
Abstract
Mosaic variegated aneuploidy is a rare recessive condition characterized by growth retardation, microcephaly, childhood cancer and constitutional mosaicism for chromosomal gains and losses. In five families with mosaic variegated aneuploidy, including two with embryonal rhabdomyosarcoma, we identified truncating and missense mutations of BUB1B, which encodes BUBR1, a key protein in the mitotic spindle checkpoint. These data are the first to relate germline mutations in a spindle checkpoint gene with a human disorder and strongly support a causal link between aneuploidy and cancer development.
Similar content being viewed by others

Main
The mitotic spindle checkpoint is a surveillance mechanism that is crucial for maintenance of correct chromosome number during cell division. Chief components of the human spindle checkpoint include MAD1, MAD2, BUB1, BUB3, BUBR1 and MPS1; many other proteins are also involved. Defects in the spindle checkpoint are implicated in the generation of aneuploidy (gains or losses of whole chromosomes), which occurs frequently in human cancers1,2,3.
Mosaic variegated aneuploidy (MVA; OMIM 257300) is a recessive condition characterized by mosaic aneuploidies, predominantly trisomies and monosomies, involving multiple different chromosomes and tissues (Fig. 1a). The proportion of aneuploid cells varies but is usually >25% and is substantially greater than in normal individuals4,5,6,7,8. Affected individuals typically present with severe intrauterine growth retardation and microcephaly. Eye anomalies, mild dysmorphism, variable developmental delay and a broad spectrum of additional congenital abnormalities and medical conditions may also occur. The risk of malignancy is high, with rhabdomyosarcoma, Wilms tumor and leukemia reported in several cases4,5,6,7,8.

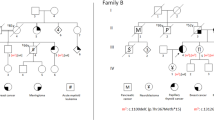
(a) Karyotype of three cells from a lymphoblastoid cell line from the affected individual from family 4, showing mosaicism for different aneuploidies. More than 90% of cells examined from multiple different passages were abnormal. (b) BUB1B mutations identified in five MVA families, with the wild-type sequences shown below. (c) Schematic representation of BUBR1 showing position of mutations, with the truncating mutations depicted above the protein and missense mutations below. (d) Alignments of BUBR1 orthologs showing conservation of missense mutations.
We hypothesized that mutations of a gene involved in the mitotic spindle checkpoint might underlie MVA. We screened the full coding sequence and intron-exon boundaries of BUB1B by conformation-sensitive gel electrophoresis (primers and conditions available on request) in eight pedigrees with MVA and identified mutations of both BUB1B alleles in five families (Fig. 1b,c and Table 1). Each family carried one missense mutation and one mutation that resulted in premature protein truncation or an absent transcript. In all cases the mutations were inherited from different parents, indicating that they were on separate alleles. None of the mutations have been reported in more than 200 BUB1B mutation analyses in colorectal cancers, lung cancers and glioblastomas, and none were present in 384 normal control chromosomes. Five of the six missense mutations were in the kinase domain and affected residues conserved in BubR1 orthologs in mouse and chicken (Fig. 1d). The final missense mutation, R550Q, was outside the kinase domain in a less-conserved region that has been implicated in inhibition of CDC20 binding to the anaphase-promoting complex9. Family 1 had two maternally inherited missense mutations, L844F and Q921H (Fig. 1c,d). Without additional data, it is difficult to determine which of these missense variants is pathogenic, and both might affect BUBR1 kinase activity and contribute to the phenotype of the affected child in this family. We did not identify a truncating or splice-site mutation in family 5, despite fully sequencing the gene. But RT-PCR analyses showed that there was no transcript from the paternal allele, indicative of a cryptic BUB1B mutation in addition to the maternally transmitted missense mutation.
All five individuals with BUB1B mutations had phenotypes typical of MVA. Detailed clinical and cytogenetic features of three of the families (families 1, 4 and 5; Table 1) have been published previously5,6. Two cases developed cancer: a child in family 1 developed an embryonal rhabdomyosarcoma of the soft palate at 7 y of age, and a child in family 4 developed an embryonal rhabdomyosarcoma of the vagina at 5 months of age. Sequencing of BUB1B in the tumor from the affected child in family 4 showed that both mutations were present, as in the constitutional DNA. There was no history of cancer, congenital abnormality or unusual medical condition in any first- or second-degree relative of any affected individual.
The mitotic spindle checkpoint delays anaphase until the kinetochores of each chromosome pair have successfully attached to the spindle. Components of the checkpoint machinery preferentially localize to kinetochores of unaligned chromosomes, where they produce a diffusible signal that delays the onset of anaphase by inhibiting the anaphase-promoting complex/cyclosome (APC/C)1,2. BUBR1 seems to have multiple roles in the spindle checkpoint. It is a potent inhibitor of APC/C, and this role is largely independent of its kinase activity9. But BUBR1 kinase activity does seem to be important in recruiting checkpoint proteins that can bind CDC20, which also results in inhibition of APC/C activity10. BUBR1 is also implicated in triggering apoptosis in polyploid cells that exit aberrantly from mitotic arrest11.
Our data provide further insight into the roles of BUBR1 in humans. Compromised BUBR1 function results in abnormal control of chromosome number that cannot be fully complemented by other spindle-checkpoint proteins. All five individuals with biallelic BUB1B mutations carried one missense mutation and one mutation that resulted in protein truncation or an absent transcript. We found no cases with either biallelic missense mutations or biallelic truncating or absent-transcript mutations. This mutation pattern could be due to chance. Alternatively, biallelic truncating or absent-transcript mutations may result in more extensive aneuploidy and embryonic lethality, whereas biallelic missense mutations may not alter BUBR1 function enough to cause the MVA phenotype. If correct, this hypothesis could explain the paucity of MVA cases from consanguineous or geographically isolated families. Our series includes only one consanguineous pedigree. We identified no BUB1B mutations in this family, and MVA is not consistent with linkage to BUB1B in this pedigree. These results indicate that MVA is a heterogeneous condition and suggest that other mitotic-spindle genes might predispose to human disorders characterized by aneuploidy.
Two Bub1b mouse models have recently been described12,13. Bub1b−/− mice die in utero. Bub1b+/− mice are developmentally normal but have defective spindle-checkpoint activation and develop lung and colon cancers in response to carcinogen12. Bub1bH/H mice, which have ∼10% normal levels of bubR1, develop aneuploidy, cataracts and growth retardation, which are characteristic of MVA, but do not have an increased cancer incidence. Bub1bH/H mice also show features of premature aging. But individuals with MVA do not have typical features of human premature aging syndromes.
Extensive mutation screening of human tumors has identified only one somatic BUB1B mutation, although BUB1B expression is epigenetically downregulated in some cancers14,15. But two of the five MVA cases due to biallelic BUB1B mutations developed rhabdomyosarcoma, indicating a markedly elevated cancer risk. The identification of BUB1B mutations in a cancer predisposition syndrome suggests that aneuploidy, secondary to functional abrogation of the mitotic spindle checkpoint, can contribute to the development of cancer.
Accession numbers. GenBank: human BUB1B, AF046079. Swiss-Prot: human BUBR1, O60566; mouse BubR1, Q9Z1S0; chicken BubR1, Q800D4; Xenopus laevis BubR1, Q8JGT8.
References
Bharadwaj, R. & Yu, H. Oncogene 32, 2016–2027 (2004).
Draviam, V.M. et al. Curr. Opin. Genet. Dev. 14, 120–125 (2004).
Lengauer, C. et al. Nature 396, 643–649 (1998).
Tolmie, J.L. et al. Hum. Genet. 80, 197–200 (1988).
Limwongse, C. et al. Am. J. Med. Genet. 82, 20–24 (1999).
Plaja, A. et al. Am. J. Med. Genet. 98, 216–223 (2001).
Kajii, T. et al. Am. J. Med. Genet. 104, 57–64 (2001).
Jacquemont, S. et al. Am. J. Med. Genet. 109, 17–21 (2002).
Tang, Z. et al. Dev. Cell 1, 227–237 (2001).
Mao, Y. et al. Cell 114, 87–98 (2003).
Shin, H.-J. et al. Cancer Cell 4, 483–497 (2003).
Dai, W. et al. Cancer Res. 64, 440–445 (2004).
Baker, D.J. et al. Nat. Genet. 36, 744–749 (2004).
Cahill, D.P. et al. Nature 392, 300–303 (1998).
Shichiri, M. et al. Cancer Res. 62, 13–17 (2002).
Acknowledgements
We thank all the families that gave informed consent to take part in this research; the many clinicians that were involved in their diagnosis and management, including D. Bain, H. Cox, A. Green, A. McEwen, T. Vendrell, P. Ellis and R. Murray; M. Stratton and A. Futreal for advice on the manuscript; and K. Tatton-Brown and T. Min for help in preparation of the figure. A.I. is supported by Tenovus the Cancer Charity. This research was supported and approved by the Institute of Cancer Research (UK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Hanks, S., Coleman, K., Reid, S. et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet 36, 1159–1161 (2004). https://doi.org/10.1038/ng1449
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1449
This article is cited by
-
RNA sequencing and target long-read sequencing reveal an intronic transposon insertion causing aberrant splicing
Journal of Human Genetics (2024)
-
Principles and dynamics of spindle assembly checkpoint signalling
Nature Reviews Molecular Cell Biology (2023)
-
Germline sequence variants contributing to cancer susceptibility in South African breast cancer patients of African ancestry
Scientific Reports (2022)
-
Physiological relevance of post-translational regulation of the spindle assembly checkpoint protein BubR1
Cell & Bioscience (2021)
-
Intellectual disability and microcephaly associated with a novel CHAMP1 mutation
Human Genome Variation (2021)
