
Abstract
Aim:
To investigate whether microRNA-21 was involved in mediating the chemoresistance of prostate cancer cells to docetaxel.
Methods:
A microarray technique was used to determine the miRNA profile in docetaxel-resistant PC3 cells. Real-time PCR was used to confirm the array results. miR-21 mimics and inhibitors were synthesized and introduced to cells using Lipofectamine 2000. Cell proliferation was examined with the CCK-8 assay. Luciferase reporter containing PDCD 3′UTR was constructed and the activity was detected by a dual luciferase assay. PDCD4 protein expression was evaluated using Western blot.
Results:
A docetaxel-resistant prostate cancer PC3 cell line (PC3R) was established . Using microarrays, miR-21 was found to be up-regulated in PC3R cells. Ectopic expression of miR-21 increased the resistance to docetaxel in PC3 wild type cells. In contrast, silencing of miR-21 in PC3R cells sensitized the cells to docetaxel. The IC50 values for miR-21-silencing cells and control cells were 28.31 and 35.89 nmol/L, respectively. PDCD4, a direct target gene of miR-21, could mediate chemoresistance to docetaxel in PC3 cells.
Conclusion:
Our findings suggest that miR-21 contributed to the resistance of PC3 cells to docetaxel, and that targeting miR-21 may offer a promising therapeutic approach in sensitizing prostate cancer to docetaxel treatment.
Similar content being viewed by others


Introduction
Prostate cancers are generally androgen-sensitive at the initial diagnosis, and most patients are therefore treated with antiandrogen therapy. However, patients eventually develop androgen-independent prostate cancers (AIPC) that will inevitably result in metastasis and death. Docetaxel is the standard of care for men diagnosed with AIPC1. Docetaxel binds to the β-tubulin subunit, causing stabilization of microtubules, which leads to mitotic arrest and subsequent apoptosis2. Docetaxel has been administered to men with AIPC in clinical trials, where it has been found to prolong survival in men with this disease3. However, there are currently no effective approaches for treating chemoresistant prostate cancers.
miRNAs are small non-coding RNAs that have been identified as post-transcriptional regulators of gene expression4. miRNAs bind to the 3′ untranslated regions (UTR) of target mRNAs, either preventing their translation or causing target degradation. Accumulating evidence has proven that miRNAs play crucial roles in multiple biological processes5. Meanwhile, deregulation of miRNAs is involved in a wide range of human diseases, including cancer6. In human cancer, miRNAs are frequently deregulated and can function as oncogenes or tumor suppressor genes7. Recent studies showed that miRNAs also participated in the resistance to chemotherapeutic agents in cancer cells8, 9, 10.
In this study, we hypothesized that altered expression of miRNAs might be involved in mediating resistance to docetaxel in prostate cancer cells. We established a docetaxel-resistant prostate cancer PC3 cell line (PC3R). Using microarrays, we found that a number of miRNAs were significantly altered in the docetaxel-resistant PC3 cells. miR-21, one of the miRNAs identified by microarrays, was up-regulated in PC3R cells. Ectopic expression of miR-21 increased the resistance of PC3 to docetaxel in the wild type cells. In contrast, silencing of miR-21 with transient transfection of its inhibitors in docetaxel-resistant PC3 cells led to sensitizing the cells to docetaxel. Furthermore, we found that PDCD4, a direct target gene of miR-21, can mediate miR-21 induction of chemoresistance to docetaxel in PC3 cells. Our findings suggest that targeting miR-21 may offer a promising therapeutic approach in sensitizing prostate cancer to chemotherapeutic agents.
Materials and methods
Cell culture
The human androgen-insensitive prostate carcinoma cell line, PC3, was obtained from the American Type Culture Collection (Rockville, MD, USA). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum at 37 °C in a humidified atmosphere with 5% CO2. PC3 cells were made resistant to docetaxel (PC3R) by culturing them under continuous exposure to docetaxel (10 nmol/L) for 30 days. The resistance factor for docetaxel was evaluated as the ratio of the 50% inhibitory concentration (IC50) for PC3R divided by the IC50 for PC3wt; the IC50 values were determined in vitro using the CCK-8 assay kit (Dojindo Corp, Japan).
miRNA microarrays
Small RNA was prepared using the miRNA Isolation Kit (Ambion, Austin, USA). miRNA microarrays were performed as described in detail on the website of CapitalBio (http://www.capitalbio.com). Briefly, fluorescein-labeled miRNAs were used for hybridization on each miRNA microarray chip containing 988 human miRNA probes in triplicate. Raw data were normalized and analyzed using the GenePix Pro 4.0 software. Statistical comparisons were done with SAM analysis.
Real-time PCR assays
The total RNA was extracted using TRIzol reagent (Invitrogen, CA, USA). Quantification of miR-21, miR-210, miR-192, and miR-296-5p was performed with a stem-loop RT-PCR miRNA kit (Ribobio, Guangzhou, China). Real-time PCR was performed using the SYBR Premix Ex Taq (TaKaRa). RNU6B was used as the internal control. All quantitative real-time PCR was performed with the 7300 real-time PCR system (Applied Biosystems, Foster City, CA).
Oligonucleotide transfection
miR-21 mimics and inhibitors were synthesized by RiboBio, Guangzhou, China. The nucleotides of miRNA inhibitors contained 2'-O-methyl modifications at each base. PDCD4 siRNA was synthesized by RiboBio, Guangzhou, China. The sense and antisense sequences were 5′-UUUCAUUCUUUAUUUCCUCAGUCCC-3′ and 5′-GGGACUGAGGAAAUAAAGAAUGAAA-3′, respectively. The transfections were performed with Lipofectamine 2000 (Invitrogen). The final concentration of small RNAs was 100 nmol/L.
Vector construction
To construct a luciferase reporter with the wild-type PDCD4 3′UTR, we placed the 3′UTR of PDCD4 downstream of a cytomegalovirus (CMV) promoter-driven firefly luciferase cassette in a pCDNA3 vector. The PDCD4 3′UTR sequences were amplified by PCR from genomic DNA using the following primers: sense, 5′-AATATAAGAACTCTTGCAGTC-3′, anti-sense: 5′-GAAGATACATTCCAATCTTGC-3′. To construct the mutated PDCD4 3'UTR, we first generated two fragments with overlapping mutation sites using two sets of primers: sense, 5′-AGTGGAATATTCTAATTTCGTACCTTTTGTAAGTG-3′, anti-sense, 5′-GCACTTACAAAAGGTACGAAATTAGAATATTCCAC-3′. These two fragments were then annealed and used as templates to amplify a mutated PDCD4 3'UTR. To create the PDCD4 expressing vector, we placed the PDCD4 coding region (CDS) downstream of a cytomegalovirus (CMV) promoter-driven cassette in a pCDNA3 vector. The sequences of PDCD4 CDS were amplified by PCR from cDNA using the following primers: sense, 5′-AGTTTTGTGGAATAGATGACC-3′, anti-sense: 5′-TGCAAGAGTTCTTATATTCAG-3′.
Dual-luciferase reporter assay
PC3 cells were cultured in 96-well plates and co-transfected with 50 ng luciferase reporter, 5 pmol miR-21 mimics and 5 ng pRL-CMV Renilla luciferase reporter. After 36 h, luciferase activity was measured using the dual-luciferase reporter assay system (Promega, Madison, USA).
Cell proliferation assay
Cell proliferation was measured using the CCK-8 assay kit (Dojindo Corp, Japan). Approximately 2500 cells were plated into each well of a 96-well plate, where 10 μL CCK-8 was added to 90 μL of culture medium. The cells were subsequently incubated for 2 h at 37 °C and the optical density was measured at 450 nm.
Western blotting
Protein expression of PDCD4 was determined using the mouse monoclonal anti-PDCD4 antibody (Santa Cruz Biotechnology, CA, USA) following standard Western blotting procedures. Protein expression was normalized for expression of beta-actin, detected using a mouse monoclonal anti-actin antibody (Sigma). The proteins were detected using ECL reagents (Pierce, IL, USA).
Statistical analysis
The results are presented as means±SEM. The data were subjected to Student's t-test (two-tailed, P<0.05 was considered significant).
Results
Exposure to docetaxel and IC50 values of PC3wt and PC3R cells
We first established the docetaxel-resistant PC3 (PC3R) using the method evaluated by the previous report11. PC3 cells were made resistant to docetaxel by culturing them under continuous exposure to docetaxel (10 nmol/L) for 30 days. After exposure to different doses of docetaxel for 2 days, the IC50 values for PC3wt and PC3R cells were 1.16 and 33.48 nmol/L, respectively (Figure 1).

The inhibitory rate of PC3wt and PC3R cells in response to docetaxel treatment.
miRNAs expression changes in docetaxel-resistant PC3 cells
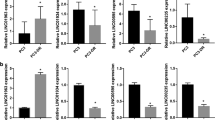
To determine whether miRNAs could be involved in docetaxel resistance in prostate cancer PC3 cells, we used microarray-based analysis to determine miRNA expression profiles in PC3R and PC3wt cells. As shown in Table 1, the expression of 10 miRNAs changed with 6 upregulated miRNAs (miR-21, miR-210, miR-30d, miR-22, and miR-30a) and 4 downregulated miRNAs (miR-565, miR-296-5p, miR-192, and miR-19a) in PC3R cells, compared with the PC3wt cells. To confirm the microarray results, quantitative RT-PCR analysis was performed to detect several of the most abundantly expressed miRNAs, including miR-21, miR-210, miR-296-5p, and miR-192. Small nuclear RNA U6 was the endogenous control used for normalization. The results were highly consistent with the miRNA microarray analysis (Figure 2).

Relative expression of four miRNA in PC3wt and PC3R cells. (A) Relative expression of miR-21 in PC3wt and PC3R cells. (B) Relative expression of miR-210 in PC3wt and PC3R cells. (C) Relative expression of miR-192 in PC3wt and PC3R cells. (D) Relative expression of miR-296-5p in PC3wt and PC3R cells. The expression was determined by quantitative real-time PCR. U6 snRNA served as the internal control. Error bars represent SEM. n=3. bP<0.05 vs PC3wt cells.
Overexpression of miR-21 increased the resistance to docetaxel in PC3wt cells
To investigate the potential mechanism and function of these altered miRNAs, we next focused on one miRNA, miR-21, an oncogenic modulator in multiple cancers12, 13, 14, 15. miR-21 is significantly upregulated in PC3R cells compared with PC3wt cells (Figure 2A), suggesting that increased expression of miR-21 might contribute to the resistance of PC3 cells to docetaxel. Therefore, we performed the ectopic expression of miR-21 by transfection with miR-21 mimics in PC3wt cells. As shown in Figure 3A, introduction of miR-21 into PC3wt cells resulted in an increase of cell proliferation compared to those transfected with the negative control. Furthermore, transfection with miR-21 showed increased cell viability upon treatment with docetaxel (Figure 3B). The IC50 values for miR-21 and control cells were 1.58 and 1.15 nmol/L, respectively, suggesting that ectopic miR-21 expression can decrease the sensitivity to docetaxel in PC3 cells.

Ectopic expression of miR-21 increased the resistance of PC3wt cells to docetaxel. (A) Cell proliferation of PC3wt cells after transfection with miR-21 mimics every 24 h for three days. Error bars represent SEM.n=3. bP<0.05. Statistical analysis was performed using Student's t-test. (B) The inhibitory rate of PC3wt cells after transfection with miR-21 mimics or control RNA for 24 h and treatment with different doses of docetaxel for another 48 h. Statistical analysis was performed using Paired t-test, P<0.01
Attenuation of the resistance to docetaxel by inhibition of miR-21 expression in PC3R cells
As miR-21 was highly expressed in PC3R cells, we next explored whether resistance to docetaxel could be attenuated by silencing miR-21 expression with miR-21 inhibitors (2'-O-methyl oligoribonucleotides). Cell proliferation was reduced in PC3R cells when miR-21 was specifically knocked down by miR-21 inhibitors (Figure 4A). We further found that PC3R cells transfected with miR-21 inhibitors showed reduced survival upon treatment with docetaxel (Figure 4B). The IC50 values for miR-21 inhibitors and control cells were 28.31 and 35.89 nmol/L, respectively. Taken together, these findings suggest that miR-21 contributes to the resistance of PC3R cells to docetaxel.

Silencing of miR-21 expression in docetaxel-resistant PC3 cells led to sensitization of the cells to docetaxel. (A) Cell proliferation of PC3R cells after transfection with miR-21 inhibitors every 24 h for three days. Error bars represent SEM. n=3. bP<0.05. Statistical analysis was performed using Student's t-test. (B) The inhibitory rate of PC3R cells after transfection with miR-21 inhibitors or control RNA for 24 h and treatment with different doses of docetaxel for another 48 h. Statistical analysis was performed using Paired t-test, P<0.05
PDCD4, a direct target gene of miR-21, can mediate miR-21-induced chemoresistance to docetaxel in PC3 cells
We next sought to identify a candidate target gene that could help mediate the miR-21-induced chemoresistance to docetaxel in PC3 cells. Recently, PDCD4, programmed cell death protein 4, was reported to be a direct target gene of miR-21 in colorectal cancer16. PDCD4 has been shown to be involved in the sensitivity to chemotherapy17. Intriguingly, we found that the expression of PDCD4 was downregualted in PC3R cells compared with PC3wt cells (Figure 5A), which was inversely correlated with the expression of miR-21. This result indicates that PDCD4 might be involved in the miR-21-induced chemoresistance to docetaxel. To determine whether PDCD4 is also regulated by miR-21 in prostate cancer cells, we constructed the full-length fragments of the PDCD4 mRNA 3'UTR (either wild-type or mutant), and placed them downstream of the luciferase reporter gene. In PC3 cells, miR-21 was found to reduce the relative luciferase activity with the wild-type 3'UTR of PDCD4, but not with the mutant-type (Figure 5B). Furthermore, the expression of PDCD4 decreased in PC3 cells when miR-21 was ectopically expressed. On the other hand, silencing of miR-21 increased PDCD4 expression in PC3R cells (Figure 5C). These results show that miR-21 can directly target PDCD4 expression in PC3 cells.

PDCD4 is a direct target gene of miR-21 in PC3 cells. (A) Western blot of PDCD4 expression in PC3wt and PC3R cells. (B) Luciferase activity assays of miR-21 co-transfected with various luciferase reporters. The luciferase activity of each sample was normalized to the Renilla luciferase activity. Error bars represent SEM. bP<0.05. Statistical analysis was performed using Student's t-test. (C) Western blot of PDCD4 expression in PC3wt transfected with miR-21 or control RNA, and PC3R cells transfected with miR-21 inhibitors or control RNA. n=3.
To explore the potential role of PDCD4 in miR-21-mediated chemoresistance to docetaxel, we synthesized a small-interfering RNA of PDCD4, which significantly reduced PDCD4 expression (Figure 6A). Remarkably, silencing of PDCD4 increased the cell viability in PC3 cells (Figure 6B, P<0.05). The IC50 values for PDCD4 siRNA and control cells were 1.78 and 1.18 nmol/L, respectively, which was similar to the phenotype induced by miR-21. On the other hand, we determined whether ectopic expression of PDCD4 could circumvent the resistance phenotype of PC3R cells. We constructed a vector containing the entire PDCD4 coding sequence. As shown in Figure 6C and 6D, ectopic expression of PDCD4 increased the PDCD4 protein levels in PC3R cells and could attenuate the resistance phenotype of PC3R cells (P<0.05). The IC50 values for PDCD4 vector and control vector were 26.07 and 30.26 nmol/L, respectively. These findings suggest that PDCD4 is indeed a functional target for miR-21-induced chemoresistance to docetaxel in PC3 cells.

PDCD4 can mediate chemoresistance to docetaxel in PC3 cells. (A) Western blot of PDCD4 expression in PC3wt transfected with PDCD4 siRNA or control RNA. (B) The inhibitory rate of PC3wt cells after transfection with PDCD4 siRNA or control RNA for 24 h and treatment with different doses of docetaxel for another 48 h. (C) Western blot of PDCD4 expression in PC3R cells transfected with PDCD4 vector or the control vector. (D) The inhibitory rate of PC3R cells after transfection with the PDCD4 vector or control vector for 24 h and treatment with different doses of docetaxel for another 48 h.
Discussion
Intrinsic chemoresistance constitutes a major problem in the therapy of malignant tumors. In previous studies, great progress has been made in the discovery of potential contributors to the resistance to chemotherapy, which allowed us to identify candidate chemoresistance-related protein-coding genes in cancers. However, little is known about the functional non-coding sequences, particularly miRNAs, in resistance to cancer therapy. In this study, we found that miR-21 expression was increased in docetaxel-resistant PC3 cells. We further identified miR-21 as an important regulator of prostate cancer cell resistance to docetaxel, which provides new evidence that miRNAs may be involved in the tumor resistance to chemotherapy.
MiR-21 has been described as an oncogenic miRNA18 and increased levels of miR-21 have been found in very diverse cancer types, including glioblastoma12 and breast13, liver14, and prostate cancers15. Recent studies have indicated that aberrant expression of miR-21 contributes to the malignant phenotype, such as proliferation, apoptosis and metastasis18. Accordantly, miR-21 has been shown to suppress a network of tumor suppressive pathways14, 19, 20. In this study, we evaluated the role of miR-21 in mediating chemoresistance to docetaxel in PC3 cells. Our results show that ectopic expression of miR-21 can decrease sensitivity to docetaxel in PC3 cells and that inhibition of miR-21 expression could attenuate the resistance to docetaxel in PC3R cells. These findings suggest that miR-21 contributes to the resistance of PC3R cells to docetaxel. It is noteworthy that the effect of miR-21 on the resistance of PC3R cells to docetaxel is limited, indicating that multiple factors might contribute to the docetaxel resistance in PC3R cells. As docetaxel is known to induce expression of the drug efflux pumps in the plasma membrane, we also examined the expression of P-gp and MRP1 genes, two main drug-efflux pumps, in PC3R and PC3 wild type cells using real time PCR. The results show that both P-gp and MRP1 are upregulated in the PC3R cells compared with that in PC3 wild type cells (Supplementary Figure S1), indicating that drug efflux pumps might also be involved in mediating chemoresistance to docetaxel in PC3R cells.
Programmed cell death 4 (PDCD4), is a novel suppressor of tumorigenesis, tumor progression and invasion21, 22, 23. PDCD4 was originally characterized as an inhibitor of cellular transformation in the mouse cell culture model24. PDCD4 is expressed in normal tissues, and there is evidence that its expression is suppressed in various tumors, including lung cancer25, breast cancer26 and hepatocellular carcinoma27. PDCD4 interacts with translation initiation factors, eIF4A and eIF4G, to inhibit translation in a mRNA-specific fashion28. Consequently, PDCD4 has been found to inhibit pro-oncogenic events, such as activation of activator protein-1 (AP-1)29, 30, anchorage-independent growth29, and invasion23. Recently, PDCD4 was also shown to be involved in the sensitivity of chemotherapy by cisplatin and paclitaxel17. The effects of PDCD4 expression on drug sensitivity are due to the down-regulation of YB-1 expression. Here, we found that miR-21 can directly down-regulate the expression of PDCD4 by targeting its 3'UTR in PC3 cells. Our results show that silencing of PDCD4 expression increased the cell viability and resistance to docetaxel in PC3 cells, suggesting that PDCD4 is a functional target for miR-21-induced chemoresistance to docetaxel. As docetaxel could induce mitotic arrest and apoptosis in prostate cancer cells, we also investigated the effects of miR-21 and PDCD4 on the docetaxel-induced mitotic arrest and apoptosis using flow cytometry and caspase 3/7 activity analysis. We found that miR-21 and PDCD4 did not significantly influence cell cycle profiles in PC3 or PC3R cells (data not shown). However, as shown in Supplementary Figure S2, silencing of miR-21 or ectopic expression of PDCD4 can increase the caspase 3/7 activities in PC3R cells, indicating that miR-21 and PDCD4 have a major effect on docetaxel-induced apoptosis in docetaxel-resistant prostate cells.
In conclusion, our data show that increased miR-21 expression plays important roles in the resistance to docetaxel in the prostate cancer PC3 cells. Meanwhile, PDCD4, a direct and functional target of miR-21, can mediate miR-21-induced chemoresistance to docetaxel in PC3 cells. The miR-21/ PDCD4 axis provides a new avenue toward understanding the mechanism of chemoresistance and may help us develop potential therapeutics against prostate cancer.
Author contribution
Ding-wei YE and Guo-hai SHI designed the research; Guo-hai SHI, Xu-dong YAO, Shi-ling ZHANG, Bo DAI, and Wen-jun XIAO performed the research; Hai-liang ZHANG and Yi-jun SHEN contributed analytic tools; Guo-hai SHI, Yao ZHU, and Yi-ping ZHU analyzed the data; Guo-hai SHI, Ding-wei YE, and Chun-guang MA wrote the paper.
References
Pazdur R, Kudelka AP, Kavanagh JJ, Cohen PR, Raber MN . The taxoids: paclitaxel (Taxol) and docetaxel (Taxotere). Cancer Treat Rev 1993; 19: 351–86.
Hagisawa S, Mikami T, Sato Y . Docetaxel-induced apoptosis in the mitotic phase: electron microscopic and cytochemical studies of human leukemia cells. Med Electron Microsc 1999; 32: 167–74.
Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004; 351: 1502–12.
Bartel DP . MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–97.
He L, Hannon GJ . MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 2004; 5: 522–31.
Calin GA, Croce CM . MicroRNA signatures in human cancers. Nat Rev Cancer 2006; 6: 857–66.
Esquela-Kerscher A, Slack FJ . Oncomirs-microRNAs with a role in cancer. Nat Rev Cancer 2006; 6: 259–69.
Song B, Wang Y, Xi Y, Kudo K, Bruheim S, Botchkina GI, et al. Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene 2009; 28: 4065–74.
Fujita Y, Kojima K, Hamada N, Ohhashi R, Akao Y, Nozawa Y, et al. Effects of miR-34a on cell growth and chemoresistance in prostate cancer PC3 cells. Biochem Biophys Res Commun 2008; 377: 114–9.
Chen R, Alvero AB, Silasi DA, Kelly MG, Fest S, Visintin I, et al. Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene 2008; 27: 4712–23.
Lo Nigro C, Maffi M, Fischel JL, Formento P, Milano G, Merlano M . The combination of docetaxel and the somatostatin analogue lanreotide on androgen-independent docetaxel-resistant prostate cancer: experimental data. BJU Int 2008; 102: 622–7.
Chan JA, Krichevsky AM, Kosik KS . MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 2005; 65: 6029–33.
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res 2005; 65: 7065–70.
Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T, et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007; 133: 647–58.
Ribas J, Ni X, Haffner M, Wentzel EA, Salmasi AH, Chowdhury WH, et al. miR-21: an androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res 2009; 69: 7165–9.
Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008; 27: 2128–36.
Shiota M, Izumi H, Tanimoto A, Takahashi M, Miyamoto N, Kashiwagi E, et al. Programmed cell death protein 4 down-regulates Y-box binding protein-1 expression via a direct interaction with Twist1 to suppress cancer cell growth. Cancer Res 2009; 69: 3148–56.
Selcuklu SD, Donoghue MT, Spillane C . miR-21 as a key regulator of oncogenic processes. Biochem Soc Trans 2009; 37: 918–25.
Zhu S, Si ML, Wu H, Mo YY . MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem 2007; 282: 14328–36.
Sayed D, Rane S, Lypowy J, He M, Chen IY, Vashistha H, et al. MicroRNA-21 targets Sprouty2 and promotes cellular outgrowths. Mol Biol Cell 2008; 19: 3272–82.
Hilliard A, Hilliard B, Zheng SJ, Sun H, Miwa T, Song W, et al. Translational regulation of autoimmune inflammation and lymphoma genesis by programmed cell death 4. J Immunol 2006; 177: 8095–102.
Jansen AP, Camalier CE, Colburn NH . Epidermal expression of the translation inhibitor programmed cell death 4 suppresses tumorigenesis. Cancer Res 2005; 65: 6034–41.
Yang HS, Matthews CP, Clair T, Wang Q, Baker AR, Li CC, et al. Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol Cell Biol 2006; 26: 1297–306.
Yang HS, Jansen AP, Nair R, Shibahara K, Verma AK, Cmarik JL, et al. A novel transformation suppressor, Pdcd4, inhibits AP-1 transactivation but not NF-kappaB or ODC transactivation. Oncogene 2001; 20: 669–76.
Chen Y, Knösel T, Kristiansen G, Pietas A, Garber ME, Matsuhashi S, et al. Loss of PDCD4 expression in human lung cancer correlates with tumour progression and prognosis. J Pathol 2003; 200: 640–6.
Afonja O, Juste D, Das S, Matsuhashi S, Samuels HH . Induction of PDCD4 tumor suppressor gene expression by RAR agonists, antiestrogen and HER-2/neu antagonist in breast cancer cells. Evidence for a role in apoptosis. Oncogene 2004; 23: 8135–45.
Zhang H, Ozaki I, Mizuta T, Hamajima H, Yasutake T, Eguchi Y, et al. Involvement of programmed cell death 4 in transforming growth factor-beta1-induced apoptosis in human hepatocellular carcinoma. Oncogene 2006; 25: 6101–12.
Cmarik JL, Min H, Hegamyer G, Zhan S, Kulesz-Martin M, Yoshinaga H, et al. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc Natl Acad Sci USA 1999; 96: 14037–42.
Yang HS, Knies JL, Stark C, Colburn NH . Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1 transactivation. Oncogene 2003; 22: 3712–20.
Yang HS, Cho MH, Zakowicz H, Hegamyer G, Sonenberg N, Colburn NH . A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol Cell Biol 2004; 24: 3894–906.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary information, Figure S1
Relative expression of P-gp and MRP1 in PC3wt and PC3R cells. (DOC 50 kb)
Supplementary information, Figure S2
The Caspase 3/7 activities in PC3R cells when silencing of miR-21 or ectopic expression of PDCD4. (DOC 33 kb)
Rights and permissions
About this article
Cite this article
Shi, Gh., Ye, Dw., Yao, Xd. et al. Involvement of microRNA-21 in mediating chemo-resistance to docetaxel in androgen-independent prostate cancer PC3 cells. Acta Pharmacol Sin 31, 867–873 (2010). https://doi.org/10.1038/aps.2010.48
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2010.48
Keywords
This article is cited by
-
Secreted miR-210-3p, miR-183-5p and miR-96-5p reduce sensitivity to docetaxel in prostate cancer cells
Cell Death Discovery (2023)
-
Engineered exosomes for targeted co-delivery of miR-21 inhibitor and chemotherapeutics to reverse drug resistance in colon cancer
Journal of Nanobiotechnology (2020)
-
Altered Expression of CD44, SIRT1, CXCR4, miR-21, miR-34a, and miR-451 Genes in MKN-45 Cell Line After Docetaxel Treatment
Journal of Gastrointestinal Cancer (2020)
-
Exosomes of pasteurized milk: potential pathogens of Western diseases
Journal of Translational Medicine (2019)
-
Human antigen R and drug resistance in tumors
Investigational New Drugs (2019)
