Fromont G, Cussenot O, Hamdy FC (2004) S100A6 (Calcyclin) is a prostate basal cell marker absent in prostate cancer and its precursors. Br J Cancer 91: 739–744
Rehman I, Cross SS, Catto JW, Leiblich A, Mukherjee A, Azzouzi AR, Leung HY, Hamdy FC (2005) Promoter hyper-methylation of calcium binding proteins S100A6 and S100A2 in human prostate cancer. Prostate 65: 322–330
Rosty C, Ueki T, Argani P, Jansen M, Yeo CJ, Cameron JL, Hruban RH, Goggins M (2002) Overexpression of S100A4 in pancreatic ductal adenocarcinomas is associated with poor differentiation and DNA hypomethylation. Am J Pathol 160: 45–50
Santamaria-Kisiel L, Rintala-Dempsey AC, Shaw GS (2006) Calcium-dependent and -independent interactions of the S100 protein family. Biochem J 396: 201–214
Sato N, Fukushima N, Matsubayashi H, Goggins M (2004) Identification of maspin and S100P as novel hypomethylation targets in pancreatic cancer using global gene expression profiling. Oncogene 23: 1531–1538
Strathdee G, Brown R (2002) Epigenetic cancer therapies: DNA methyltransferase inhibitors. Expert Opin Investig Drugs 11: 747–754
Strathdee G, Sim A, Brown R (2004) Control of gene expression by CpG island methylation in normal cells. Biochem Soc Trans 32: 913–915
Wang G, Platt-Higgins A, Carroll J, de Silva Rudland S, Winstanley J, Barraclough R, Rudland PS (2006) Induction of metastasis by S100P in a rat mammary model and its association with poor survival of breast cancer patients. Cancer Res 66: 1199–1207
Wicki R, Franz C, Scholl FA, Heizmann CW, Schafer BW (1997) Repression of the candidate tumor suppressor gene S100A2 in breast cancer is mediated by site-specific hypermethylation. Cell Calcium 22: 243–254
Xiong Z, Laird PW (1997) COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res 25: 2532–2534
Zhang L, Fogg DK, Waisman DM (2004) RNA interference-mediated silencing of the S100A10 gene attenuates plasmin generation and invasiveness of Colo 222 colorectal cancer cells. J Biol Chem 279: 2053–2062
Zimmer DB, Chaplin J, Baldwin A, Rast M (2005) S100-mediated signal transduction in the nervous system and neurological diseases. Cell Mol Biol (Noisy-le-grand) 51: 201–214
Acknowledgements
This work was funded by the Samantha Dickson Brain Tumour Trust, Charlies challenge and the Katie Trust. Cell lines D384 Med, D425 Med were kind gifts from Dr D Bigner (Duke University, Durham, NC, USA). MHH-MED-1 and MEB-MED8A were kind gifts from Professor T Pietsch (University of Bonn Medical Centre, Bonn, Germany). UW228-2 and UW228-3 were kind gifts from Dr J Silber (University of Washington, Seattle, WA, USA). The remaining cell lines were obtained from the ATCC (Manassas, VA, USA). Three normal cerebellar DNAs were kind gifts from Dr M Fruhwald (University of Munster, Munster, Germany). Normal blood DNA controls were obtained from the North Cumbria Community Genetics Project, UK.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Lindsey, J., Lusher, M., Anderton, J. et al. Epigenetic deregulation of multiple S100 gene family members by differential hypomethylation and hypermethylation events in medulloblastoma. Br J Cancer 97, 267–274 (2007). https://doi.org/10.1038/sj.bjc.6603852
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6603852
Keywords
This article is cited by
-
S100A14 suppresses metastasis of nasopharyngeal carcinoma by inhibition of NF-kB signaling through degradation of IRAK1
Oncogene (2020)
-
P11 promoter methylation predicts the antidepressant effect of electroconvulsive therapy
Translational Psychiatry (2018)
-
Epigenome-wide association study reveals decreased average methylation levels years before breast cancer diagnosis
Clinical Epigenetics (2015)
-
Irradiation-induced angiogenesis is associated with an MMP-9-miR-494-syndecan-1 regulatory loop in medulloblastoma cells
Oncogene (2014)
-
Maintaining the unmethylated state
Clinical Epigenetics (2013)
