
Abstract
Inflammation-related changes in the concentrations of kynurenine pathway metabolites occur in depression secondary to medical conditions but are not firmly established in primary mood disorders. Reductions in hippocampal and amygdalar volume that putatively reflect dendritic atrophy are widely reported in major depressive disorder (MDD). Here we tested whether the relative serum concentrations of putatively neuroprotective (kynurenic acid (KA)) and neurotoxic (3-hydroxykynurenine (3HK) and quinolinic acid (QA)) kynurenine pathway metabolites were altered in primary MDD and whether these metabolites were associated with hippocampal and amygdalar volume. A total of 29 moderately to severely depressed unmedicated subjects who met DSM-IV criteria for MDD and 20 healthy controls (HCs) completed a structural MRI scan and provided blood sample for kynurenine metabolite analysis, performed using high-performance liquid chromatography with tandem mass spectrometry. Cytokine concentrations were measured with ELISA and gray matter volumes were measured with the automated segmentation software, FreeSurfer. An a priori defined variable of interest, the KA/QA ratio, a putative neuroprotective index, trended lower in the MDD versus the HC group and correlated negatively with anhedonia but positively with the total hippocampal and amygdala volume in the MDD subjects. The post hoc data reduction methods yielded three principal components. Component 1 (interleukin-1 receptor antagonist, QA, and kynurenine) was significantly elevated in MDD participants versus the HCs, whereas component 2 (KA, tryptophan, and kynurenine) was positively correlated with hippocampal and amygdala volume within the MDD group. Our results raise the possibility that an immune-related imbalance in the relative metabolism of KA and QA predisposes to depression-associated dendritic atrophy and anhedonia.
Similar content being viewed by others
INTRODUCTION
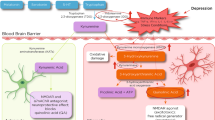
Major depressive disorder (MDD) has been associated with reductions in hippocampal and amygdalar volume that are thought to reflect dendritic atrophy occurring in the context of excitotoxicity, decreased neurogenesis, and impaired neurotrophic function (Koolschijn et al, 2009; Pereira et al, 2007; Petrik et al, 2012; Savitz and Drevets, 2009; Stockmeier et al, 2004). At least a subgroup of individuals with MDD also display putative signs of inflammation such as elevated circulating concentrations of C-reactive protein (CRP), interleukin-6 (IL-6), and interleukin-1β (IL-1β) (Dowlati et al, 2010; Howren et al, 2009). Inflammation can also lead to the activation of the tryptophan (TRP)-degrading enzyme indoleamine 2,3 dioxygenase (IDO), ultimately increasing the formation of kynurenine (KYN) metabolites, including kynurenic acid (KA), a putatively neuroprotective antagonist of N-methyl-D-aspartate (NMDA) receptors that also decreases glutamate levels via inhibition of α7 nicotinic receptors 3-hydroxykynurenine (3HK), a free radical generator, and quinolinic acid (QA), an NMDA receptor agonist that also exerts neurotoxic effects via lipid peroxidation, and disruption of the blood–brain barrier (Dantzer et al, 2011; Maes et al, 2011; Perkins and Stone, 1982; Schwarcz et al, 2012; Schwarcz et al, 1983) (Figure 1).

Main branches of the kynurenine pathway. Each box represents a metabolite resulting from the oxidation of tryptophan. Putative neurotoxic metabolites are colored red whereas KA, which is putatively neuroprotective, is colored green. The black italicized text shows the enzymes that catalyze each step in the metabolic pathway. The blue and red stars indicate that the metabolite is able to cross the blood–brain barrier. The effects on NMDA receptor activity listed for some metabolites have been established in vitro, but the extent to which the metabolite concentrations achieve sufficiently high levels in vivo to impact the function of this receptor system remains unclear. NMDA, N-methyl-D-aspartate glutamate receptor. A full text version of this figure is available at Neuropsychopharmacology Journal online.
A causal association between neuroactive cytokine release and changes in brain structure and/or function has been reported in putative animal models of depression. IL-1 plays a key role in the development of depression-like behavior and in the dendritic atrophy, synaptic loss, suppressed neurogenesis, and decreased brain-derived neurotrophic factor (BDNF) concentrations that are apparent in the hippocampi of rodents subjected to various inflammatory challenges (Barrientos et al, 2009; Goshen et al, 2008; Koo and Duman, 2008; Williamson and Bilbo, 2013; Yirmiya and Goshen, 2011). The effect of IL-1 may be partly mediated by the production of neurotoxic kynurenine metabolites. IL-1 increases the expression of IDO, kynurenine 3-monooxygenase (KMO), and kynureninase—that is, the enzymes that produce potentially neurotoxic kynurenine metabolites (Zunszain et al, 2012). Moreover, IL-1-driven synthesis of 3HK decreases human hippocampal cell neurogenesis in vitro (Zunszain et al, 2012). Consistent with these data, IDO transcripts were upregulated in the hippocampi of rats displaying comorbid pain and depression in response to inflammation in the paw, and these comorbid symptoms were IDO dependent (Kim et al, 2012).
Other cytokines also play an important role in activating IDO. Dendritic cells express the highest level of IDO activity in response to interferon-γ (IFN-γ) (Fallarino et al, 2002), thus aiding in the inhibition of the local replication of pathogens. Similarly, IDO is upregulated by the tumor necrosis factor-α (TNF-α) secretion associated with viral infections such as the Epstein–Barr virus (Liu et al, 2014). Kynurenine pathway metabolites are also regulators of T-cell tolerance and maintenance, an effect that occurs in the context of transforming growth factor-β (TGF-β) signaling (Orabona et al, 2012; Puccetti and Grohmann, 2007).
Nevertheless, it is not yet known whether the relationship between inflammation and hippocampal physiology extends to patients with MDD in vivo. Reductions in the gray matter (GM) volume of limbic structures in depressed humans appear to reflect dendritic atrophy, a hypothesis confirmed in subjects with MDD post-mortem (Stockmeier et al, 2004) and more recently in a dual ex vivo MRI and histopathological study of rats exposed to antipsychotic medications (Vernon et al, 2013).
In our study, we therefore hypothesized that reductions in the hippocampal and the amygdalar volumes measured with T1-weighted MRI could be secondary to inflammation and activation of the kynurenine metabolite pathway. If this is the case, these volumes should be associated with increased concentrations of potentially neurotoxic kynurenine metabolites and/or decreased concentrations of potentially neuroprotective kynurenine metabolites. Because the effects of ‘neurotoxic’ metabolites on GM volumes may depend on the corresponding concentration of ‘neuroprotective’ metabolites, we maximized sensitivity by focusing a priori on the ratios of ‘neuroprotective’ to ‘neurotoxic metabolites’—KA versus 3HK (KA/3HK) as well as KA versus QA (KA/QA)—considered ‘neuroprotective indices’ (Johansson et al, 2013; Kocki et al, 2012). In addition, in order to reduce the probability of type I error while maintaining a hypothesis-free approach, we conducted a principal components analysis (PCA) and used the extracted principal components in a post hoc analysis. Here we show that a higher neuroprotective index (KA/QA) and a principal component that was highly associated with KA correlated positively with hippocampal and amygdalar volumes in unmedicated moderately to severely depressed subjects with MDD.
MATERIALS AND METHODS
Subjects
Subjects provided written informed consent after receiving a full explanation of the study procedures and risks, as approved by the local IRB.
All depressed subjects (n=29) and healthy controls (n=20) were interviewed with the Structured Clinical Interview for the DSM-IV-TR (First et al, 1996). In addition, unstructured psychiatric interviews with board-certified psychiatrists were obtained on all depressed participants.
The depressed subjects met DSM-IV-TR criteria for primary MDD in a current major depressive episode and had a Hamilton Depression Rating Scale (HDRS, 24-item) score in the moderately to severely depressed range (Table 1). The MDD patients did not receive any psychotropic medications for at least 3 weeks (8 for fluoxetine) before the blood draw and MRI scanning (mean length of time off medication: 42.3±42.3 months; 9 participants were treatment naive). Exclusion criteria were as follows: serious suicidal ideation or behavior; bipolar disorder; medical conditions or concomitant medications likely to influence CNS or immunological function including cardiovascular, respiratory, endocrine, and neurological diseases; a history of drug or alcohol abuse within 6 months or a history of drug or alcohol dependence within 1 year (DSM-IV-TR criteria), and general MRI exclusion criteria such as magnetic implants or claustrophobia. Details are provided in the Supplementary Data File, including Supplementary Tables S1 and S2.
The healthy control (HC) individuals met the same exclusion criteria except that they had no personal or family (first-degree relatives) history of psychiatric illness assessed using the Structured Clinical Interview for the DSM-IV-TR and the Family Interview for Genetic Studies (FIGS) (Maxwell, 1992).
MRI
The scans were acquired on a 3 Tesla GE Discovery MR750 MRI scanner (GE Medical Systems) with a receive-only 32 elements surface coil brain array (Nova Medical) using a magnetization-prepared, rapid gradient echo (MP-RAGE) pulse sequence with sensitivity encoding (SENSE) optimized for tissue contrast resolution: (TR=5 ms, TE=2.01 ms, FOV 240 × 192 mm; voxel size=0.94 × 0.94 × 0.90 mm; prep=725 ms, delay=1400 ms, flip=8° SENSE acceleration R=2). The automated segmentation program, FreeSurfer (http://surfer.nmr.mgh.harvard.edu/), was used to obtain unbiased GM volumes of the hippocampus, amygdala, and whole brain. The T1-weighted anatomical images were processed using the default analysis settings of FreeSurfer. Visual inspection of the FreeSurfer-based segmentations was performed to confirm accurate segmentation of the hippocampus and amygdala. The accuracy of FreeSurfer-derived volumetric measures estimates have been validated by histological analysis (Rosas et al, 2002) and compare favorably with manual measurements (Kuperberg et al, 2003). Representative examples of the hippocampal and amygdala masks from a single subject are shown in Supplementary Figures S1 and S2.
Kynurenine Pathway Metabolites
A blood sample was obtained from each subject within 3 days of completing the MRI scan. Subjects fasted overnight and the blood sample was drawn between 0800 and 1100 h. Serum samples were collected with BD Vacutainer serum tubes, processed according to the standard BD Vacutainer protocol, and stored at −80 °C.
Concentrations of TRP, KYN, KA, 3HK, and QA were measured blind to diagnosis by Brains Online, LLC (www.brainsonline.org/home). The metabolite concentrations were determined by high-performance liquid chromatography (HPLC) with tandem mass spectrometry (MS/MS) detection using their standard protocols. The lower limit of quantification and the coefficients of variation for the analytes are provided in Supplementary Table S4.
ELISAs
Commercially available colorimetric sandwich ELISA kits were used to quantify plasma levels of BDNF (EMD Millipore, Billerica, MA), IL-6, CRP, and IL-1RA (eBioscience, San Diego, CA) blind to diagnosis. Plasma samples were stored at −80 °C until use and thawed on ice the day of the assays. To remove any precipitate, samples were centrifuged for 15 min at 3000 r.p.m. Each sample was run in duplicate according to the manufacturer’s instructions using the provided reagents. Two control plasma samples were run on each plate to determine interassay variation and were used to normalize the data across plates. The respective mean interassay coefficient of variation and lower detection limit for each kit are as follows: BDNF 6%, 7.8 pg/ml; IL-6 7%, 0.03 pg/ml; CRP 14%, 3 pg/ml; and IL-1RA 2%, 30 pg/ml
Statistical Analysis
We took two approaches to statistical analysis, an a priori approach and an agnostic approach. Regarding the a priori analysis, analysis of variance was used to test for differences in KA/3HK and KA/QA between the HC and MDD groups after controlling for age, sex, and body mass index (BMI). Second, linear regression was used to test for associations between KA/3HK and KA/QA and hippocampal and amygdala volume within the MDD group, controlling for intracranial volume. Analysis of variance was used to test for differences between the MDD and HC groups in hippocampal and amygdala volumes using sex, age, BMI, and intracranial volumes as covariates.
Regarding the agnostic approach, we conducted the PCA on the kynurenine pathway metabolites and other immune markers, followed by linear regression analyses using the extracted principal components as independent variables.
The PCA was performed using Statistical Package for the Social Sciences (SPSS) (V17) (oblimin rotation). The Kaiser–Meyer–Olkin measure was used to verify sampling adequacy for the analysis (KMO=0.54). Bartlett’s test of sphericity χ2=63, 1, p<0.001, indicated that the correlations between kynurenine metabolites and cytokines were sufficiently large for PCA. Three components had eigenvalues over Kaiser’s criterion of 1, and in combination explained 68% of the variance. Table 2 shows the factor loadings after rotation. The items that cluster on the same components suggest that component 1 (IL-1RA, QA, and KYN) represents inflammation-related activation of the QA branch of the kynurenine pathway. Component 2 (KA, TRP, and KYN) appears to represent the activity of the KA branch of the kynurenine pathway, whereas component 3 (3HK, BDNF, and TRP) is more difficult to interpret but conceivably suggests an interaction between BDNF and 3HK.
Analysis of variance with sex, age, and BMI as covariates was used to test for mean differences between the MDD and HC groups in the three principal components derived from the PCA.
In order to test for associations between the principal components and GM volumes of the hippocampus and amygdala, we conducted linear regression analyses with components 1, 2, and 3 as independent variables, intracranial volume as a covariate, and hippocampal volume or amygdala volume as the dependent variables.
RESULTS
After excluding subjects who failed quality control checks of the metabolite analyses or the FreeSurfer segmentations or who had missing data points, 29 MDD subjects and 20 HCs had both valid MRI and metabolite data available for analysis. Correlations between the kynurenine metabolite measurements and the cytokine measurements are provided in Supplementary Table S3.
The MDD group had significantly smaller total hippocampal volumes and total amygdalar volumes than the HCs, although these differences were no longer statistically significant after regressing out sex, age, BMI, and intracranial volumes.
Regarding the a priori analyses, the ratios of KA/3HK and KA/QA, putative neuroprotective indices (Johansson et al, 2013; Kocki et al, 2012), were lower in the MDD group relative to the HCs (KA/3HK: t=2.42, p=0.019; KA/QA: t=2.04, p=0.047). However, after regressing out sex, age, and BMI, these differences no longer remained statistically significant (Figure 2).

Scatterplots showing the distributions of kynurenine pathway metabolites and the principal components in MDD subjects and HCs. (a) Kynurenic acid. (b) 3-hydroxykynurenine. (c) Quinolinic acid. (d) Principal component 1 (IL-1RA, QA, and KYN). After controlling for sex, age, and BMI, the MDD group showed higher levels of component 1 relative to the healthy controls (F=4.5, p=0.039). (e) Principal component 2 (KA, TRP, KYN). (f) Principal component 3 (3HK, BDNF, and TRP). The MDD subjects are represented by blue squares and the HC subjects by pink circles. The black lines show the mean concentration and the error bars show the SEM. A full text version of this figure is available at Neuropsychopharmacology Journal online.
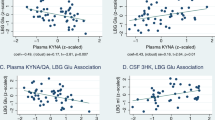
Within the MDD group the KA/QA ratio was positively correlated with larger hippocampal (β-weight=0.42, t=2.5, p=0.022) and amygdala volumes (β-weight=0.37, t=2.2, p=0.041; Figure 3) but the KA/3HK ratio did not predict either hippocampal or amygdala volumes (p>0.1). Neither KA/QA nor KA/3HK was significantly associated with GM volumes in the HC group. Regarding the clinical ratings, KA/QA was significantly correlated (negatively) with the SHAPS score (r=−0.41, p=0.039) but not the HAM-D score (r=−0.25, p=0.183). Higher scores on the SHAPS are indicative of greater anhedonia. KA/3HK was not significantly correlated with the HAM-D or the SHAPS.

(a) Scatterplot showing the correlation between total hippocampal GM volume (X axis) and principal component 2 in subjects with MDD (β-weight=0.43, t=2.4, p=0.024). Hippocampal volumes are provided in cubic millimeters. The linear trendline is displayed in black. (b) Scatterplot showing the correlation between total amygdala GM volume (X axis) and principal component 2 in subjects with MDD (β-weight=0.38, t=2.2, p=0.038). Amygdala volumes are provided in cubic millimeters. (c) Scatterplot showing the correlation between total hippocampal GM volume (X axis) and the serum concentration of the ratio of kynurenic acid (KA) to quinolinic acid (QA) (Y axis) in subjects with MDD (β-weight=0.42, t=2.5, p=0.022). (d) Scatterplot showing the relationship between total amygdalar GM volume (X axis) and the ratio of serum KA to QA (Y axis) in subjects with MDD (β-weight=0.37, t=2.2, p=0.041).
After controlling for sex, age, and BMI, the MDD group showed higher levels of component 1 relative to the HCs (F=4.5, p=0.039; Figure 2). There was no significant group difference in components 2 or 3.
In the combined MDD and HC sample, component 2 was significantly associated with total hippocampal volume (β-weight=0.30, t=2.3, p=0.028) and trended significant for total amygdala volume (β-weight=0.25, t=1.8, p=0.073) after controlling for intracranial volume. The association between component 2 and GM volume was present in the MDD group alone (Figure 3), hippocampus (β-weight=0.43, t=2.4, p=0.024) and amygdala (β-weight=0.38, t=2.2, p=0.038), but was not significant in the HC group alone. There were no significant associations between components 1 and 3 and GM volume in either the MDD or HC groups. There were also no significant associations between the principal components and the clinical rating scales.
DISCUSSION
The main finding of this study is that within the MDD group, both the KA/QA ratio and principal component 2, which largely reflects KA, were associated with larger hippocampal and amygdalar volumes in clinically depressed, unmedicated patients.
The positive association between KA/QA and principal component 2 with hippocampal and amygdala volumes is arguably consistent with a heuristic model that construes dendritic atrophy and/or decreased hippocampal neurogenesis as a consequence of an inflammation-driven decrease in the ratio of KA to QA metabolism. Whereas QA is a neurotoxin that in elevated concentrations can act as an agonist of the NMDA receptor, and 3HK is a potent free radical donor that promotes oxidative stress, and KA acts as an antagonist of both the glycine co-agonist site of the NMDA receptor and the α7 nicotinic receptor situated on glutamatergic neuronal terminals in the hippocampus and other brain regions, reducing glutamate release (Hilmas et al, 2001; Kessler et al, 1989; Perkins and Stone, 1982; Potter et al, 2010; Schwarcz et al, 1983).
Preclinical, in vitro, and post-mortem data demonstrate that elevated concentrations of ‘neurotoxic’ kynurenine metabolites are associated with neuronal damage and/or suppression of neurogenesis—particularly in the hippocampus. For instance, a rat model of pneumococcal meningitis showed that acute infection resulted in the accumulation of 3HK in the hippocampus and that the concentration of 3HK was positively correlated (r=0.5) with the extent of apoptosis in this region (Bellac et al, 2006). Another study demonstrated that neurons exposed to extracellular glutamate show a reduction in dendritic growth that is prevented by administration of KA (Monnerie et al, 2003). IL-1 was shown not only to decrease hippocampal neurogenesis in human hippocampal progenitor cells but also to upregulate the expression of the enzymes, IDO, KMO, and kynureninase and, crucially, treatment with a KMO inhibitor reversed the effects of IL-1 on hippocampal neurogenesis (Zunszain et al, 2012). The reduction in GM volume and the decrease in neuronal density that has been found in some depressed patients post-mortem may be related to higher brain concentrations of microglia-derived QA, as was recently reported in subjects with MDD (Steiner et al, 2008, 2011).
Conversely, psychiatric medication may increase the synthesis of the potentially neuroprotective compound, KA, and/or decrease the synthesis of 3HK. (Kocki et al (2012) reported that 24–48 h of exposure to selective serotonin reuptake inhibitors or tricyclic antidepressant medications stimulated the de novo synthesis of KA and decreased 3HK production in astroglial cultures, thus resulting in an increase in the KA to 3HK ratio. Patients with schizophrenia had lower plasma concentrations of KA and higher plasma concentrations of 3HK than HCs, an effect that was ameliorated by 6 weeks of treatment with antipsychotic treatment such that a significant increase in the KA/3HK ratio was observed after treatment (Myint et al, 2011). Interestingly, atypical antipsychotic medications have been reported to attenuate the typical pattern of hippocampal volume loss observed over time in schizophrenic patients (Koolschijn et al, 2010). In addition, administration of valproic acid, which we previously demonstrated to be associated with larger amygdala volumes in patients with bipolar disorder (BD) (Savitz et al, 2010), reportedly increased the levels of KA in the rat brain (Maciejak et al, 2013). In mice, peripheral administration of low-dose ketamine after injection with lipopolysaccharide (LPS) abrogated the LPS-induced depressive behavior, an effect that was due to ketamine’s antagonistic effect at the NMDA receptor rather than suppression of cytokine release per se (Walker et al, 2013). Interestingly, burn victims with posttraumatic stress disorder (PTSD) who were treated with ketamine were found to have larger hippocampal volumes than nonketamine-treated patients (Winter and Irle, 2004).
Nevertheless, the above model that characterizes KA as ‘neuroprotective’ and 3HK and QA as ‘neurotoxic’ is heuristic and should be interpreted with caution as there are contradictory reports in the literature. For instance, the QA and the KA enzymatic pathways were reported to be equally activated in patients receiving IFN-α treatment, with QA and KA contributing approximately equally to symptom changes (Raison et al, 2010). Consistent with these data, a recent study reported an upregulation of KA in the saliva samples of HCs and schizophrenic subjects after a psychological stress challenge (Chiappelli et al, 2014). Conceivably, the relative effects of KA versus 3HK and QA are dependent on the type of the disorder under study (eg, schizophrenia versus primary MDD), the nature of the inflammatory stimulus, and as yet uncharacterized molecular actors. Reports indicative of elevations of KA in schizophrenia or BD with psychosis (Olsson et al, 2012; Wonodi et al, 2011) may be particularly relevant as elevations in KA concentrations may potentiate dopaminergic neurotransmission potentially explaining the putative link with psychosis (Olsson et al, 2009).
Second, relative to HCs, unmedicated, depressed subjects with primary MDD had higher levels of principal component 1 that is largely reflective of IL-1RA and QA. Although elevations in pro-inflammatory cytokines and kynurenine metabolites such as QA have been widely reported in patients with depression secondary to a general medical condition (Capuron et al, 2003; Raison et al, 2010), the kynurenine pathway has been understudied in primary mood disorders. The increase in principal component 1 is partially consistent with prior reports in the MDD (Gabbay et al, 2010; Myint et al, 2007) and BD (Johansson et al, 2013) literature that are indicative of an increased production of ‘neurotoxic’ versus ‘neuroprotective’ metabolites in depressed patients.
A third finding of interest was the inverse association between KA/QA and the degree of anhedonia in subjects with MDD. The link between inflammation-induced sickness behavior and anhedonia is well characterized (Dantzer et al, 2008; Maes et al, 2012), and a previous study hypothesized that it is primarily MDD patients with melancholic features who show alterations in immune function (Gabbay et al, 2010). As CRP and pro-inflammatory cytokines were not significantly correlated with the SHAPS score, our finding additionally raises the possibility that the relative metabolism of KA to QA might influence the development of anhedonic symptoms over and above the severity of inflammation per se.
Another finding of interest was the composition of principal component 3 (inverse relationship between BDNF and 3HK) given that inflammation-induced synthesis of 3HK decreases human hippocampal cell neurogenesis in vitro (Zunszain et al, 2012) and that BDNF is a key regulator of hippocampal neurogenesis and neuroplasticity (Gray et al, 2013; Vithlani et al, 2013). Future studies of the potential interactions between BDNF and 3HK may provide greater insight.
The neuroanatomical specificity of our findings warrants follow-up. Regression analyses showed no association between KA/QA or component 2 and either whole-brain GM volume or intracranial volume (data not shown), thus raising the possibility that any putative effect of cytokines or kynurenine metabolites on brain structure could be limited to brain regions that are known to display reductions in GM volume in MDD. Future functional imaging may conceivably clarify any differential impact of inflammation on hippocampal-centric and amygdalar-centric circuits.
The main limitation of our study is that we examined kynurenine metabolites in serum rather than in brain, and thus it is theoretically possible that our data are not reflective of central kynurenine metabolite concentrations. KYN and 3HK are, however, able to cross the blood–brain barrier into the brain parenchyma via the neutral amino acid transporter (Fukui et al, 1991), where KYN and 3HK may be metabolized into QA by microglial cells and KYN metabolized into KA by astrocytes (Schwarcz et al, 2012). This finding raises the possibility that peripheral measures of kynurenine pathway metabolites may be at least partially reflective of metabolite concentrations within the brain parenchyma. In support of this hypothesis, Raison et al (2010) reported that INFα-treated hepatitis C patients showed both plasma and CSF increases in KYN and QA that correlated with depressive symptoms. Furthermore, peripheral administration of KYN has been shown to induce behavioral analogs of depression in rodents (O'Connor et al, 2009), and Laugeray et al (2011) reported that mice subjected to chronic mild stress showed a peripheral increase in kynurenine pathway activity that was inversely correlated with KA concentration in the amygdala.
Second, this was a cross-sectional study that was designed to investigate the relationship between kynurenine pathway metabolites and pro-inflammatory cytokines and GM volume of two regions widely implicated in MDD: the hippocampus and amygdala. The data reported here are correlative in nature and, unlike some of the animal studies cited above, do not provide information concerning cause and effect relationships.
Third, because we were only able to measure circulating levels of kynurenine pathway metabolites, it remains unclear whether the hippocampal and amygdalar concentrations of KA, 3HK, and QA in the brains of the volunteers were sufficient to affect NMDA or α7 nicotinic receptor function.
In sum, our results raise the possibility that dysregulation of the innate immune system contributes to the neuromorphometric abnormalities observed in some patients with MDD. The potential existence of a positive association between the peripheral KA/QA ratio and principal component 2 and hippocampal and amygdalar GM volume is of both theoretical and clinical interest. Firstly, it raises the possibility that at least with respect to the kynurenine pathway, peripheral concentrations of metabolites may be reflective of depression-related changes in brain structure. Second, it has been hypothesized by other researchers that immune dysregulation predisposes to mood disorders via its effect on glutamatergic neurotransmission. In other words, abnormal NMDA receptor signaling may be the unifying mechanism underlying the glutamate and inflammation hypotheses of depression (Miller, 2013; Steiner et al, 2012; Walker et al, 2013). Although we cannot demonstrate that KA and QA are at high enough physiological concentrations in the hippocampus and amygdala to affect glutamate release, the correlations between the neuroprotective ratios and GM volumes reported here suggest that the potential effects of KA and QA on glutamatergic neurotransmission are worthy of future study. Third, our results support the hypothesis that pharmacological agents such as KMO inhibitors (the KMO enzyme metabolizes KYN into 3HK) that shunt the catabolism of KYN away from 3HK and toward KA may exert neuroprotective and/or antidepressant effects and therefore may constitute novel targets for drug development (Haroon et al, 2012; Stone and Darlington, 2002).
FUNDING AND DISCLOSURE
Jonathan Savitz has received research funding from Janssen Pharmaceuticals for an independent study. Wayne C Drevets is an employee of Janssen Pharmaceuticals of Johnson & Johnson (Titusville, NJ, USA) and has received within the past 3 years salary from University of Oklahoma Health Sciences Center, and lecture honoraria or consulting fees from Johns-Hopkins University, The University of Michigan, University of Illinois at Chicago, University of Kansas-Wichita, Washington University School of Medicine, St Louis, The Taiwanese Society of Biological Psychiatry and Neuropsychopharmacology, Janssen Pharmaceuticals, and Myriad/ Rules Based Medicine. Dr Dantzer has received consulting fees from Ironwood Pharma (Cambridge, MA), and an honorarium from Pfizer, France. The other authors declare no conflict of interest.
References
Barrientos RM, Frank MG, Hein AM, Higgins EA, Watkins LR, Rudy JW et al (2009). Time course of hippocampal IL-1 beta and memory consolidation impairments in aging rats following peripheral infection. Brain Behav Immun 23: 46–54.
Bellac CL, Coimbra RS, Christen S, Leib SL (2006). Pneumococcal meningitis causes accumulation of neurotoxic kynurenine metabolites in brain regions prone to injury. Neurobiol Dis 24: 395–402.
Capuron L, Neurauter G, Musselman DL, Lawson DH, Nemeroff CB, Fuchs D et al (2003). Interferon-alpha-induced changes in tryptophan metabolism. relationship to depression and paroxetine treatment. Biol Psychiatry 54: 906–914.
Chiappelli J, Pocivavsek A, Nugent KL, Notarangelo FM, Kochunov P, Rowland LM et al (2014). Stress-induced increase in kynurenic acid as a potential biomarker for patients with schizophrenia and distress intolerance. JAMA Psychiatry 71: 761–768.
Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9: 46–56.
Dantzer R, O'Connor JC, Lawson MA, Kelley KW (2011). Inflammation-associated depression: from serotonin to kynurenine. Psychoneuroendocrinology 36: 426–436.
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK et al (2010). A meta-analysis of cytokines in major depression. Biol Psychiatry 67: 446–457.
Fallarino F, Vacca C, Orabona C, Belladonna ML, Bianchi R, Marshall B et al (2002). Functional expression of indoleamine 2,3-dioxygenase by murine CD8 alpha(+) dendritic cells. Int Immunol 14: 65–68.
First MB, Spitzer RL, Gibbon M, Williams JBW (1996) Structured Clinical Interview for DSM-IV Axis I Disorders, Clinician Version (SCID-CV). American Psychiatric Press: Washington, DC.
Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith QR (1991). Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem 56: 2007–2017.
Gabbay V, Klein RG, Katz Y, Mendoza S, Guttman LE, Alonso CM et al (2010). The possible role of the kynurenine pathway in adolescent depression with melancholic features. J Child Psychol Psychiatry 51: 935–943.
Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T et al (2008). Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry 13: 717–728.
Gray JD, Milner TA, McEwen BS (2013). Dynamic plasticity: the role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience 239: 214–227.
Haroon E, Raison CL, Miller AH (2012). Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 37: 137–162.
Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX (2001). The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci 21: 7463–7473.
Howren MB, Lamkin DM, Suls J (2009). Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med 71: 171–186.
Johansson AS, Owe-Larsson B, Asp L, Kocki T, Adler M, Hetta J et al (2013). Activation of kynurenine pathway in ex vivo fibroblasts from patients with bipolar disorder or schizophrenia: cytokine challenge increases production of 3-hydroxykynurenine. JPsychiatr Res 47: 1815–1823.
Kessler M, Terramani T, Lynch G, Baudry M (1989). A glycine site associated with N-methyl-D-aspartic acid receptors: characterization and identification of a new class of antagonists. J Neurochem 52: 1319–1328.
Kim H, Chen L, Lim G, Sung B, Wang S, McCabe MF et al (2012). Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J Clin Invest 122: 2940–2954.
Kocki T, Wnuk S, Kloc R, Kocki J, Owe-Larsson B, Urbanska EM (2012). New insight into the antidepressants action: modulation of kynurenine pathway by increasing the kynurenic acid/3-hydroxykynurenine ratio. J Neural Transm 119: 235–243.
Koo JW, Duman RS (2008). IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci USA 105: 751–756.
Koolschijn PC, van Haren NE, Cahn W, Schnack HG, Janssen J, Klumpers F et al (2010). Hippocampal volume change in schizophrenia. J Clin Psychiatry 71: 737–744.
Koolschijn PC, van Haren NE, Lensvelt-Mulders GJ, Hulshoff Pol HE, Kahn RS (2009). Brain volume abnormalities in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Hum Brain Mapp 30: 3719–3735.
Kuperberg GR, Broome MR, McGuire PK, David AS, Eddy M, Ozawa F et al (2003). Regionally localized thinning of the cerebral cortex in schizophrenia. Arch Gen Psychiatry 60: 878–888.
Laugeray A, Launay JM, Callebert J, Surget A, Belzung C, Barone PR (2011). Evidence for a key role of the peripheral kynurenine pathway in the modulation of anxiety- and depression-like behaviours in mice: focus on individual differences. Pharmacol Biochem Behav 98: 161–168.
Liu WL, Lin YH, Xiao H, Xing S, Chen H, Chi PD et al (2014). Epstein-Barr virus infection induces indoleamine 2,3-dioxygenase expression in human monocyte-derived macrophages through p38/mitogen-activated protein kinase and NF-kappaB pathways: impairment in T cell functions. J Virol 88: 6660–6671.
Maciejak P, Szyndler J, Turzynska D, Sobolewska A, Kolosowska K, Lehner M et al (2013). The kynurenine pathway: a missing piece in the puzzle of valproate action? Neuroscience 234: 135–145.
Maes M, Berk M, Goehler L, Song C, Anderson G, Galecki P et al (2012). Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med 10: 66.
Maes M, Leonard BE, Myint AM, Kubera M, Verkerk R (2011). The new ‘5-HT’ hypothesis of depression: cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog Neuropsychopharmacol Biol Psychiatry 35: 702–721.
Maxwell M (1992) Family Interview for Genetic Studies (FIGS): Manual for FIGS. Clinical Neurogenetics Branch, Intramural Research Program, National Institute of Mental Health: Bethesda, MD.
Miller AH (2013). Conceptual confluence: the kynurenine pathway as a common target for ketamine and the convergence of the inflammation and glutamate hypotheses of depression. Neuropsychopharmacology 38: 1607–1608.
Monnerie H, Shashidhara S, Le Roux PD (2003). Effect of excess extracellular glutamate on dendrite growth from cerebral cortical neurons at 3 days in vitro: involvement of NMDA receptors. J Neurosci Res 74: 688–700.
Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B (2007). Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord 98: 143–151.
Myint AM, Schwarz MJ, Verkerk R, Mueller HH, Zach J, Scharpe S et al (2011). Reversal of imbalance between kynurenic acid and 3-hydroxykynurenine by antipsychotics in medication-naive and medication-free schizophrenic patients. Brain Behav Immun 25: 1576–1581.
O'Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N et al (2009). Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry 14: 511–522.
Olsson SK, Andersson AS, Linderholm KR, Holtze M, Nilsson-Todd LK, Schwieler L et al (2009). Elevated levels of kynurenic acid change the dopaminergic response to amphetamine: implications for schizophrenia. Int J Neuropsychopharmacol 12: 501–512.
Olsson SK, Sellgren C, Engberg G, Landen M, Erhardt S (2012). Cerebrospinal fluid kynurenic acid is associated with manic and psychotic features in patients with bipolar I disorder. Bipolar Disord 14: 719–726.
Orabona C, Pallotta MT, Grohmann U (2012). Different partners, opposite outcomes: a new perspective of the immunobiology of indoleamine 2,3-dioxygenase. Mol Med 18: 834–842.
Pereira AC, Huddleston DE, Brickman AM, Sosunov AA, Hen R, McKhann GM et al (2007). An in vivo correlate of exercise-induced neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci USA 104: 5638–5643.
Perkins MN, Stone TW (1982). An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res 247: 184–187.
Petrik D, Lagace DC, Eisch AJ (2012). The neurogenesis hypothesis of affective and anxiety disorders: are we mistaking the scaffolding for the building? Neuropharmacology 62: 21–34.
Potter MC, Elmer GI, Bergeron R, Albuquerque EX, Guidetti P, Wu HQ et al (2010). Reduction of endogenous kynurenic acid formation enhances extracellular glutamate, hippocampal plasticity, and cognitive behavior. Neuropsychopharmacology 35: 1734–1742.
Puccetti P, Grohmann U (2007). IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nat Rev Immunol 7: 817–823.
Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G et al (2010). CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry 15: 393–403.
Rosas HD, Liu AK, Hersch S, Glessner M, Ferrante RJ, Salat DH et al (2002). Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology 58: 695–701.
Savitz J, Drevets WC (2009). Bipolar and major depressive disorder: neuroimaging the developmental-degenerative divide. Neurosci Biobehav Rev 33: 699–771.
Savitz J, Nugent AC, Bogers W, Liu A, Sills R, Luckenbaugh DA et al (2010). Amygdala volume in depressed patients with bipolar disorder assessed using high resolution 3T MRI: the impact of medication. Neuroimage 49: 2966–2976.
Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ (2012). Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 13: 465–477.
Schwarcz R, Whetsell WO Jr, Mangano RM (1983). Quinolinic acid: an endogenous metabolite that produces axon-sparing lesions in rat brain. Science 219: 316–318.
Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C et al (2008). Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res 42: 151–157.
Steiner J, Bogerts B, Sarnyai Z, Walter M, Gos T, Bernstein HG et al (2012). Bridging the gap between the immune and glutamate hypotheses of schizophrenia and major depression: potential role of glial NMDA receptor modulators and impaired blood-brain barrier integrity. World J Biol Psychiatry 13: 482–492.
Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z et al (2011). Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation 8: 94.
Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY et al (2004). Cellular changes in the postmortem hippocampus in major depression. Biol Psychiatry 56: 640–650.
Stone TW, Darlington LG (2002). Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov 1: 609–620.
Vernon AC, Crum WR, Lerch JP, Chege W, Natesan S, Modo M et al (2013). Reduced cortical volume and elevated astrocyte density in rats chronically treated with antipsychotic drugs-linking magnetic resonance imaging findings to cellular pathology. Biol Psychiatry 75: 982–990.
Vithlani M, Hines RM, Zhong P, Terunuma M, Hines DJ, Revilla-Sanchez R et al (2013). The ability of BDNF to modify neurogenesis and depressive-like behaviors is dependent upon phosphorylation of tyrosine residues 365/367 in the GABA(A)-receptor gamma2 subunit. J Neurosci 33: 15567–15577.
Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B et al (2013). NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology 38: 1609–1616.
Williamson LL, Bilbo SD (2013). Chemokines and the hippocampus: a new perspective on hippocampal plasticity and vulnerability. Brain Behav Immun 30: 186–194.
Winter H, Irle E (2004). Hippocampal volume in adult burn patients with and without posttraumatic stress disorder. Am J Psychiatry 161: 2194–2200.
Wonodi I, Stine OC, Sathyasaikumar KV, Roberts RC, Mitchell BD, Hong LE et al (2011). Downregulated kynurenine 3-monooxygenase gene expression and enzyme activity in schizophrenia and genetic association with schizophrenia endophenotypes. Arch Gen Psychiatry 68: 665–674.
Yirmiya R, Goshen I (2011). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25: 181–213.
Zunszain PA, Anacker C, Cattaneo A, Choudhury S, Musaelyan K, Myint AM et al (2012). Interleukin-1beta: a new regulator of the kynurenine pathway affecting human hippocampal neurogenesis. Neuropsychopharmacology 37: 939–949.
Acknowledgements
We acknowledge Marieke van der Hart at Brains Online for excellence in HPLC sample analysis. We also thank all the research participants and wish to acknowledge the contributions of Brenda Davis, Debbie Neal, Chibing Tan, and Ashlee Taylor from the laboratory of TKT towards the transport, processing, and handling of all blood samples. This study was funded by a grant from the National Institute of Mental Health to JS (K01MH096077). JS, WCD, TAV, BEW, PFSB, and JB received support from The William K Warren Foundation. CMS and TKT received support from the Oklahoma Tobacco Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Rights and permissions
About this article

Cite this article
Savitz, J., Drevets, W., Smith, C. et al. Putative Neuroprotective and Neurotoxic Kynurenine Pathway Metabolites Are Associated with Hippocampal and Amygdalar Volumes in Subjects with Major Depressive Disorder. Neuropsychopharmacol 40, 463–471 (2015). https://doi.org/10.1038/npp.2014.194
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.194
This article is cited by
-
Review of research progress on intestinal microbiota based on metabolism and inflammation for depression
Archives of Microbiology (2024)
-
First trimester human umbilical cord perivascular cells (HUCPVC) modulate the kynurenine pathway and glutamate neurotransmission in an LPS-induced mouse model of neuroinflammation
Journal of Inflammation (2023)
-
Molecular pathways of major depressive disorder converge on the synapse
Molecular Psychiatry (2023)
-
Towards a multilevel model of major depression: genes, immuno-metabolic function, and cortico-striatal signaling
Translational Psychiatry (2023)
-
Kynurenine pathway and its role in neurologic, psychiatric, and inflammatory bowel diseases
Molecular Biology Reports (2023)
