
Abstract
DNA mismatch repair (MMR) is a highly conserved biological pathway that plays a key role in maintaining genomic stability. The specificity of MMR is primarily for base-base mismatches and insertion/deletion mispairs generated during DNA replication and recombination. MMR also suppresses homeologous recombination and was recently shown to play a role in DNA damage signaling in eukaryotic cells. Escherichia coli MutS and MutL and their eukaryotic homologs, MutSα and MutLα, respectively, are key players in MMR-associated genome maintenance. Many other protein components that participate in various DNA metabolic pathways, such as PCNA and RPA, are also essential for MMR. Defects in MMR are associated with genome-wide instability, predisposition to certain types of cancer including hereditary non-polyposis colorectal cancer, resistance to certain chemotherapeutic agents, and abnormalities in meiosis and sterility in mammalian systems.
Similar content being viewed by others
Introduction
DNA damage accumulates in cells over time as a result of exposure to exogenous chemicals and physical agents (i.e., benzo[a]pyrene, polychlorinated biphenyls, dioxin, cigarette smoke, asbestos, ultraviolet light, radon), as well as endogenous reactive metabolites including reactive oxygen and nitrogen species (ROS and NOS). Another source of DNA damage is errors that occur during normal DNA metabolism or aberrant DNA processing reactions, including DNA replication, recombination, and repair. Nucleotide misincorporation generates DNA base-base mismatches during DNA synthesis at variable rates, depending on many factors, including the specific DNA polymerases. In general, the replicative DNA polymerases have relatively high replication fidelity (see McCulloch and Kunkel, this issue), while translesion DNA polymerases, which specifically bypass sites of DNA damage, have lower replication fidelity (see Andersen et al. and Gan et al. in this issue). DNA damage, if unrepaired, has the potential to generate mutations in somatic or germline cells, which can alter cellular phenotype and cause dysfunction and disease. To prevent such deleterious effects and safeguard the integrity of the genome, cells possess multiple mechanisms to repair DNA damage and thus prevent mutations. One such system is the critical pathway known as DNA mismatch repair (MMR).
MMR corrects DNA mismatches generated during DNA replication, thereby preventing mutations from becoming permanent in dividing cells 1, 2, 3. Because MMR reduces the number of replication-associated errors, defects in MMR increase the spontaneous mutation rate 4. Inactivation of MMR in human cells is associated with hereditary and sporadic human cancers 1, 3, 5, and the MMR system is required for cell cycle arrest and/or programmed cell death in response to certain types of DNA damage 6, 7. Thus, MMR plays a role in the DNA damage response pathway that eliminates severely damaged cells and prevents both mutagenesis in the short term and tumorigenesis in the long term.
The prototypical Escherichia coli MMR pathway has been extensively studied and is well characterized both biochemically and genetically. Thus, E. coli MMR is a useful and important framework for understanding eukaryotic MMR. In this review, the biochemistry and genetics of E. coli MMR will be described briefly by way of introduction, and the remainder of the discussion will focus on the cellular functions of MMR and their roles in cancer avoidance in mammalian cells. For areas of research on human MMR not discussed in this paper or an additional discussion of MMR in other species, readers are referred to the following excellent reviews: 1, 2, 3, 8, 9, 10, 11.
Mechanism of mismatch correction
DNA MMR in E. coli
E. coli MMR requires the following protein components: MutS, MutL, MutH, DNA helicase II (MutU/UvrD), four exonucleases (ExoI, ExoVII, ExoX, and RecJ), single-stranded DNA binding protein (SSB), DNA polymerase III holoenzyme, and DNA ligase 12, 13. MutS, MutL, and MutH initiate MMR and play specialized biological roles in MMR in E. coli.
MutS recognizes base-base mismatches and small nucleotide insertion/deletion (ID) mispairs, and thus MutS has been called the “mismatch recognition” protein 3. MutS possesses intrinsic ATPase activity. High-resolution structures of MutS bound to DNA have been determined by X-ray crystallography 14, 15. These structures revealed that MutS binds to a mismatch as a homodimer. Interestingly, the mismatch-binding site is comprised of sequence-wise identical but structurally and functionally different domains from the two subunits, indicating asymmetry in the protein-DNA complex. Hence, the MutS homodimer acts as a virtual heterodimer when bound to a DNA mismatch. This characteristic is mimicked by eukaryotic MutS homologs (MSH), which function as heterodimers instead of homodimers (see below). MMR in E. coli is ATP-dependent, and requires the functional MutS ATPase.
MutL interacts physically with MutS, enhances mismatch recognition, and recruits and activates MutH. Defects in MutL completely inhibit MMR in E. coli. Despite the fact that a functional human MutL homolog, MutLα, possesses an endonuclease activity that is essential for mammalian MMR 16, no hydrolytic activity has been detected in MutL. However, MutL may play a role as a molecular matchmaker that facilitates assembly of a functional MMR complex 3, 17, because it stimulates the loading and the processivity of helicase II (or UvrD) at the MMR initiation site 18, 19. Like MutS, MutL functions as a homodimer and possesses ATPase activity 20. Mutations in the ATP-binding domain lead to a dominant negative mutator phenotype 21. MutL mutants that are defective in ATP hydrolysis but proficient in ATP binding can activate MutH but cannot stimulate MutH in response to a mismatch or MutS, suggesting that ATP hydrolysis by MutL is essential for mediating the activation of MutH by MutS 22. Recent studies show that MutL interacts physically with the clamp loader subunits of DNA polymerase III 23, 24, suggesting that MutL may promote binding of DNA polymerase III to MMR intermediates. These observations suggest that MMR is coupled with DNA replication.
In E. coli, DNA is methylated at the N6 position of adenine in dGATC sequences. In replicating DNA, the daughter strand is transiently unmethylated, and it is the presence of hemimethylated dGATC sequences that molecularly distinguishes the newly synthesized daughter strand from the parental DNA strand. In MMR, hemi-methylated dGATC sites determine the strand specificity of repair. MutH, which recognizes hemimethylated dGATC sequences, functions as a monomer and belongs to a family of type-II restriction endonucleases 25, 26. Upon its recruitment and activation by MutS and MutL in the presence of ATP, MutH specifically incises the unmethylated daughter strand of hemimethylated dGATC 3, 22, and this strand-specific nick provides the initiation site for mismatch-provoked excision.
The first step of the MMR pathway is binding of a MutS homodimer to the mismatch. Subsequently, a hemi-methylated dGATC site 5′ or 3′ to the mismatch is located and cleaved by the concerted action of MutS, MutL, MutH, and ATP. Three models have been proposed to address how mismatch binding by MutS leads to cleavage of the hemimethylated dGATC site (see the section of Unsolved Fundamental Problems in MMR for details). The strand-specific nick generated by MutH at hemimethylated dGATC is a starting point for excision of the mispaired base. In the presence of MutL, helicase II (UvrD) loads at the nick and unwinds the duplex from the nick towards the mismatch 18, generating single-strand DNA, which is rapidly bound by single-stranded DNA-binding protein (SSB) and protected from nuclease attack 27. Depending on the position of the strand break relative to the mismatch, ExoI or ExoX (3′→5′ exonuclease), or ExoVII or RecJ (5′→3′ exonuclease) excises the nicked strand from the nicked site (the dGATC site) up to and slightly past the mismatch. The resulting single-stranded gap undergoes repair DNA resynthesis and ligation by DNA polymerase III holoenzyme, SSB, and DNA ligase 3.
These early studies on E. coli MMR demonstrate three key features of this important pathway: first, repair is strand specific (i.e., restricted to the newly synthesized DNA strand); second, repair is bi-directional, proceeding 5′→3′ or 3′→5′ from the nick to the site of the mismatch; and third, MMR has broad substrate specificity including base-base mismatches and small ID mispairs. All of these properties require functional MutS, MutL, and MutH. Because the mechanism of MMR is highly conserved throughout evolution, E. coli MMR is an excellent model for MMR in eukaryotic cells.
MMR in human cells
MMR is a highly conserved biological pathway with strong similarities between human MMR and prototypical E. coli MMR 2, 3. These similarities include substrate specificity, bidirectionality, and nick-directed strand specificity. The role of hemi-methylated dGATC sites as a signal for strand discrimination is not conserved from E. coli MMR to human MMR, but because the hemi-methylated dGATC site directs MutH-dependent nicking, and because human MMR is presumed to be nick-directed in vivo, both systems are thought to discriminate daughter and template strands using a strand-specific nick.
Several human MMR proteins have been identified based on their homology to E. coli MMR proteins (Table 1). These include human homologs of MutS 28, 29, 30, 31, 32, MutL 33, 34, 35, 36, EXO1 37, 38, 39, single-strand DNA-binding protein RPA 27, 40, proliferating cellular nuclear antigen (PCNA) 41, 42, 43, DNA polymerase δ (pol δ) 44, and DNA ligase I 45. Although E. coli MutS and MutL proteins are homodimers, human MutS and MutL homologues are heterodimers 32, 34, 46. hMSH2 heterodimerizes with hMSH6 or hMSH3 to form hMutSα or hMutSβ, respectively, both of which are ATPases that play a critical role in mismatch recognition and initiation of repair 2. hMutSα preferentially recognizes base-base mismatches and ID mispairs of 1 or 2 nucleotides, while hMutSβ preferentially recognizes larger ID mispairs. At least 4 human MutL homologs (hMLH1, hMLH3, hPMS1, and hPMS2) have been identified 33, 35, 36, 47. hMLH1 heterodimerizes with hPMS2, hPMS1, or hMLH3 to form hMutLα, hMutLβ, or hMutLγ, respectively 2. hMutLα is required for MMR and hMutLγ plays a role in meiosis, but no specific biological role has been identified for hMutLβ 2. hMutLα possesses an ATPase activity and defects in this activity inactivate MMR in human cells. In a reconstituted human MMR system, hMutLα regulates termination of mismatch-provoked excision 45. Recent studies show that MutLα possesses a PCNA/replication factor C (RFC)-dependent endonuclease activity which plays a critical role in 3′ nick-directed MMR involving EXO1 16.
PCNA interacts with MSH2 and MLH1 and is thought to play roles in the initiation and DNA resynthesis steps of MMR 41, 43. PCNA also interacts with MSH6 and MSH3 48, 49, 50, 51 via a conserved PCNA interaction motif termed the PIP box 52. It has been proposed that PCNA may help localize MutSα and MutSβ to mispairs in newly replicated DNA 53, 54. Although PCNA is absolutely required during 3′ nick-directed MMR, it is not essential during 5′ nick-directed MMR 55. This observation might be explained by the fact that EXO1, a 5′→3′ exonuclease, is involved in both 5′ and 3′ directed MMR. Like PCNA, EXO1 also interacts with MSH2 and MLH1 37, 38, 39, 56, 57, 58. While EXO1 can readily carry out 5′ directed mismatch excision in the presence of MutSα or MutSβ and RPA 45, 59, its role in catalyzing 3′ nick-directed excision requires the MutLα endonuclease, which is activated by PCNA and RFC 16, 60. Although it has been suggested that EXO1 possesses a cryptic 3′→5′ exonuclease activity 60, 61, current data do not support that hypothesis. Instead, recent studies suggest the following steps for EXO1-catalyzed 3′ nick-directed repair: (1) after recognition of the 3′ nick and the mismatch, MutLα endonuclease makes an incision 5′ to the mismatch in a manner dependent on PCNA and RFC; and (2) EXO1 performs 5′→3′ excision from the MutLα-incision site through and beyond the site of the mismatch 16. However, exo1 null mutants in mice and yeast have a weak mutator phenotype 56, 62; thus, it is likely that additional as yet unidentified exonucleases are involved in eukaryotic MMR.
Other protein components involved in human MMR include single-strand DNA-binding protein RPA, RFC, high mobility group box 1 protein (HMGB1), and DNA pol δ. RPA seems to be involved in all stages of MMR: it binds to nicked heteroduplex DNA before MutSα and MutLα, stimulates mismatch-provoked excision, protects the ssDNA gapped region generated during excision, and facilitates DNA resynthesis 27, 45, 60, 63. Furthermore, RPA is phosphorylated after pol δ is recruited to the gapped DNA substrate. Recent studies indicate that phosphorylation reduces the affinity of RPA for DNA, that unphosphorylated RPA stimulates mismatch-provoked DNA excision more efficiently than phosphorylated RPA, and that phosphorylated RPA facilitates MMR-associated DNA resynthesis more efficiently than unphosphorylated RPA 63. These results are consistent with the fact that a high-affinity RPA-DNA complex might be required to protect nascent ssDNA and to displace DNA-bound MutSα/MutLα 27, 45, while a lower-affinity RPA-DNA complex might facilitate DNA resynthesis by pol δ 63. HMGB1 is a mismatch-binding protein and has a DNA-unwinding activity 64, 65, 66. It interacts with MSH2 and MSH6 in vitro 67. Recent studies show that HMGB1 can substitute for RPA in an in vitro reconstituted MMR system 45. Additional studies are needed to precisely define the function of HMGB1 in MMR.
Unsolved fundamental problems in MMR
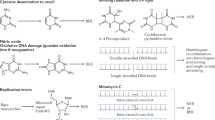
Despite great progress in identifying MMR proteins and genes and application of state-of-the-art biochemical and genetic approaches to analyze the mechanism of MMR in prokaryotic and eukaryotic cells, several key questions about this pathway remain unanswered. One of these questions concerns the mechanism by which MMR proteins facilitate the communication between two physically distant DNA sites: the mismatch and the strand discrimination signal. It is generally agreed that the strand discrimination signal is a strand-specific nick in both prokaryotic and eukaryotic cells (see above), although the source of the nicking activity, at least for the leading strand, is not known in eukaryotic cells. Previous studies have proposed several alternative models for this process, which can be classified into “cis-” or “moving” and “trans-” or “stationary” models (Figure 1). The “stationary” model (Figure 1, right) proposes that interactions among MMR proteins induce DNA bending or looping that brings the two distant sites together, while MutS (or the MSH heterodimers, i.e. MutSα and MutSβ) remains bound at the mismatch 19, 22. In this model, the MutS (or MSH heterodimers) ATPase activity acts in a proofreading role to verify mismatch binding and authorize the downstream excision 22. Support for the stationary model came from the following experiments. Junop et al. showed that recognition of a mismatch by MutS on a DNA molecule activated MutH cleavage of a GATC site located on a separate DNA molecule without a mismatch 22. Consistent with this observation, a second study demonstrated that mismatch-provoked excision could be initiated when a biotin-streptavidin blockade was placed between the mismatch and pre-existing nick 68. The “cis” or “moving” models suggest that MutS-MutL (or MutSα/β-MutLα) complexes load at a mismatch site and then move away from the site to search for the strand break, where exonucleases can be recruited to initiate excision.

Models for signaling downstream MMR events following mismatch recognition. A schematic diagram for signaling between the mismatch and the strand discrimination signal is shown. Here, a 5′ nick is the strand discrimination signal. Similar models apply for 3′ nick-directed MMR. The “stationary” or “trans” model (right) emphasizes that MutS or its homolog (MSH) proteins remain bound at the mismatch. It is the protein-protein interactions that induce DNA bending or looping that brings the two distant sites together. The two DNA sites can cooperate in a “trans” configuration. In two “cis” or “moving” models, one called the “translocation” model (left) and the other called the “molecular switch” or “sliding clamp” model (middle), it is hypothesized that the MSH proteins bind to the mismatch and then move away from the site to search for the strand discrimination signal. The translocation model suggests that ATP hydrolysis drives unidirectional movement of the MSH proteins, resulting in the formation of an α-like loop. In the molecular switch model (center), binding of an MSH protein (in its ADP-bound state) to the mismatch triggers an ADP to ATP exchange that promotes bi-directional sliding of the protein away from the mismatch, thereby emptying the mismatch site for an incoming MSH protein. Mismatch excision begins when an MSH protein reaches the strand break.
There are two moving models, one called the “translocation” model and the other called the “molecular switch” or “sliding clamp” model (Figure 1). In the translocation model 69, ATP reduces the mismatch-binding affinity of MutS or the MSH heterodimers, and ATP hydrolysis drives unidirectional translocation of MutS proteins along the DNA helix. DNA is threaded through the protein complex until the latter reaches a strand discrimination signal in either orientation, a process that forms a DNA loop (Figure 1, left). In the molecular switch model, MutS or the MSH heterodimer binds to mismatched DNA in an ADP-bound state. The mismatch binding by MutS or the MSH heterodimer triggers a conformational change that allows an ADP to ATP exchange, which promotes a second conformational change that allows MutS or the MSH heterodimers to form a sliding clamp 70, 71, 72, 73. In this model (Figure 1, middle), it is the binding of ATP, not ATP hydrolysis, that signals downstream events including formation of ternary complex with MutL (or MLH heterodimers) and sliding of the ternary complex from the mismatch to the strand break 70, 71, 72, 73.
Recent studies by Pluciennik and Modrich 74 argue in favor of a moving rather than a stationary mechanism, because their data demonstrate that a dsDNA break 75 or a protein “roadblock” between the mismatch and the nick inhibits in vitro MMR with recombinant E. coli proteins. It is not clear why two “roadblock” experiments 68, 74 obtained distinct results. In a reconstituted human MMR reaction, Zhang et al. 45 show that multiple MutSα-MutLα complexes are essential for processing a single mismatch, providing evidence to support the molecular switch model. Additional studies are needed to address these unresolved questions about the molecular mechanism of MMR.
MMR mediates DNA damage signaling
MMR deficiency and drug resistance
DNA-damaging agents such as the alkylating agents N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), temozolomide, or procarbazine are cytotoxic agents that kill most of the replicating cells. Many cancer therapeutics are genotoxic and cytotoxic agents that induce apoptotic cell death. Interestingly, many cells that acquire resistance to such agents are deficient in MMR. For example, the human lymphoblastoid cell line MTI, which has a defect in hMSH6, was derived by culturing TK6 cells in the presence of a high concentration of MNNG. The resulting MNNG-resistant MT1 cells are defective in strand-specific MMR 76. Many human colorectal cancer cell lines are also resistant to alkylating agents and have associated defects in MMR. The causal relationship between drug resistance and MMR is demonstrated by the fact that hMLH1-defective MNNG-resistant cells lose drug resistance when the hMLH1 defect is genetically complemented with wild-type hMLH1 on chromosome 3 77. It has also been observed that defects in MSH2 and PMS2 confer resistance to alkylating agents (reviewed in 78). The mechanism by which MMR influences drug cytotoxicity is discussed further below.
Resistance to methotrexate (MTX) has also been associated with phenotypic changes in MMR in human cells. This occurs by the unusual mechanism of co-amplification of the human chromosomal region that encodes dihydrofolate reductase (DHFR, the target of MTX) and hMSH3 79, 80. Amplification of DHFR lowers sensitivity to MTX by overexpressing the target of the drug. However, overexpression of hMSH3 sequesters hMSH2 in the hMutSβ heterodimer, effectively preventing formation of the hMutSα (hMSH2/hMSH6) heterodimer, which leads to degradation of uncomplexed hMSH6, significant dysregulation of MMR and hypermutability 81, 82. Overexpression of DHFR combined with genome-wide hypermutability and defective MMR are likely responsible for the MTX resistance of HL60 and other tumor cells.
MMR proteins promote DNA damage-induced cell cycle arrest and apoptosis
Cell cycle arrest is an important mechanism for preventing DNA damage-induced genomic instability. A large number of studies have characterized the so-called G2 or S phase checkpoints, and identified proteins required for cell cycle arrest, including ATM, ATR, p53, p73, Chk1, and Chk2. However, it was a somewhat unexpected finding that hMutSα- and hMutLα-deficient cells are defective in cell cycle arrest in response to multiple types of DNA damaging agents 6, 7, 83. While the molecular basis of this effect is not precisely known, it has been reported that MMR-deficient cells fail to phosphorylate p53 and p73 in response to DNA damage 84, 85. This implicates ATM, ATR, and/or c-Abl, because these kinases phosphorylate p53 and p73 during the response to DNA damage 85, 86. In support of this, it has been reported that hMutSα and hMutLα interact physically with ATM, ATR-ARTIP, c-Abl, and p73 in cells treated with DNA damaging agents/drugs 83, 87, 88, 89. These observations implicate hMutSα and hMutLα in a signaling cascade that leads from DNA damage to cell cycle arrest and/or apoptosis. They also at least in part explain the fact that drug-induced cytotoxicity is lost in MMR-deficient cells, as discussed above 6. Very recently, EXO1 has been shown to be essential for upstream induction of DNA damage response, possibly by reducing ssDNA formation and recruiting RPA and ATR to the damage site 90. It remains to be seen if MutSα and/or MutLα act to recruit EXO1 in DNA damage response as they do in MMR.
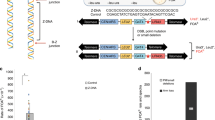
Two models have been proposed to describe the role of MMR in DNA damage signaling. The “futile DNA repair cycle” model (Figure 2, left) proposes that strand-specific MMR, which targets only newly replicated DNA, engages in a futile DNA repair cycle when it encounters DNA lesions in the template strand, and this futile cycling activates DNA damage signaling pathways to induce cell cycle arrest and apoptosis 6. Support for this model came from both in vivo and in vitro experiments. Stojic et al. 86 showed that exposure to MNNG induces DNA breaks/gaps, cell cycle arrest, and persistent nuclear foci at sites of DNA damage. The DNA damage-associated repair foci contain both damage signaling and DNA repair proteins, including ATR, γ-H2AX, and RPA. York and Modrich 91 showed that nicked circular heteroduplex plasmid DNA containing a single O6-methylguanine (O6-me-G)-thymine (T) mispair cannot be repaired by the MMR system when the lesion (O6-me-G) and the nick are on opposite strands; this suggests a futile repair process. An alternative model, referred to as the direct signaling model (Figure 2, right), argues that hMutSα/hMutLα directly trigger DNA damage signaling by recruiting ATM or ATR/ARTIP to the lesion, which activates a checkpoint response. This model is supported by an elegant study from the Hsieh laboratory showing that ATR and ATRIP form a complex with MutSα/MutLα in the presence of O6-me-G/T, which activates the ATR kinase and phosphorylates Chk1 89. Because mammalian MMR proteins interact with a broad spectrum of DNA lesions 6, this model is consistent with the notion that MutSα/MutLα acts as a sensor for DNA damage in mammalian cells. Both models provide a reasonable explanation for decreased DNA damage-induced apoptotic signaling and increased drug resistance in MMR-deficient cells.

Models for MMR-dependent DNA damage signaling. The “futile DNA repair cycle” model (left) suggests that DNA adducts (solid black circle) induce misincorporation, which triggers the strand-specific MMR reaction. Since MMR only targets the newly synthesized strand for repair, the offending adduct in the template strand cannot be removed, and will provoke a new cycle of MMR upon repair resynthesis. Such a futile repair cycle persists and activates the ATR and/or ATM damage signaling network to promote cell cycle arrest and/or programmed cell death. The direct signaling model proposes that recognition of DNA adducts by MSH-MLH complexes allows the proteins to recruit ATR and/or ATM to the site, activating the downstream damage signaling.
Role of MMR in other DNA metabolic pathways
MMR proteins have also been implicated in homeologous recombination, immunoglobulin class switching, somatic hypermutation, interstrand-crosslink repair, and trinucleotide repeat (TNR) expansion. Homeologous recombination (recombination between related but non-identical DNA sequences) generates mispairs/heteroduplexes, and induces genomic instability via chromosomal translocations, deletions, or inversions 92, 93. The frequency of homeologous recombination is much lower than that of homologous recombination in normal cells, but the frequency of homeologous recombination is dramatically elevated in MMR-deficient cells, suggesting that MMR suppresses homeologous recombination 3, 94. MutS and MutL inhibit DNA strand exchange between divergent sequences in vitro, most likely by binding to the mismatches generated during strand exchange 95. Recent studies in yeast reveal that suppression of homeologous recombination is mediated by MutSα and a RecQ family helicase, SGS1 96, 97. Consistent with this notion, yeast strains defective in sgs1 fail to suppress homeologous recombination 97, 98. It has been postulated that MutSα recruits SGS1 to DNA mismatches, where it unwinds the heteroduplex and blocks homeologous recombination 96. Although suppression of homeologous recombination by MMR proteins in human cells is less well understood, two human SGS1 homologous proteins, BLM and RECQ1, interact with MutSα 99, 100. This suggests that a similar mechanism is used to suppress homeologous recombination in yeast and human cells.
Recent studies also implicate MMR in repair of inter-strand crosslinks (ICLs), in a process that involves protein components from homologous recombination, double-strand break repair, and nucleotide excision repair 101, 102, 103. The precise nature of this involvement is not yet clear, and the specific MMR proteins that participate remain somewhat controversial. However, hMutSβ appears to directly bind ICLs in vitro 102, and hMutLα interacts specifically with the helicase domain of Fanconi Anemia protein FANC-J to facilitate ICL repair 101.
The studies discussed above suggest that MMR promotes genomic stability. However, during immunoglobulin class switching and somatic hypermutation, it appears that MMR proteins play a highly specialized role in promoting genetic variation. Immunoglobulin class switching and somatic hypermutation are mechanisms for increasing antibody diversity during antigen-stimulated B-cell differentiation. During this process, activation-induced cytidine deaminase (AID) deaminates cytosine residues to uracil, generating G:U mispairs, which can be recognized and processed by MMR 104, 105. However, during repair resynthesis, the high-fidelity replicative pol δ and ɛ are thought to be replaced by the translesion polymerases, which are error-prone and crucially introduce base substitutions and frameshift mutations 106. Additionally, MMR proteins play an important role in class switch recombination, an event where the IgM constant region is substituted by downstream constant sequences. In this capacity, MMR proteins utilize strand breaks generated by uracil DNA glycosylase to repair the AID-induced G:U mispairs in a strand-indiscriminate manner, leading to double-strand DNA breaks. It is these breaks that stimulate class switch recombination 107. B cells from mice deficient in MMR genes (MSH2, MSH6, MLH1, PMS2, or EXO1) display a low level of somatic hypermutation and reduced class switch recombination 8, 62, 108, 109.
MMR proteins also promote TNR expansion, a phenomenon associated with a number of neurological disorders in humans, including Huntington's disease, myotonic dystrophy, and fragile X syndrome 110 (also see Kovtun and McMurray in this issue). TNRs such as (CAG)n form single-stranded DNA loop/hairpin structures in vitro 111. Surprisingly, transgenic CAG repeats undergo expansion in wild-type, but not in knockout mice defective in MMR genes MSH2 and MSH3 112, 113, suggesting that the expansion mutations of the transgenic CAG repeats require functional MMR proteins MSH2 and MSH3. In vitro biochemical studies indeed show that MutSβ (the MSH2-MSH3 heterodimer) specifically binds to the (CAG)n hairpin structure 113. One model proposes that MutSβ inhibits repair or resolution of the (CAG)n hairpin, thus stimulating (CAG)n expansion 113. However, many of these studies were conducted using transgenic mouse models for TNR expansion. Thus, the role of MMR in human neurological diseases involving TNR expansion is at present unclear, and additional genetic and biochemical studies are needed to define the mechanism of TNR expansion in human cells.
MMR deficiency leads to cancer development
MMR defects in hereditary non-polyposis colorectal cancer (HNPCC) and other cancers
In the early 1990s, it was shown that HNPCC and some cases of sporadic colon cancer are caused by defects in human MMR 3. For HNPCC, Kolodner and co-workers, and Vogelstein and co-workers independently identified germ-line mutations in hMSH2 at chromosome 2p16-p21 in HNPCC families 29, 30. Genetic analyses of HNPCC kindreds revealed a large increase in frequency of insertion and deletion mutations in simple repeat (microsatellite) sequences, a phenomenon known as microsatellite instability (MSI) 114. MSI was also observed at lower incidence in sporadic colon cancers 114, 115, 116. Additional studies showed 100- to 700-fold decreased stability of poly(GT) tracts in yeast strains with single or double knockouts in MSH2, MLH1, or PMS1 117. Biochemical studies by Modrich and co-workers 118 and Kunkel and co-workers 119 also showed that extracts of MSI-positive tumor cells were severely defective in repair of base-base and ID mispairs. Thus, genetic and biochemical evidence converged to support the hypothesis that defective MMR plays a causal role in carcinogenesis leading to HNPCC, and strongly implicated such defects in some sporadic human cancers such as colorectal cancer.
The second locus linked to HNPCC was hMLH1 at 3p21-p23 33, 36, 120. Furthermore, HNPCC has also been linked to mutations in two additional MutL homolog genes hPMS1 and hPMS2, on 2q and 7p, respectively 35. Defects in hMLH1 represent the majority of all HNPCC cases 35, 36, with mutations in hMSH2 accounting for a large fraction of all the remaining HNPCC cases for which a genetic defect has been identified. In contrast, germ-line mutations in hMSH3 have not yet been linked to HNPCC, and mutations in hPMS1, hPMS2, and hMSH6 are relatively rare in HNPCC patients 1, 2, 3. These observations are consistent with the fact that hMSH2 and hMLH1 are essential MMR components, while hPMS1, hPMS2, hMSH6, and hMSH3 play important but partially redundant and/or dispensable roles in MMR.
Genetic evidence described above shows that defects in MMR correlate with HNPCC, and biochemical studies provide compelling additional evidence for this hypothesis. In particular, cell lines from HNPCC patients and sporadic tumors with MSI are defective in strand-specific MMR, and these cells can be divided into at least two complementation groups, corresponding to hMutLα 34 and hMutSα 32. Importantly, purified hMutLα or hMutSα specifically complements the biochemical defect in these cells. Similar evidence was obtained from chromosome or gene transfer experiments: expressions of the exogenous MMR genes complement the biochemical defect and stabilize simple repetitive sequences in the transfected cells (reviewed in 121).
These genetic and biochemical complementation studies essentially prove the causal role of MMR defects in HNPCC. However, many questions remain, including whether or not mutations in hMLH3 or hEXOI also cause HNPCC, a possibility that remains controversial 47, 122, 123. Mice defective in EXOI exhibit a mild defect in genomic stability, are partially defective in strand-specific MMR, and have increased rates of some cancers 62. Thus, it seems possible that germ-line mutations in hEXOI could potentially have similar phenotypic effects in humans. Additional studies are needed to clarify this point.
Although MSI was first correlated with MMR defects in tumors from HNPCC patients, MSI is also associated with a wide variety of non-HNPCC and non-colonic tumors (reviewed in Ref. 124). These include endometrial, ovarian, gastric, cervical, breast, skin, lung, prostate, and bladder tumors as well as glioma, leukemia, and lymphoma. Biochemical studies confirmed that MSI cell lines from sporadic leukemia, endometrial, ovarian, prostate, and bladder cancers are defective in strand-specific MMR 32, 125, 126. Interestingly, MSI in sporadic non-colonic tumors is often associated with hypermethylation of the promoter of hMLH1 (see below for details), and few mutations in MMR genes have been identified in these cells. These findings suggest that MMR defects are a likely cause of MSI in non-colonic sporadic cancers, although other mechanisms may also be involved in causing the MSI mutator phenotype.
Mouse models for MMR demonstrate roles in cancer and meiosis
Knockout mouse models have been developed for MSH2, MSH3, MSH6, MLH1, MLH3, PMS1, PMS2, and EXO1 (reviewed in 8, 62, 127, 128) and their phenotypes have been somewhat informative. Most of the knockout mice have a mutator phenotype, are MSI-positive, and are cancer-prone. However, the primary cancer susceptibility of MSH2, MLH1, and PMS2 knockout mice is lymphoma, not colorectal cancer as in humans, and secondary cancer susceptibilities are to gastrointestinal tumors, skin neoplasms, and/or sarcomas (Table 2).
MSH2−/− deficient mice are fertile 129, 130, are MSI-positive, develop lymphoma within 1 year of age, and have a significantly shorter lifespan than wild-type mice (i.e., 50% mortality by 6 months of age). The phenotype of MSH6−/− deficient mice is similar to that of MSH2−/− deficient mice, but lacking MSI 129, 131, a phenotype resembling that of atypical HNPCC with an hMSH6 defect as the tumors in these MSH6-defective individuals have longer latency and low MSI 132. Cells from MSH3−/− mice are defective in repair of ID mispairs but can repair base-base mismatches. MSH3−/− mice are either tumor free 133 or develop tumors at a very late age 134, essentially consistent with the fact that no MSH3 mutations have been identified in HNPCC patients. However, in MSH3−/− and MSH6−/− double deficient mice, the tumor predisposition phenotype is indistinguishable from MSH2−/− or MLH1−/− mice 133, 134, suggesting that MSH3 cooperates with MSH6 in tumor suppression.
Sterility is a characteristic feature of MLH mutant mice (except PMS1) 135, 136, 137. These animals are also susceptible to cancer and display genomic instability, reflecting defective MMR. However, male and female MLH1 and MLH3 knockout mice 135, 136, 137, and male PMS2 knockout mice are completely sterile 138. PMS1 knockout mice are exceptional, because they are fertile, they lack cancer susceptibility, and, apart from a very small increase in mutations in mononucleotide repeats, they appear to be MSI-negative 127. EXO1 defective mice are also sterile 62. It is clear that the loss of fertility in these knockout mice is caused by abnormal meiosis 62, 128, 135, 136, 137.
The characteristics of all MMR knockout mouse models are summarized in Table 2. These data strongly support the ideas that MMR is a basic genome surveillance mechanism and that defects in MMR can promote cancer development. The effects of MMR defects on carcinogenesis appear to be tissue- and species-specific, in a manner that is poorly understood. The effects of MMR defects on meiosis in humans remain poorly characterized. However, MMR clearly plays a critical role during meiosis and/or gamete formation in mice.
Epigenetic silencing of MMR gene expression leads to cancers
Mutations in MMR genes cause genomic instability and MSI in HNPCC and in a subset of sporadic colorectal cancers. However, in a significant fraction of MSI-positive sporadic colon tumors that have an MMR defect, mutations have not been identified in MMR genes. Epigenetic silencing of hMLH1 via promoter hypermethylation strongly down-regulates MMR in many of these cases 139, 140. In contrast, hypermethylation of the hMSH2 gene is rarely observed in tumors with MSI. In fact, it has been reported that more than 95% of MSI-H sporadic tumors demonstrate mutation and/or epigenetic silencing of hMLH1 141. While most studies demonstrate epigenetic silencing of the hMLH1 promoter in sporadic tumors, hypermethylation of the hMLH1 promoter was also recently demonstrated in an HNPCC patient who does not have a germ-line mutation in any MMR gene 142. Interestingly, recent studies suggest that this effect may be heritable 143, 144, 145. Direct evidence that hMLH1 promoter hypermethylation down-regulates hMLH1 gene expression was obtained by treating cells with 5-aza-deoxycytidine, which reversed promoter hypermethylation, and restored hMLH1 gene expression and normal MMR capacity 146, 147.
MMR deficiency and mutations in coding repeat sequences
Previous studies demonstrate that defects in MMR increase the mutation rate in genes containing a simple repeat sequence in coding regions, often referred to as target genes 148. Thus, defects in MMR confer a mutator phenotype. It is presumed that such a mutator phenotype has genome-wide consequences and could increase the frequency of additional genome-destabilizing and cancer-promoting mutations; however, this is a difficult hypothesis to test experimentally. One approach is to selectively analyze the stability of di- and tri-nucleotide tracts within coding regions. For example, Markowitz et al. 149 reported two mutation “hotspots” in the type II transforming growth factor-β receptor (TGF-β RII) gene in MMR-deficient tumor cells from a patient with sporadic colorectal cancer. One of these mutational hotspots fell within a 6-bp GT dinucleotide repeat and the other fell within an (A)10 mononucleotide repeat 149. Both of these hotspots were sites of frequent frameshift mutations that truncated the TGF-β RII gene product. Similar observations have been made in other colorectal tumor cells and many other MSI-positive tumor cells including glioma, gastric, uterine, cervical and squamous head and neck tumors, as well as ulcerative colitis-associated cancer and cecum cancer. Furthermore, in some of these tumor cells, somatic frameshift mutations have been documented in many other genes including Bax, insulin-like growth factor 2 receptor (IGF2-R), transcription factor E2F-4, APC, PTEN, hMSH3, hMSH6, Mre11, MBD4/MED, ACTRII, AIM2, APAF-1, AXIN-2, BCL-10, BLM, Caspase-5, CDX-2, CHK-1, FAS, GRB-14, cell cycle protein hG4-1, KIAA0977, ubiquinone oxidoreductase gene NADH, OGT, Rad50, RHAMM, RIZ, SEC63, SLC23AT, TCF-4, and WISP-3 (reviewed in Ref. 150). These data are consistent with the idea that similar mutations occur on a genome-wide basis and at a much higher rate in MMR-deficient cells than in wild-type cells. Because the genes noted above play critical roles in regulating cell growth or genomic stability, loss-of-function mutations in these genes may be crucial steps in the multi-step pathway of carcinogenesis.
Conclusion and perspectives
The discovery that defects in MMR play a causal role in HNPCC and many MSI-positive sporadic cancers brought immediate clinical relevance to research in the field of eukaryotic MMR. This discovery led to intensive research on and better understanding of the biological roles of MMR in eukaryotic cells, which relate to cancer prevention and therapy. Although the primary role of MMR is to improve replication fidelity by correcting replication-associated base-base mismatches or ID mispairs, important secondary roles are to modulate DNA recombination and facilitate DNA damage signaling. Thus, it is abundantly clear that defects in MMR are “permissive” for carcinogenesis.
The identification that MMR-deficient cells are resistant to certain chemotherapeutic drugs such as temozolomide, procarbazine, or cisplatin has significant impacts on cancer treatments, especially for patients with tumors defective in MMR. It is also known that MMR deficiency can be acquired during chemotherapy by selective mutations in MMR genes 6. Therefore, the risks for chemotherapy are two-fold. First, for patients with MMR-deficient tumors, chemotherapeutic treatments could selectively kill patients' MMR-proficient cells (e.g., blood cells) that undergo proliferation, thereby leading to rapid deaths of the cancer patients. Second, if a patient's tumor is not caused by loss of MMR function, chemotherapeutic treatments may kill the tumor cells; at the same time, the treatment may induce or select for a mutation in MMR genes, which could lead to a secondary cancer. Therefore, novel chemotherapeutic or alternative approaches are needed for cancer patients with or without MSI-positive tumors. Such approaches might include targeted gene therapy, which could selectively restore drug sensitivity in tumor cells defective in MMR or treatment with agents that stimulate apoptosis downstream of MMR in tumor cells. Additional research on mechanisms that selectively kill MMR-deficient cells is also warranted. Such efforts should also include more basic research on the molecular mechanisms of eukaryotic MMR. Understanding these mechanisms will support efforts for developing new therapeutic approaches for patients with HNPCC or other MSI-positive MMR-deficient tumors.
References
Kolodner RD, Marsischky GT . Eukaryotic DNA mismatch repair. Curr Opin Genet Dev 1999; 9:89–96.
Kunkel TA, Erie DA . DNA mismatch repair. Annu Rev Biochem 2005; 74:681–710.
Modrich P, Lahue R . Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem 1996; 65:101–133.
Tiraby JG, Fox MS . Marker discrimination in transformation and mutation of pneumococcus. Proc Natl Acad Sci USA 1973; 70:3541–3545.
Lynch HT, de la Chapelle A . Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet 1999; 36:801–818.
Li GM . The role of mismatch repair in DNA damage-induced apoptosis. Oncol Res 1999; 11: 393–400.
Stojic L, Brun R, Jiricny J . Mismatch repair and DNA damage signalling. DNA Repair (Amst) 2004; 3:1091–1101.
Buermeyer AB, Deschenes SM, Baker SM, Liskay RM . Mammalian DNA mismatch repair. Annu Rev Genet 1999; 33:533–564.
Jiricny J . The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 2006; 7:335–346.
Yang W . Structure and function of mismatch repair proteins. Mutat Res 2000; 460:245–256.
Schofield MJ, Hsieh P . DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol 2003; 57:579–608.
Lahue RS, Au KG, Modrich P . DNA mismatch correction in a defined system. Science 1989; 245:160–164.
Burdett V, Baitinger C, Viswanathan M, Lovett ST, Modrich P . In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proc Natl Acad Sci USA 2001; 98:6765–6770.
Obmolova G, Ban C, Hsieh P, Yang W . Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 2000; 407:703–710.
Lamers MH, Perrakis A, Enzlin JH, Winterwerp HH, de Wind N, Sixma TK . The crystal structure of DNA mismatch repair protein MutS binding to a G•T mismatch. Nature 2000; 407:711–717.
Kadyrov FA, Dzantiev L, Constantin N, Modrich P . Endonucleolytic function of MutLalpha in human mismatch repair. Cell 2006; 126:297–308.
Sancar A, Hearst JE . Molecular matchmakers. Science 1993; 259:1415–1420.
Dao V, Modrich P . Mismatch-, MutS-, MutL-, and helicase II-dependent unwinding from the single-strand break of an incised heteroduplex. J Biol Chem 1998; 273:9202–9207.
Guarne A, Ramon-Maiques S, Wolff EM, et al. Structure of the MutL C-terminal domain: a model of intact MutL and its roles in mismatch repair. Embo J 2004; 23:4134–4145.
Ban C, Yang W . Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell 1998; 95:541–552.
Aronshtam A, Marinus MG . Dominant negative mutator mutations in the mutL gene of Escherichia coli. Nucleic Acids Res 1996; 24:2498–2504.
Junop MS, Obmolova G, Rausch K, Hsieh P, Yang W . Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Mol Cell 2001; 7:1–12.
Li F, Liu Q, Chen YY, et al. Escherichia coli mismatch repair protein MutL interacts with the clamp loader subunits of DNA polymerase III. Mutat Res 2007 Jul 25; doi:10.1016/j.mrfmmm.2007.07.008.
Lopez de Saro FJ, Marinus MG, Modrich P, O'Donnell M . The beta sliding clamp binds to multiple sites within MutL and MutS. J Biol Chem 2006; 281:14340–14349.
Ban C, Yang W . Structural basis for MutH activation in E.coli mismatch repair and relationship of MutH to restriction endonucleases. Embo J 1998; 17:1526–1534.
Lee JY, Chang J, Joseph N, Ghirlando R, Rao DN, Yang W . MutH complexed with hemi- and unmethylated DNAs: coupling base recognition and DNA cleavage. Mol Cell 2005; 20:155–166.
Ramilo C, Gu L, Guo S, et al. Partial reconstitution of human DNA mismatch repair in vitro: characterization of the role of human replication protein A. Mol Cell Biol 2002; 22:2037–2046.
Reenan RA, Kolodner RD . Isolation and characterization of two Saccharomyces cerevisiae genes encoding homologs of the bacterial HexA and MutS mismatch repair proteins. Genetics 1992; 132:963–973.
Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993; 75:1027–1038.
Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993; 75:1215–1225.
Palombo F, Gallinari P, Iaccarino I, et al. GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science 1995; 268:1912–1914.
Drummond JT, Li GM, Longley MJ, Modrich P . Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science 1995; 268:1909–1912.
Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994; 368:258–261.
Li GM, Modrich P . Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc Natl Acad Sci USA 1995; 92:1950–1954.
Nicolaides NC, Papadopoulos N, Liu B, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994; 371:75–80.
Papadopoulos N, Nicolaides NC, Wei YF, et al. Mutation of a mutL homolog in hereditary colon cancer. Science 1994; 263:1625–1629.
Schmutte C, Marinescu RC, Sadoff MM, Guerrette S, Overhauser J, Fishel R . Human exonuclease I interacts with the mismatch repair protein hMSH2. Cancer Res 1998; 58:4537–4542.
Tishkoff DX, Amin NS, Viars CS, Arden KC, Kolodner RD . Identification of a human gene encoding a homologue of Saccharomyces cerevisiae EXO1, an exonuclease implicated in mismatch repair and recombination. Cancer Res 1998; 58:5027–5031.
Tishkoff DX, Boerger AL, Bertrand P, et al. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc Natl Acad Sci USA 1997; 94:7487–7492.
Lin YL, Shivji MK, Chen C, Kolodner R, Wood RD, Dutta A . The evolutionarily conserved zinc finger motif in the largest subunit of human replication protein A is required for DNA replication and mismatch repair but not for nucleotide excision repair. J Biol Chem 1998; 273:1453–1461.
Gu L, Hong Y, McCulloch S, Watanabe H, Li GM . ATP-dependent interaction of human mismatch repair proteins and dual role of PCNA in mismatch repair. Nucleic Acids Res 1998; 26:1173–1178.
Johnson RE, Kovvali GK, Guzder SN, et al. Evidence for involvement of yeast proliferating cell nuclear antigen in DNA mismatch repair. J Biol Chem 1996; 271:27987–27990.
Umar A, Buermeyer AB, Simon JA, et al. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell 1996; 87:65–73.
Longley MJ, Pierce AJ, Modrich P . DNA polymerase delta is required for human mismatch repair in vitro. J Biol Chem 1997; 272:10917–10921.
Zhang Y, Yuan F, Presnell SR, et al. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell 2005; 122:693–705.
Prolla TA, Christie DM, Liskay RM . Dual requirement in yeast DNA mismatch repair for MLH1 and PMS1, two homologs of the bacterial mutL gene. Mol Cell Biol 1994; 14:407–415.
Lipkin SM, Wang V, Jacoby R, et al. MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat Genet 2000; 24:27–35.
Bowers J, Tran PT, Joshi A, Liskay RM, Alani E . MSH-MLH complexes formed at a DNA mismatch are disrupted by the PCNA sliding clamp. J Mol Biol 2001; 306:957–968.
Clark AB, Valle F, Drotschmann K, Gary RK, Kunkel TA . Functional interaction of proliferating cell nuclear antigen with MSH2-MSH6 and MSH2-MSH3 complexes. J Biol Chem 2000; 275:36498–36501.
Flores-Rozas H, Clark D, Kolodner RD . Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat Genet 2000; 26:375–378.
Kleczkowska HE, Marra G, Lettieri T, Jiricny J . hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev 2001; 15:724–736.
Warbrick E . The puzzle of PCNA's many partners. Bioessays 2000; 22:997–1006.
Lau PJ, Kolodner RD . Transfer of the MSH2.MSH6 complex from proliferating cell nuclear antigen to mispaired bases in DNA. J Biol Chem 2003; 278:14–17.
Shell SS, Putnam CD, Kolodner RD . The N terminus of Saccharomyces cerevisiae Msh6 is an unstructured tether to PCNA. Mol Cell 2007; 26:565–578.
Guo S, Presnell SR, Yuan F, Zhang Y, Gu L, Li GM . Differential requirement for proliferating cell nuclear antigen in 5¢ and 3¢ nick-directed excision in human mismatch repair. J Biol Chem 2004; 279:16912–16917.
Amin NS, Nguyen MN, Oh S, Kolodner RD . exo1-Dependent mutator mutations: model system for studying functional interactions in mismatch repair. Mol Cell Biol 2001; 21:5142–5155.
Nielsen FC, Jager AC, Lutzen A, Bundgaard JR, Rasmussen LJ . Characterization of human exonuclease 1 in complex with mismatch repair proteins, subcellular localization and association with PCNA. Oncogene 2004; 23:1457–1468.
Tran PT, Erdeniz N, Symington LS, Liskay RM . EXO1-A multi-tasking eukaryotic nuclease. DNA Repair (Amst) 2004; 3:1549–1559.
Genschel J, Modrich P . Mechanism of 5′-directed excision in human mismatch repair. Mol Cell 2003; 12:1077–1086.
Dzantiev L, Constantin N, Genschel J, Iyer RR, Burgers PM, Modrich P . A defined human system that supports bidirectional mismatch-provoked excision. Mol Cell 2004; 15:31–41.
Genschel J, Bazemore LR, Modrich P . Human exonuclease I is required for 5′ and 3′ mismatch repair. J Biol Chem 2002; 277:13302–13311.
Wei K, Clark AB, Wong E, et al. Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev 2003; 17:603–614.
Guo S, Zhang Y, Yuan F, et al. Regulation of replication protein A functions in mismatch repair by phosphorylation. J Biol Chem, 2006; 281:21607–21616.
Fleck O, Kunz C, Rudolph C, Kohli J . The high mobility group domain protein Cmb1 of Schizosaccharomyces pombe binds to cytosines in base mismatches and opposite chemically altered guanines. J Biol Chem 1998; 273:30398–30405.
Javaherian K, Liu JF, Wang JC . Nonhistone proteins HMG1 and HMG2 change the DNA helical structure. Science 1978; 199:1345–1346.
Javaherian K, Sadeghi M, Liu LF . Nonhistone proteins HMG1 and HMG2 unwind DNA double helix. Nucleic Acids Res 1979; 6:3569–3580.
Yuan F, Gu L, Guo S, Wang C, Li GM . Evidence for involvement of HMGB1 protein in human DNA mismatch repair. J Biol Chem 2004; 279:20935–20940.
Wang H, Hays JB . Signaling from DNA mispairs to mismatch-repair excision sites despite intervening blockades. Embo J 2004; 23:2126–2133.
Allen DJ, Makhov A, Grilley M, et al. MutS mediates heteroduplex loop formation by a translocation mechanism. Embo J 1997; 16:4467–4476.
Fishel R . Mismatch repair, molecular switches, and signal transduction. Genes Dev 1998; 12:2096–2101.
Gradia S, Acharya S, Fishel R . The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell 1997; 91:995–1005.
Jiang J, Bai L, Surtees JA, Gemici Z, Wang MD, Alani E . Detection of high-affinity and sliding clamp modes for MSH2-MSH6 by single-molecule unzipping force analysis. Mol Cell 2005; 20:771–781.
Mendillo ML, Mazur DJ, Kolodner RD . Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. J Biol Chem 2005; 280:22245–22257.
Pluciennik A, Modrich P . From the cover: protein roadblocks and helix discontinuities are barriers to the initiation of mismatch repair. Proc Natl Acad Sci USA 2007; 104:12709–12713.
Au KG, Welsh K, Modrich P . Initiation of methyl-directed mismatch repair. J Biol Chem 1992; 267:12142–12148.
Kat A, Thilly WG, Fang WH, Longley MJ, Li GM, Modrich P . An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc Natl Acad Sci USA 1993; 90:6424–6428.
Koi M, Umar A, Chauhan DP, et al. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res 1994; 54:4308–4312.
Fink D, Nebel S, Norris PS, et al. The effect of different chemotherapeutic agents on the enrichment of DNA mismatch repair-deficient tumour cells. Br J Cancer 1998; 77:703–708.
Fujii H, Shimada T . Isolation and characterization of cDNA clones derived from the divergently transcribed gene in the region upstream from the human dihydrofolate reductase gene. J Biol Chem 1989; 264:10057–10064.
Linton JP, Yen J-YJ, Selby E, et al. Dual bidirectional promoters at the mouse DHFR locus: cloning and characterization of two mRNA classes of the divergently transcribed Rep-1 gene. Mol Cell Biol 1989; 9:3058–3072.
Drummond JT, Genschel J, Wolf E, Modrich P . DHFR/MSH3 amplification in methotrexate-resistant cells alters the hMutSalpha/hMutSbeta ratio and reduces the efficiency of base-base mismatch repair. Proc Natl Acad Sci USA 1997; 94:10144–10149.
Marra G, Iaccarino I, Lettieri T, Roscilli G, Delmastro P, Jiricny J . Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc Natl Acad Sci USA 1998; 95:8568–8573.
Brown KD, Rathi A, Kamath R, et al. The mismatch repair system is required for S-phase checkpoint activation. Nat Genet 2003; 33:80–84.
Duckett DR, Drummond JT, Murchie AI, et al. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc Natl Acad Sci USA 1996; 93:6443–6447.
Gong JG, Costanzo A, Yang HQ, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 1999; 399:806–809.
Stojic L, Mojas N, Cejka P, et al. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev 2004; 18:1331–1344.
Kim WJ, Rajasekaran B, Brown KD . MLH1 and ATM-dependent MAP kinase signaling is activated through C-Abl in response to the alkylator N-methyl-N′-nitro-N-nitrosoguanidine. J Biol Chem, 2007; 282:32021–32031.
Shimodaira H, Yoshioka-Yamashita A, Kolodner RD, Wang JY . Interaction of mismatch repair protein PMS2 and the p53-related transcription factor p73 in apoptosis response to cisplatin. Proc Natl Acad Sci USA 2003; 100:2420–2425.
Yoshioka K, Yoshioka Y, Hsieh P . ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell 2006; 22:501–510.
Schaetzlein S, Kodandaramireddy NR, Ju Z, et al. Exonuclease-1 deletion impairs DNA damage signaling and prolongs lifespan of telomere-dysfunctional mice. Cell 2007; 130:863–877.
York SJ, Modrich P . Mismatch repair-dependent iterative excision at irreparable O6-methylguanine lesions in human nuclear extracts. J Biol Chem 2006; 281:22674–22683.
Kolodner RD, Putnam CD, Myung K . Maintenance of genome stability in Saccharomyces cerevisiae. Science 2002; 297:552–557.
Vogelstein B, Kinzler KW . Cancer genes and the pathways they control. Nat Med 2004; 10:789–799.
Harfe BD, Jinks-Robertson S . DNA mismatch repair and genetic instability. Annu Rev Genet 2000; 34:359–399.
Worth L Jr, Clark S, Radman M, Modrich P . Mismatch repair proteins MutS and MutL inhibit RecA-catalyzed strand transfer between diverged DNAs. Proc Natl Acad Sci USA 1994; 91:3238–3241.
Goldfarb T, Alani E . Distinct roles for the Saccharomyces cerevisiae mismatch repair proteins in heteroduplex rejection, mismatch repair and nonhomologous tail removal. Genetics 2005; 169:563–574.
Sugawara N, Goldfarb T, Studamire B, Alani E, Haber JE . Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc Natl Acad Sci USA 2004; 101:9315–9320.
Myung K, Datta A, Chen C, Kolodner RD . SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat Genet 2001; 27:113–116.
Doherty KM, Sharma S, Uzdilla L, et al. RECQ1 helicase interacts with human mismatch repair factors that regulate genetic recombination. J Biol Chem, 2005; 280:28085–28094.
Pedrazzi G, Bachrati CZ, Selak N, et al. The Bloom¢s syndrome helicase interacts directly with the human DNA mismatch repair protein hMSH6. Biol Chem 2003; 384:1155–1164.
Peng M, Litman R, Xie J, Sharma S, Brosh RM Jr, Cantor SB . The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. Embo J 2007; 26:3238–3249.
Zhang N, Liu X, Li L, Legerski R . Double-strand breaks induce homologous recombinational repair of interstrand cross-links via cooperation of MSH2, ERCC1-XPF, REV3, and the Fanconi anemia pathway. DNA Repair (Amst) 2007; 6:1670–1678.
Zhang N, Lu X, Zhang X, Peterson CA, Legerski RJ . hMutSbeta is required for the recognition and uncoupling of psoralen interstrand cross-links in vitro. Mol Cell Biol 2002; 22:2388–2397.
Gu L, Wu J, Qiu L, Jennings CD, Li GM . Involvement of DNA mismatch repair in folate deficiency-induced apoptosis small star, filled. J Nutr Biochem 2002; 13:355–363.
Wilson TM, Vaisman A, Martomo SA, et al. MSH2-MSH6 stimulates DNA polymerase eta, suggesting a role for A:T mutations in antibody genes. J Exp Med 2005; 201:637–645.
Casali P, Pal Z, Xu Z, Zan H . DNA repair in antibody somatic hypermutation. Trends Immunol 2006; 27:313–321.
Schrader CE, Guikema JE, Linehan EK, Selsing E, Stavnezer J . Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J Immunol 2007; 179:6064–6071.
Bardwell PD, Woo CJ, Wei K, et al. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat Immunol 2004; 5:224–229.
Winter DB, Phung QH, Umar A, et al. Altered spectra of hypermutation in antibodies from mice deficient for the DNA mismatch repair protein PMS2. Proc Natl Acad Sci USA 1998; 95:6953–6958.
Pearson CE, Nichol Edamura K, Cleary JD . Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet 2005; 6:729–742.
Gacy AM, Goellner G, Juranic N, Macura S, McMurray CT . Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 1995; 81:533–540.
Manley K, Shirley TL, Flaherty L, Messer A . Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet 1999; 23:471–473.
Owen BA, Yang Z, Lai M, et al. (CAG)(n)-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat Struct Mol Biol 2005; 12:663–670.
Aaltonen LA, Peltomaki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993; 260:812–816.
Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M . Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993; 363:558–561.
Thibodeau SN, Bren G, Schaid D . Microsatellite instability in cancer of the proximal colon. Science 1993; 260:816–819.
Strand M, Prolla TA, Liskay RM, Petes TD . Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 1993; 365:274–276.
Parsons R, Li GM, Longley MJ, et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 1993; 75:1227–1236.
Umar A, Boyer JC, Thomas DC, et al. Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J Biol Chem 1994; 269:14367–14370.
Lindblom A, Tannergard P, Werelius B, Nordenskjold M . Genetic mapping of a second locus predisposing to hereditary non-polyposis colon cancer. Nat Genet 1993; 5:279–282.
Li GM . DNA mismatch repair and cancer. Front Biosci 2003; 8:d997–d1017.
Liberti SE, Rasmussen LJ . Is hEXO1 a cancer predisposing gene? Mol Cancer Res 2004; 2:427–432.
Sun X, Zheng L, Shen B . Functional alterations of human exonuclease 1 mutants identified in atypical hereditary nonpolyposis colorectal cancer syndrome. Cancer Res 2002; 62:6026–6030.
Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998; 58:5248–5257.
Gu L, Cline-Brown B, Zhang F, Qiu L, Li GM . Mismatch repair deficiency in hematological malignancies with microsatellite instability. Oncogene 2002; 21:5758–5764.
Gu L, Wu J, Zhu BB, Li GM . Deficiency of a novel mismatch repair activity in a bladder tumor cell line. Nucleic Acids Res 2002; 30:2758–2763.
Prolla TA, Baker SM, Harris AC, et al. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet 1998; 18:276–279.
Wei K, Kucherlapati R, Edelmann W . Mouse models for human DNA mismatch-repair gene defects. Trends Mol Med 2002; 8:346–353.
de Wind N, Dekker M, Berns A, Radman M, te Riele H . Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 1995; 82:321–330.
Reitmair AH, Schmits R, Ewel A, et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet 1995; 11:64–70.
Edelmann W, Yang K, Umar A, et al. Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell 1997; 91:467–477.
Kolodner RD, Tytell JD, Schmeits JL, et al. Germ-line msh6 mutations in colorectal cancer families. Cancer Res 1999; 59:5068–5074.
de Wind N, Dekker M, Claij N, et al. HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nat Genet 1999; 23:359–362.
Edelmann W, Umar A, Yang K, et al. The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Res 2000; 60:803–807.
Baker SM, Plug AW, Prolla TA, et al. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet 1996; 13:336–342.
Edelmann W, Cohen PE, Kane M, et al. Meiotic pachytene arrest in MLH1-deficient mice. Cell 1996; 85:1125–1134.
Lipkin SM, Moens PB, Wang V, et al. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat Genet 2002; 31:385–390.
Baker SM, Bronner CE, Zhang L, et al. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell 1995; 82:309–319.
Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 1997; 57:808–811.
Grady WM, Markowitz SD . Genetic and epigenetic alterations in colon cancer. Annu Rev Genomics Hum Genet 2002; 3:101–128.
Thibodeau SN, French AJ, Cunningham JM, et al. Microsatellite instability in colorectal cancer: different mutator phenotypes and the principal involvement of hMLH1. Cancer Res 1998; 58:1713–1718.
Gazzoli I, Loda M, Garber J, Syngal S, Kolodner RD . A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res 2002; 62:3925–3928.
Chan TL, Yuen ST, Kong CK, et al. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat Genet 2006; 38:1178–1183.
Hitchins MP, Wong JJ, Suthers G, et al. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med 2007; 356:697–705.
Suter CM, Martin DI, Ward RL . Germline epimutation of MLH1 in individuals with multiple cancers. Nat Genet 2004; 36:497–501.
Veigl ML, Kasturi L, Olechnowicz J, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci USA 1998; 95:8698–8702.
Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 1998; 95:6870–6875.
Kinzler KW, Vogelstein B . Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 1997; 386:761, 763.
Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995; 268:1336–1338.
Duval A, Hamelin R . Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res 2002; 62:2447–2454.
Acknowledgements
The author acknowledges research support from the National Institutes of Health (GM072756 and CA115942) and the Kentucky Lung Cancer Research Program, USA. The author regrets the lack of citations for many important observations mentioned in the text, but their omission is made necessary by restrictions in the preparation of this review. The author holds the James-Gardner Endowed Chair in Cancer Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, GM. Mechanisms and functions of DNA mismatch repair. Cell Res 18, 85–98 (2008). https://doi.org/10.1038/cr.2007.115
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2007.115
Keywords
This article is cited by
-
DNA mismatch repair system regulates the expression of PD-L1 through DNMTs in cervical cancer
Cancer Cell International (2024)
-
APOBEC3-mediated mutagenesis in cancer: causes, clinical significance and therapeutic potential
Journal of Hematology & Oncology (2023)
-
Germline-related molecular phenotype in Metazoa: conservation and innovation highlighted by comparative transcriptomics
EvoDevo (2023)
-
Synergistic effect of cryptotanshinone and temozolomide treatment against human glioblastoma cells
Scientific Reports (2023)
-
The DNA damage response in advanced ovarian cancer: functional analysis combined with machine learning identifies signatures that correlate with chemotherapy sensitivity and patient outcome
British Journal of Cancer (2023)
